
Abstract
Pro-protein-convertase-subtilisin-kexin-9 (PCSK9) enhances the degradation of the low-density lipoprotein receptor (LDLR) that plays a major role in cholesterol homeostasis. Recent advances have revealed a large number of genetic variants of PCSK9 that may modulate plasma cholesterol levels either positively or negatively, therefore influencing the risk of atherosclerosis. Recognition of these mutants may have clinical implication in assessing severity of disease, prognosis, or response to drug therapy. PCSK9’s expression, secretion, and plasma levels maybe modulated by the proprotein convertase furin, by natural inhibitors (annexin-A2), or influenced by lipid-altering agents such as statins, fibrates, ezetimibe, and berberine. It is now a prime target for therapy, prompting the development of various approaches to reduce its LDLR degrading activity, including antibody neutralization, anti-sense oligonucleotides such as phosphorothioates, locked nucleic acids, and RNA interference, and eventually small molecule inhibitors. Which one will be clinically applicable will depend on long-term effects, cost, and ease of administration.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The discovery that autosomal co-dominant familial hypercholesterolemia (FH), in majority attributed to mutations of the LDL-receptor (LDLR) or more rarely of apolipoprotein (apo) B (dominant), could also be associated with mutations in the proprotein convertase-subtilisin/kexin type 9 (PCSK9) gene was a breakthrough [1]. The importance of these findings was amplified by the demonstration that PCSK9 acted by enhancing the degradation of the LDLR [2]. The demonstration that loss-of-function (LOF) mutations could be associated with low plasma cholesterol concentrations and protect against coronary artery disease (CAD), in contrast to the opposite effects for the gain-of-function (GOF) mutations [3], further kindled the interest in PCSK9. Altogether, these observations changed our clinical perspective for the diagnosis and management of FH but also added to our knowledge of the LDLR pathway and fostered a large number of research initiatives. This review focuses on recent advances of the influence of genetic variations of PCSK9 on LDL cholesterol (LDL-C) levels and on the risk and progression of atherosclerosis.
PCSK9 Protein and Gene Structure
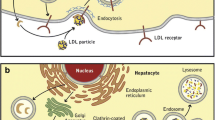
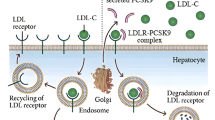
The unraveled structures of both the gene and the protein greatly enhanced our understanding of PCSK9’s role in cholesterol homeostasis [1, 4, 5]. The PCSK9 gene on the short arm of chromosome 1 (1p32.3) is 25-kb long and comprises 12 exons and 11 introns. It encodes a 692-amino acid serine protease PCSK9, formerly called NARC1 (for neural apoptosis regulated convertase-1), which is a member of the proprotein convertase family of enzymes. These convertases act as molecular scissors for the tissue-specific processing of multiple precursor proteins. The domain organization of PCSK9 is depicted in Fig. 1 [6, 7]. The N-terminal signal peptide necessary for secretion is followed by a pro-domain that inhibits the catalytic activity when cleaved. The catalytic domain, responsible for its autocatalytic intramolecular processing in the endoplasmic reticulum (ER) (the only known catalytic function of PCSK9), is followed by a cysteine-histidine rich domain (CHRD). After cleavage, the N-terminal inhibitory pro-segment remains associated with the catalytic domain of PCSK9 and forms a stable secreted heterodimeric pro-segment–PCSK9 complex. The catalytic domain contains the main binding structure of PCSK9 to the epidermal growth factor–like repeat A domain (EGF-A) of the LDLR. The major function of the heterodimeric pro-segment PCSK9 is to degrade the LDLR intracellularly and extracellularly, acting as a chaperone that binds to the LDLR to promote its lysosomal degradation [8].

The PCSK9 protein and mutations influencing plasma low-density lipoprotein cholesterol (LDL-C). The PCSK9 protein is in the center, the key amino acids of the catalytic pocket containing the nucleophiles aspartic acid (D), histidine (H), asparagine (N) and serine (S) are depicted; it takes an active conformation after cleavage of the pro-segment. The position and the amino acid substitutions of the mutations that are associated with high LDL-C levels are shown above the protein, those that are associated with a reduced plasma LDL-C are shown below. The color code indicates the origin of the mutation: France (blue), Great Britain (orange), Canada (green), Norway (red), Italy (dark red), Japan (purple), New Zealand (dark green), and Portugal (light blue). The mutations associated with low LDL-C reported by Lakoski et al. [18] in the Dallas Heart Study are shown in gray. Asterisk indicates that the E670G coding the single nucleotide protein reported in whites also influences LDL-C levels (increased with the rare GG genotype). (Data from Davignon and Dufour [6] and Leigh et al. [7]. See also the LOVD database at http://www.ucl.ac.uk/ldlr/Current/index.php?select_db=PCSK9)
In adults, PCSK9 is produced primarily by the liver and intestine, but only the liver releases it into the circulation [9]. It is degraded in plasma where it is cut at Arg218 by furin, another proprotein convertase, generating an inactive 474-amino acid protein [10]. The gene is polymorphic and a large number of LOF and GOF mutations have been reported in the past few years [7, 11] (also see http://www.ucl.ac.uk/ldlr/Current/index.php?select_db=PCSK9). The fact that lowered plasma LDL-C and apolipoprotein (apo) B from birth with LOF mutations in PCSK9 were associated with reduced coronary artery disease (CAD) risk in both whites and blacks [3] markedly increased the excitement for this protein.
PCSK9 Single Nucleotide Polymorphisms/Variants/Mutations Influencing LDL-C Levels and Atherosclerosis
To date, more than 50 amino acid variants of PCSK9 have been shown to affect plasma cholesterol levels in humans (Fig. 1) [4, 11, 12]. GOF mutations result in mild to severe hypercholesterolemia. In the most severe Anglo-Saxon mutation, D374Y, total cholesterol (TC) reaches values as high as 13.1 mmol/L (506 mg/dL) [13]. The onset of CAD in patients with D374Y maybe 10 years sooner than in heterozygous FH patients with severe LDLR mutations [14]. In a recent transgenic mouse study by Herbert et al. [15•], the expression of human PCSK9 gene (WT) or of the human D374Y mutant gene and their endogenous regulatory sequences at physiologic levels produced a phenotype that closely matched that found in heterozygous D374Y patients. Both transgenes increased serum cholesterol level and reduced hepatic LDLR protein; however, the effects were greater in D374Y mice. Extensive atherosclerotic plaques were found only in the D374Y on a high-cholesterol diet. They also secreted more triglyceride-rich lipoproteins into the circulation than WT mice, suggesting that reduced LDLR activity is not the sole cause of their hypercholesterolemia and propensity to dietary-induced atherosclerosis. This is the first study to use the endogenous PCSK9 promoter, and not apoE or albumin promoters as in previous studies, to drive PCSK9 D374Y expression.
On the other hand, a retrospective study has shown a significantly reduced risk of CAD in carriers of PCSK9 LOF variants R46L (partial LOF) and Y142X or C679X (complete LOF). Together, the latter two nonsense mutations were associated with a 28% reduction of plasma LDL-C and an 88% reduction in the frequency of coronary events in blacks, whereas in whites the R46L was associated with a 15% reduction in LDL-C and a 47% reduction in the risk of CAD [3]. Although that study supported the cardioprotective role of long-term reduction of cholesterol levels, a direct protective effect of reduced PCSK9 was not excluded. Furthermore, a compound heterozygous black American woman exhibiting two inactivating mutations (Y142X and ΔR97) in PCSK9 had strikingly low plasma levels of LDL-C (0.36 mmol/L; 14 mg/dL) and no immunodetectable circulating PCSK9 [12]. Another woman homozygous for the C679X mutation in Zimbabwe had a plasma LDL-C of 0.41 mmol/L (15.8 mg/dL) [16]. All these findings support the hypothesis that levels and/or higher activity of plasma PCSK9 modulate LDL-C and TC levels, suggesting that long-term lowering of PCSK9 might be beneficial in reducing the incidence of CAD [17]. Recently, the discovery of new mutations and methods to regulate PCSK9 led to the deeper analysis of the influence of PSCK9 on various lipid and metabolic parameters and to the probing of the therapeutic benefits of silencing PCSK9 in cardiovascular diseases at large.
Lakoski et al. [18] in a study of plasma PCSK9 in the large multi-ethnic population of the Dallas Heart study (n = 3,138) found that individuals with LOF mutations and reduced levels of LDL-C had significantly lower plasma levels of PCSK9 after adjusting for age and gender (P < 0.0001). A 100-ng/mL increase in plasma PCSK9 level was associated with a 4.5-mg/dL (0.12 mmol/L) increase in plasma LDL-C concentration in women and a 3.2-mg/dL (0.08 mmol/L) increase in LDL-C in men. Women had approximately 15% higher plasma concentrations than men and post-menopausal women higher levels than pre-menopausal women, regardless of estrogen status. They also observed that plasma triglycerides, insulin, and glucose correlated with plasma PCSK9. They did not find any relationship between apoE polymorphism and plasma concentrations of PCSK9. They observed the usual genotypic effect of increasing levels of LDL-C from E2/2 to E4/4 across a population subset of 3,096 individuals, but no difference among those genotypes for PCSK9 plasma levels. However, they did not look at the impact of PCSK9 variants on LDL-C as a function of variation in apoE. Two recent reports addressed this question.
Huang et al. [19••] investigated the associations of six PCSK9 variants with LDL-C over 20 years in 1,750 blacks and 1,828 whites from the Coronary Artery Risk Development In Young Adults (CARDIA) study. This longitudinal study of normal young adults aimed at studying development of cardiovascular disease risk factors revealed that for blacks, LDL-C levels at age 18 years were significantly lower (P < 0.001) among those with one of the three variants L253F, C679X, and Y142X, (81.5 mg/dL; 2.1 mmol/L) or A443T (95.5 mg/dL; 2.47 mmol/L) compared with non-carriers (109.6 mg/dL; 2.83 mmol/L). White carriers of the R46L variant had lower mean LDL-C concentrations at age 18 years (84.4 mg/dL; 2.18 mmol/L) than non-carriers (100.9 mg/dL; 2.6 mmol/L; P < 0.001), and the increase in LDL-C with age was similar to that of non-carriers. Importantly, the four cholesterol-lowering variants in black men were associated with lower carotid intima-media thickness and lower prevalence of coronary artery calcifications (CAC) measured once at year 20 in participants aged 38 to 50 years. Interestingly, the authors observed that apoE genotypes exerted their respective effects on LDL-C in an additive manner to that of the PCSK9 variants. This is the first demonstration of an apoE by PCSK9 LOF mutation interaction in a cohort of normal individuals and the relative protective effect of the apoE2 allele. The tracking of LDL-C over time in carriers of LOF mutations has been shown also in the Bogalusa Heart Study in a lower age range (4–38 years) in both whites (n = 1,086, R46L LOF) and blacks (n = 478, Y142X and C679X LOF) [20]. These results also showed that these LOF variants are associated with significantly lower LDL-C levels starting in childhood.
Norata et al. [21••] investigated the effects of two common PCSK9 mutations (E670G and I474V) on the intima media thickness of the common carotid artery and the possible relation to apoE polymorphism. Their database included 1,541 middle-aged individuals selected from the general population enrolled in the Progression of Lesion in the Intima of the Carotid artery (PLIC) study. They confirmed their major findings in 776 individuals from a second free-living population enrolled in the Ventimiglia in Sicilia study for whom a DNA sample was available. The E670G polymorphism of the PCSK9 gene was associated with increased IMT progression in the general population but the I474V was neutral. When the presence of 670G allele was stratified according to the apoE gene alleles, apoE2–PCSK9-670EE carriers showed a more favorable plasma lipid profile and decreased intima media thickness compared with the apoE4–PCSK9-670G carriers. This is the first demonstration of an apoE by PCSK9 gain-of-function mutation interaction in a general population and supports the relative protective effect of the apoE2 allele. It is consistent with the findings of Chen et al. [22] both for the effect of the E670E variation as part of a haplotype on LDL-C and on severity of CAD in the Lipoprotein Coronary Atherosclerosis Study (LCAS) in Houston.
There is no consensus, however, on the effect of the E670G mutation on plasma cholesterol or LDL-C levels and one should keep in mind that variation in the effect of PCSK9 on a lipid parameter maybe population-specific or influenced by other genetic or environmental factors. Hsu et al. [23] failed to find a cholesterol-raising effect of the E670G in a Taiwanese population sample, in contrast to the findings Norata [21••] observed in an Italian population. Moreover, in their observation, the polymorphism was associated with lower LDL-C levels. A gender effect may also be responsible for some discrepancies. Evans and Beil [24] determined that the PCSK9 670G allele was associated with increased LDL-C levels in men, but not in women, of European origin. The study of Kotowski et al. [11] failed to find such an association between plasma levels of LDL-C and the E670G substitution in the Dallas Heart Study. Finally, one must consider an age effect as a source of phenotypic modulation. Polisecki et al. [25], in the elderly population of the Prospective Study of Pravastatin in the Elderly (PROSPER) study (n = 5,783, 43% with a history of vascular disease at baseline), found significantly reduced LDL-C associated with the T allele of the G to T substitution accounting for the LOF R46L mutation (3.5% of sample; 10% reduction). There was a non-significant (19% unadjusted and 9% adjusted) decreased risk of vascular disease at baseline, with no on-trial effect. In this study, the 6% carriers of the E670G G allele did not exhibit a significant relationship with baseline LDL-C, response to pravastatin, or vascular disease risk.
Peripheral Vascular Disease
The impact of PCSK9 variation on peripheral arterial disease (PAD; defined by an ankle-brachial index <0.9 or a history of leg claudication) was evaluated by Folsom et al. [26•] in the Atherosclerosis Risk in Communities (ARIC) study, which assessed risk factors and PCSK9 variants Y142X and C679X (relevant to blacks) and R46L (relevant to whites) in a cohort of individuals 45 to 64 years of age in the years from 1987 to 1989 (n = 13,634). Greater LDL-C was a risk factor for prevalent and incident PAD. Carriers of one of the race-specific PCSK9 variants (2.4% of blacks and 3.1% of whites) had lower prevalence of PAD compared with non-carriers (2.3% vs 4.6%). The corresponding age-adjusted and sex-adjusted odds ratio of PAD was 0.47 (95% CI, 0.24–0.92). In contrast with the cross-sectional findings, however, there was no association between PCSK9 variants and incident PAD, providing only mixed evidence that variation in PCSK9 may contribute to genetic risk of PAD. A longitudinal study focused on patients with established PAD in a vascular clinic with extensive assessment of severity of disease might help to clarify this issue.
Cerebrovascular Disease
Two independent studies, one Belgian and one Finnish, were combined to obtain clinical and autopsy data [27]. They provided evidence that PCSK9 variations associate with the risk of large-vessel atherosclerosis (LVA) stroke subtype and suggest that the risk is mediated by the severity of intracranial atherosclerosis. The Belgium Stroke Study included 237 middle-aged (45–60 years of age) patients with small-vessel occlusion (SVO) and large-vessel atherosclerosis stroke (LVA), and 326 gender-matched and ethnicity-matched controls (>60 years of age) without a history of stroke. In multi-variate analysis, the minor allele (G) of the E670G PCSK9 polymorphism was a significant predictor of LVA (odds ratio = 3.52; 95% CI, 1.25–9.85; P = 0.017). In the Finnish cross-sectional population-based study of consecutive autopsy series of 604 men and women (mean age of 62.5 years), the G-allele carriers tended to have more severe allele copy number–dependent (P = 0.095) atherosclerosis in the circle of Willis and in its branches. The genetic variation is not associated with small vessel occlusion, a phenomenon that is unrelated to atherosclerosis. LDL-C values were not reported. These findings support the involvement of the E670G in determining serum lipids (higher frequency of hyperlipidemia in affected) and atherosclerosis (gene–dosage effect).
PCSK9 Plasma Levels Modulators
Because PCSK9 degrades the LDLR, contributes to cholesterol homeostasis, and may impact on atherosclerotic vascular disease, it is a potential therapeutic target [4]. This notion has prompted a major effort to identify natural and synthetic agents that affect the PCSK9 gene and its protein expression and metabolism. In human studies, the accent was put recently on plasma levels and effects of lipid-lowering agents.
Natural Modulators
To date, two natural modulators have been shown to interact with PCSK9. As mentioned previously, furin is capable of cleaving PCSK9 at residue Arg218, resulting in an inhibition of PCSK9-induced degradation of the LDLR [10]. Recently, Mayer et al. [28] demonstrated that the C-terminal Cys-His rich domain (CHRD) of PCSK9 interacts specifically with the N-terminal repeat R1 of annexin-A2 (AnxA2) and inhibits the degradation of the LDLR induced by PCSK9, thereby identifying the first natural inhibitor of PCSK9. AnxA2 is a calcium-dependent phospholipid-binding protein whose function is to help organize exocytosis of intracellular proteins to the extracellular domain and that is involved in pleiotropic functions such as cell motility, linkage of membrane-associated protein complexes to the actin cytoskeleton, endocytosis, fibrinolysis, ion channel formation, and cell matrix interactions [29]. Mayer et al. [28] reported that the LOF mutant Q554E binds at least threefold better to the R1 repeat of AnxA2, leading to a tighter binding of PCSK9 to AnxA2, resulting in loss of ability of PCSK9 to induce the degradation of the LDLR. This discovery should prompt the search for mutations of annexin-A2 in orphan forms of inherited hypercholesterolemia and should pave the way toward the development of PCSK9 inhibitory molecules [5].
Artificial Modulators and Therapeutic Agents
Dubuc et al. [30] previously demonstrated that PCSK9 mRNA is upregulated in vitro by statins via a pathway involving reduced intracellular cholesterol concentration and increased SREBP-2 levels [31, 32]. Recently, in a cross-sectional study, Dubuc et al. [33••] evaluated plasma levels of PCSK9 and their relationship with plasma lipids and lipoproteins in normal (n = 254) and hypercholesterolemic (n = 200) individuals treated or not treated with statins or a statin plus ezetimibe (two hypocholesterolemic agents with complementary modes of action). In hypercholesterolemic participants, PCSK9 levels were higher than in controls (99.3 ± 31.7 ng/mL; P < 0.04) and increased in proportion to the statin dose, combined or not with ezetimibe. In treated patients (n = 139), those with FH due to LDL receptor gene mutations had higher PCSK9 values than patients without FH (147.01 ± 42.5 ng/mL vs 127.2 ± 40.8 ng/mL; P < 0.005). This finding is in line with the observation of Careskey et al. [34] that plasma PCSK9 increases as LDL-C goes down during 16 weeks of atorvastatin therapy. Dubuc et al. [33••] also showed that plasma PCSK9 is further increased when ezetimibe and statins are used in combination. However, studies examining the effect of another common lipid-lowering medication, fenofibrate, on PCSK9 plasma levels seem to be contradictory. Troutt et al. [35] studied the effect of fenofibrate on circulating PCSK9 protein levels in patients with an atherogenic dyslipidemia treated with fenofibrate or placebo for 12 weeks. They observed that fenofibrate (200 mg/d) significantly increased circulating PCSK9 levels by 25% compared with baseline. Interestingly, fenofibrate-induced increases in serum PCSK9 levels were highly correlated with fenofibrate-induced changes in HDL-C and triglyceride levels, as well as with fenofibrate-induced changes in LDL-C levels. Mayne et al. [36] also observed an increase in PCSK9 plasma level in 19 patients on the American Heart Association NCEP step-1 diet after a 24-week treatment with gemfibrozil or fenofibrate (17.01%; P = 0.031). In contrast, Lambert et al. [37] measured circulating PCSK9 concentrations in 115 diabetic patients from the Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study before and after a 6-week fenofibrate treatment and found an 8.5% decrease (P = 0.041 vs pretreatment). These divergent findings are difficult to reconcile. In vitro studies with immortalized hepatocytes by Kourimate et al. [38] showing that fenofibric acid represses PCSK9 both by reducing PCSK9 expression and increasing furin expression (a PCSK9 inhibitor) are in favor of the lowering effect. Differences in methods and the patient subset studied might contribute to better understanding.
Berberine
Berberine (BBR), is a new cholesterol-lowering agent derived from a Chinese herb (Coptis sinensis) that upregulates LDLR expression via mRNA stabilization, implicating novel regulatory proteins located downstream of the extracellular signal regulated kinase (ERK) pathway and able to interact with sequences in the proximal section of the LDLR mRNA 3′ untranslated region (UTR) [39]. Cameron et al. [40] reported that in HepG2 cells, berberine reduces PCSK9 expression and secretion in a dose-dependent manner. Also, it suppresses the PCSK9 mRNA–raising effect of mevastatin while increasing its LDLR-raising effect. In hepatic cell lysates, however, berberine and mevastatin both increase LDLR protein, acting in synergy when combined. Recently, Li et al. [41•] identified the mRNA binding proteins that regulate the stability of LDLR mRNA induced by berberine through AU-rich (ie, a region with frequent A and U bases in mRNA) elements. Heterogeneous nuclear ribonucleoprotein D (hnRNP D), hnRNP I, and KH-type splicing regulatory protein (KSRP) were found to be key modulators of LDLR mRNA stability in liver cells. These interact with AU-rich elements of the LDLR 3′UTR with sequence specificity. Silencing the expression of these proteins increased LDLR mRNA and protein levels. This work provides new insight on the regulation of LDLR gene expression at the post-transcriptional level. Importantly, Li et al. [42••], in studying mechanisms underlying the transcriptional suppression of PCSK9 by berberine in HepG2 cells, also identified a highly conserved hepatocyte nuclear factor 1-α (HNF1α) binding site upstream from the sterol regulatory element (SRE) as a critical sequence motif for PCSK9 transcription and its regulation by berberine. They discovered that HNF1α, from among an array of transcription factors, was the predominant transactivator of PCSK9, and mutation of its binding site reduced PCSK9 activity by more than 90%. They found that HNF1α works cooperatively with SRE, as HNF1α site mutations significantly attenuated the activity of nuclear SREBP-2 to transactivate the PCSK9 promoter. A coordinate modest reduction of HNF1α and nuclear SREBP-2 by berberine led to a strong suppression of PCSK9 transcription through these two critical regulatory sequences. These findings further improve our understanding of the role of PCSK9 in cholesterol homeostasis and of the regulatory networks that modulate its transcription. It provides a new tool for the development of selective repressors of PCSK9 via inhibition of its transcriptional enhancers and may help to improve statin efficacy.
Interfering Antibody, RNA Interference, and Anti-Sense Oligonucleotides
PCSK9 deficiency causes a 40% to 50% decrease in total plasma cholesterol and a threefold increase in hepatic LDLR in mice [9, 43, 44]. These observations and the lifetime reduction of LDL-C in individuals with PCSK9 LOF mutations reported above suggest that pharmacologic inhibition of PCSK9 would result in increased LDLR activity and enhanced plasma LDL-C clearance. This hypothesis is reinforced by the demonstration of a hypersensitivity to statin in PCSK9-deficient mice, suggesting a possible synergic interaction between a statin’s cholesterol-lowering effects and an eventual PCSK9 inhibitor [44]. Recently, inhibition of PCSK9 expression using anti-sense oligonucleotides (ASO) [45], locked nucleic acids [46], and RNA interference (RNAi) [47], or inhibition of PCSK9 activity on LDLR using neutralizing antibodies [48] was tested with success in animals models, including mice and monkeys, and a very recent early report evaluated a PCSK9 mAb in humans, with positive results on LDL-C lowering (http://www.fiercebiotech.com/press-releases/regeneron-provides-initial-data-two-antibody-product-candidates-0).These therapeutic approaches have been reviewed recently [5]. ASO administered in subcutaneous injections in mice on a fat-rich diet during 6 weeks provoked a 90% reduction in PCSK9 mRNA, a 53% reduction in total plasma cholesterol, and a 38% reduction in LDL-C [45]. This was the first demonstration that injectable ASOs could be a useful therapeutic approach to reduce PCSK9 hepatic levels and as a possible complement to statin treatment. Moreover, RNAi in injectable lipidoid nanoparticles (LNP) dispensed in mice and rat produced a 50% to 70% reduction in PCSK9 mRNA and significant LDL-C reductions in cynomolgus monkeys without affecting HDL-C or TG levels [47]. This gene-silencing effect lasted for 3 weeks. Finally, smaller ASOs from the “locked nucleic acid” technology have shown safety and efficacy in mice and non-human primates, and a neutralizing effect that lasts for 21 days with an injection twice weekly [46]. These results are very promising and studies in humans are undergoing.
PCSK9 Polymorphisms and Response to Statin Therapy
Statins are effective at lowering LDL-C and reducing cardiovascular disease risk, but variability in response is not well understood. To address this, Thompson et al. [49] genotyped 5,745 individuals from the Treating to New Targets (TNT) trial in a candidate gene approach to identify associations with response to atorvastatin treatment. Twenty-three candidate genes previously associated with statin response were analyzed and three single nucleotide polymorphisms in apoE were most highly associated with LDL-C response, followed by one in PCSK9 with a similar effect size. Single nucleotide polymorphisms in HMGCR were also significant, although the effect was less than with those in apoE and PCSK9. rs7412/apoE had the most significant association (P = 6 × 10(−30)), and its high significance in the whole-genome study (P = 4 × 10(−9)) confirmed the suitability of this population for detecting effects. Age and gender were found to influence LDL-C response to a similar extent as the most pronounced genetic effect. This study confirmed previous results of Berge et al. [50], suggesting that a disruption in the normal function of PCSK9 could result in increased number of LDLRs and improved response to statin therapy. The authors screened 38 unrelated hypocholesterolemic individuals as well as 25 unrelated familial hypercholesterolemia (FH) heterozygotes who responded particularly well to statin therapy for mutations in the 12 exons of the PCSK9 gene by DNA sequencing. Six of the 38 (15.8%) hypocholesterolemic individuals were heterozygous for one of the three mutations R46L, G106R, or R237W in PCSK9. In the group of 25 FH heterozygotes who responded particularly well to statin therapy, three (8.8%) were heterozygous for mutations R46L or N157K in PCSK9.
Conclusions
The discovery of PCSK9 has stimulated a large number of research initiatives, resulting in an improved understanding of LDLR biology and cholesterol homeostasis. PCSK9 is becoming a hub in a network of metabolic interactions that are just beginning to be unraveled. The field is expanding exponentially as the focus moves from mechanisms to clinical and therapeutic applications. This review, which highlights but a few of the recent advances in PCSK9 physiology and pathology, is a witness to the growing interest for and importance of this field.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Abifadel M, Varret M, Rabes JP, et al.: Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet 2003, 34:154–156.
Maxwell KN, Breslow JL: Adenoviral-mediated expression of Pcsk9 in mice results in a low-density lipoprotein receptor knockout phenotype. Proc Natl Acad Sci U S A 2004, 101:7100–7105.
Cohen JC, Boerwinkle E, Mosley TH Jr, Hobbs HH: Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med 2006, 354:1264–1272.
Seidah NG, Prat A: The proprotein convertases are potential targets in the treatment of dyslipidemia. J Mol Med 2007, 85:685–696.
Seidah NG: PCSK9 as a therapeutic target of dyslipidemia. Expert Opinion on Therapeutic Targets 2009, 13:19–28.
Davignon J, Dufour R: Primary Hyperlipidemias. Oxford: Clinical Publishing; 2007.
Leigh SE, Leren TP, Humphries SE: Commentary PCSK9 variants: a new database. Atherosclerosis 2009, 203:32–33.
Poirier S, Mayer G, Poupon V, et al.: Dissection of the endogenous cellular pathways of PCSK9-induced low density lipoprotein receptor degradation: evidence for an intracellular route. J Biol Chem 2009, 284:28856–28864.
Zaid A, Roubtsova A, Essalmani R, et al.: Proprotein convertase subtilisin/kexin type 9 (PCSK9): hepatocyte-specific low-density lipoprotein receptor degradation and critical role in mouse liver regeneration. Hepatology 2008, 48:646–654.
Benjannet S, Rhainds D, Hamelin J, et al.: The proprotein convertase (PC) PCSK9 is inactivated by furin and/or PC5/6A: functional consequences of natural mutations and post-translational modifications. J Biol Chem 2006, 281:30561–30572.
Kotowski IK, Pertsemlidis A, Luke A, et al.: A spectrum of PCSK9 alleles contributes to plasma levels of low-density lipoprotein cholesterol. Am J Hum Genet 2006, 78:410–422.
Zhao Z, Tuakli-Wosornu Y, Lagace TA, et al.: Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet 2006, 79:514–523.
Timms KM, Wagner S, Samuels ME, et al.: A mutation in PCSK9 causing autosomal-dominant hypercholesterolemia in a Utah pedigree. Hum Genet 2004, 114:349–353.
Naoumova RP, Tosi I, Patel D, et al.: Severe hypercholesterolemia in four British families with the D374Y mutation in the PCSK9 gene: long-term follow-up and treatment response. Arterioscler Thromb Vasc Biol 2005, 25:2654–2660.
• Herbert B, Patel D, Waddington SN, et al.: Increased secretion of lipoproteins in transgenic mice expressing human D374Y PCSK9 under physiological genetic control. Arterioscler Thromb Vasc Biol 2010, 30:1333–1339. This is the first study to use the endogenous promoter to drive PCSK9 D374Y mutant expression and reproduce in mice the human phenotype of this highly deleterious defect.
Hooper AJ, Marais AD, Tanyanyiwa DM, Burnett JR: The C679X mutation in PCSK9 is present and lowers blood cholesterol in a Southern African population. Atherosclerosis 2007, 193:445–448.
Brown MS, Goldstein JL: Biomedicine. Lowering LDL—not only how low, but how long? Science 2006, 311:1721–1723.
Lakoski SG, Lagace TA, Cohen JC, et al.: Genetic and metabolic determinants of plasma PCSK9 levels. J Clin Endocrinol Metab 2009, 94:2537–2543.
•• Huang CC, Fornage M, Lloyd-Jones DM, et al.: Longitudinal association of PCSK9 sequence variations with low-density lipoprotein cholesterol levels: the Coronary Artery Risk Development in Young Adults Study. Circ Cardiovasc Genet 2009, 2:354–361. This longitudinal study of normal young adults demonstrates the impact of LOF mutations on CAD risk factors in blacks and whites, shows the tracking with time of the cholesterol-lowering effect, and reveals an apoE by PCSK9 LOF mutation interaction.
Hallman DM, Srinivasan SR, Chen W, et al.: Relation of PCSK9 mutations to serum low-density lipoprotein cholesterol in childhood and adulthood (from The Bogalusa Heart Study). Am J Cardiol 2007, 100:69–72.
•• Norata GD, Garlaschelli K, Grigore L, et al.: Effects of PCSK9 variants on common carotid artery intima media thickness and relation to ApoE alleles. Atherosclerosis 2010, 208:177–182. This work demonstrates that E670G polymorphism of the PCSK9 gene is associated with increased intima media thickness progression in the general population and reveals an interaction with apoE polymorphism.
Chen SN, Ballantyne CM, Gotto AM Jr, et al.: A common PCSK9 haplotype, encompassing the E670G coding single nucleotide polymorphism, is a novel genetic marker for plasma low-density lipoprotein cholesterol levels and severity of coronary atherosclerosis. J Am Coll Cardiol 2005, 45:1611–1619.
Hsu LA, Teng MS, Ko YL, et al.: The PCSK9 gene E670G polymorphism affects low-density lipoprotein cholesterol levels but is not a risk factor for coronary artery disease in ethnic Chinese in Taiwan. Clin Chem Lab Med 2009, 47:154–158.
Evans D, Beil FU: The E670G SNP in the PCSK9 gene is associated with polygenic hypercholesterolemia in men but not in women. BMC Med Genet 2006, 7:66.
Polisecki E, Peter I, Robertson M, et al.: Genetic variation at the PCSK9 locus moderately lowers low-density lipoprotein cholesterol levels, but does not significantly lower vascular disease risk in an elderly population. Atherosclerosis 2008, 200:95–101.
• Folsom AR, Peacock JM, Boerwinkle E: Variation in PCSK9, low LDL cholesterol, and risk of peripheral arterial disease. Atherosclerosis 2009, 202:211–215. This work reports an association of peripheral arterial disease prevalence, but not incidence, with PCSK9 LOF mutations.
Abboud S, Karhunen PJ, Lutjohann D, et al.: Proprotein convertase subtilisin/kexin type 9 (PCSK9) gene is a risk factor of large-vessel atherosclerosis stroke. PLoS ONE 2007, 2:e1043.
Mayer G, Poirier S, Seidah NG: Annexin A2 is a C-terminal PCSK9-binding protein that regulates endogenous low density lipoprotein receptor levels. J Biol Chem 2008, 283:31791–31801.
Hayes MJ, Longbottom RE, Evans MA, Moss SE: Annexinopathies. Subcell Biochem 2007, 45:1–28.
Dubuc G, Chamberland A, Wassef H, et al.: Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol 2004, 24:1454–1459.
Maxwell KN, Soccio RE, Duncan EM, et al.: Novel putative SREBP and LXR target genes identified by microarray analysis in liver of cholesterol-fed mice. J Lipid Res 2003, 44:2109–2119.
Horton JD, Shah NA, Warrington JA, et al.: Combined analysis of oligonucleotide microarray data from transgenic and knockout mice identifies direct SREBP target genes. Proc Natl Acad Sci U S A 2003, 100:12027–12032.
•• Dubuc G, Tremblay M, Pare G, et al.: A new method for measurement of total plasma PCSK9: clinical applications. J Lipid Res 2010, 51:140–149. This work explores clinical applications of a new method that measures total plasma PCSK9 and demonstrates an additive effect of ezetimibe in raising plasma levels of PCSK9.
Careskey HE, Davis RA, Alborn WE, et al.: Atorvastatin increases human serum levels of proprotein convertase subtilisin/kexin type 9. J Lipid Res 2008, 49:394–398.
Troutt JS, Alborn WE, Cao G, Konrad RJ: Fenofibrate treatment increases human serum proprotein convertase subtilisin kexin type 9 levels. J Lipid Res 2010, 51:345–351.
Mayne J, Dewpura T, Raymond A, et al.: Plasma PCSK9 levels are significantly modified by statins and fibrates in humans. Lipids Health Dis 2008, 7:22.
Lambert G, Ancellin N, Charlton F, et al.: Plasma PCSK9 concentrations correlate with LDL and total cholesterol in diabetic patients and are decreased by fenofibrate treatment. Clin Chem 2008, 54:1038–1045.
Kourimate S, Le May C, Langhi C, et al.: Dual mechanisms for the fibrate-mediated repression of proprotein convertase subtilisin/kexin type 9. J Biol Chem 2008, 283:9666–9673.
Abidi P, Zhou Y, Jiang JD, Liu J: Extracellular signal-regulated kinase-dependent stabilization of hepatic low-density lipoprotein receptor mRNA by herbal medicine berberine. Arterioscler Thromb Vasc Biol 2005, 25:2170–2176.
Cameron J, Ranheim T, Kulseth MA, et al.: Berberine decreases PCSK9 expression in HepG2 cells. Atherosclerosis 2008, 201:266–273.
• Li H, Chen W, Zhou Y, et al.: Identification of mRNA binding proteins that regulate the stability of LDL receptor mRNA through AU-rich elements. J Lipid Res 2009, 50:820–831. This work identifies mRNA binding proteins that regulate the stability of LDLR mRNA induced by berberine through AU-rich elements, helping to explain its mechanism of action.
•• Li H, Dong B, Park SW, et al.: Hepatocyte nuclear factor 1α plays a critical role in PCSK9 gene transcription and regulation by the natural hypocholesterolemic compound berberine. J Biol Chem 2009, 284:28885–28895. This study identifies HNF1α as a critical sequence motif for PCSK9 transcription and its regulation by berberine and reveals coordinate interactions between HNF1, SRE, and SREBP-2 in transactivation of the PCSK9 promoter.
Park SW, Moon YA, Horton JD: Post-transcriptional regulation of low density lipoprotein receptor protein by proprotein convertase subtilisin/kexin type 9a in mouse liver. J Biol Chem 2004, 279:50630–50638.
Rashid S, Curtis DE, Garuti R, et al.: Decreased plasma cholesterol and hypersensitivity to statins in mice lacking Pcsk9. Proc Natl Acad Sci U S A 2005, 102:5374–5379.
Graham MJ, Lemonidis KM, Whipple CP, et al.: Antisense inhibition of proprotein convertase subtilisin/kexin type 9 reduces serum LDL in hyperlipidemic mice. J Lipid Res 2007, 48:763–767.
Gupta N, Fisker N, Asselin MC, et al.: A locked nucleic acid antisense oligonucleotide (LNA) silences PCSK9 and enhances LDLR expression in vitro and in vivo. PLoS ONE 2010, 5:e10682.
Frank-Kamenetsky M, Grefhorst A, Anderson NN, et al.: Therapeutic RNAi targeting PCSK9 acutely lowers plasma cholesterol in rodents and LDL cholesterol in nonhuman primates. Proc Natl Acad Sci U S A 2008, 105:11915–11920.
Chan JC, Piper DE, Cao Q, et al.: A proprotein convertase subtilisin/kexin type 9 neutralizing antibody reduces serum cholesterol in mice and nonhuman primates. Proc Natl Acad Sci U S A 2009, 106:9820–9825.
Thompson JF, Hyde CL, Wood LS, et al.: Comprehensive whole-genome and candidate gene analysis for response to statin therapy in the Treating to New Targets (TNT) cohort. Circ Cardiovasc Genet 2009, 2:173–181.
Berge KE, Ose L, Leren TP: Missense mutations in the PCSK9 gene are associated with hypocholesterolemia and possibly increased response to statin therapy. Arterioscler Thromb Vasc Biol 2006, 26:1094–1100.
Disclosure
No potential conflicts of interest relevant to this article were reported.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Davignon, J., Dubuc, G. & Seidah, N.G. The Influence of PCSK9 Polymorphisms on Serum Low-Density Lipoprotein Cholesterol and Risk of Atherosclerosis. Curr Atheroscler Rep 12, 308–315 (2010). https://doi.org/10.1007/s11883-010-0123-6
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11883-010-0123-6