
Abstract
Proprotein convertase subtilisin/kexin type 9 (PCSK9) is a serine protease that binds to low-density lipoprotein receptors (LDL-Rs), leading to their accelerated degradation and increased low-density lipoprotein cholesterol (LDL-C) levels. Therefore, PCSK9 levels play a critical role in cholesterol metabolism by reducing LDL-R levels and thus increasing levels of plasma LDL-C. Recently, investigational agents inhibiting PCSK9 have been shown to lower LDL-C and also, potentially, an important secondary target, lipoprotein(a). Therefore, several pharmaceutical companies have initiated drug-development programs that target PCSK9 and are built on a solid foundation of basic science, genetic studies, and epidemiological observations. PCSK9 inhibition with monoclonal antibodies demonstrated LDL-C lowering of up to 57 % when the PCSK9 antibodies are used as monotherapy and up to 73 % when added to background lipid-lowering therapy. In addition, long-term cardiovascular outcome studies are currently under way to confirm the longer term safety and efficacy of PCSK9 inhibitors and to determine whether PCSK9 inhibition lowers the incidence of major cardiovascular events. PCSK9 inhibitors may provide safe and effective lipid-lowering therapy, especially for patients with inadequate LDL-C lowering on lipid-lowering treatments, those who are statin intolerant or have contraindications to statin therapy, and those with hereditary hypercholesterolemia/familial hypercholesterolemia and severely elevated LDL-C.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cholesterol is synthesized in the liver via the rate-limiting enzyme 3-hydroxy-3-methyl-glutaryl-CoA reductase. It is then transported in the blood in lipoprotein particles such as low-density lipoprotein (LDL), high-density lipoprotein (HDL), and lipoprotein(a) [Lp(a)]. Of these, LDL proteins are the primary carriers of cholesterol in the blood. Elevated levels of LDL cholesterol (LDL-C) have been clearly associated with the leading cause of death and disability in the United States and in developed societies worldwide—atherosclerotic cardiovascular disease (ASCVD)—specifically, coronary artery disease and associated vascular diseases [1–4]. Further, levels of LDL-C have a curvilinear association with coronary heart disease (CHD) events [5, 6].
Given the favorable effects demonstrated in large cardiovascular outcomes trials (primarily with statins) on ASCVD morbidity and mortality [7], LDL-C reduction is the primary therapeutic target to decrease cardiovascular (CV) events in patients with hypercholesterolemia. Moreover, the proven beneficial effects of statins on LDL-C reduction are closely linked to an increased number of LDL receptors (LDL-R) [8].
Additionally, elevated Lp(a) has been shown to be a risk factor for ASCVD and as such is a potential target for lipid-lowering therapy [9, 10]. However, conventional treatments with statins and other lipid-lowering pharmacotherapy have either limited or no efficacy in lowering Lp(a) levels, or, in the case of therapies such as mipomersen or lomitapide, have side effect profiles that limit their use [11].
First identified in 2003 by Seidah and colleagues, the serine protease proprotein convertase subtilisin/kexin type 9 (PCSK9) has been associated with LDL-R and serum LDL-C levels [12–14]. This review summarizes the discovery of PCSK9, its function, and the potentially significant role of drugs that inhibit PCSK9 as possible new, effective agents for lipid lowering.
Interaction Between PCSK9 and LDL-C Metabolism: Discovery, Basic Science, and Pathophysiology
PCSK9 was discovered slightly more than a decade ago [14]. The investigation into the presence and function of PCSK9 has become an exciting example of the rapid translation of basic science from the bench to clinical investigation, and perhaps, in the future, to evidence-based pharmacotherapy.
PCSK9 is synthesized in the liver and then released into the blood, where it plays a central role in the life cycle and number of LDL-Rs, which function in the regulation of plasma LDL-C levels [15]. Expressed on the surface of hepatocytes, LDL-Rs bind LDL particles, removing them from the circulation. The LDL–LDL-R complex is internalized by the cell where the molecules separate and the LDL-R is recycled to the cell surface, the LDL protein is degraded, and the cholesterol that is released is stored and used in the cell. The recycling of the LDL-R to the hepatocyte surface is crucial to maintaining plasma LDL-C levels.
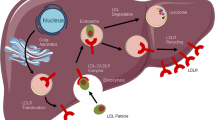
The PCSK9 enzyme binds to LDL-Rs on the surface of the hepatocyte in such a manner that PCSK9 and associated LDL-Rs cannot detach [15]. The entire PCSK9–LDL-R complex is internalized and subsequently degraded in lysosomes. Therefore, PCSK9 functions to prevent the recycling of the LDL-Rs, decreasing the number of LDL-Rs on the surface of hepatocytes and reducing the capacity of hepatocytes to bind and bring into the liver more LDL particles (Fig. 1). Consequently, the rate of LDL-C clearance from the blood is decreased, leading to higher measurable LDL-C levels.

LDL-C metabolism in the presence or absence of PCSK9 [15]. a. PCSK9 is synthesized in the liver and circulates in the blood. Upon binding to LDL-Rs on hepatocytes in complex with LDL, the entire complex is internalized and undergoes degradation in lysosomes, leading to a decreased number of LDL-Rs on the cell surface b. Monoclonal antibodies (mAbs) can bind to PCSK9, preventing it from binding to LDL-R. The LDL–LDL-R complex is endocytosed, but (in the absence of PCSK9) LDL-R is not degraded and is recycled back to the surface of the cell. LDL, low-density lipoprotein; LDL-C, LDL cholesterol; LDL-R, LDL receptor; PCSK9, proprotein convertase subtilisin/kexin type 9. Figure adapted from Catapano AL, Papadopoulos N. The safety of therapeutic monoclonal antibodies: implications for cardiovascular disease and targeting the PCSK9 pathway. Atherosclerosis. 2013:228(1);18–28. Under a Creative Commons license, http://creativecommons.org/licenses/by-nc-nd/3.0/legalcode. Minor deletions were made
Observations Demonstrating the Association Between PCSK9 Levels and LDL-C Levels
Mutations in PCSK9 appear to be directly related to levels of LDL-C. Abifadel and colleagues demonstrated that in French families with autosomal-dominant hypercholesterolemia (ADH), one of two gain-of-function (GOF) mutations is present in the PCSK9 gene [16]. Several additional GOF mutations have subsequently been identified in patients with ADH [17]. Studies suggest that although patients with ADH and GOF PCSK9 mutations respond to statins, they may be more refractory to statin therapy [18, 19]. Although the GOF PCSK9 mutations appear to be rare in patients with ADH compared with patients who have mutations in LDL-R and apolipoprotein B (apo B) [17], they have been reported in up to 12.5 % of patients with ADH [16]). Together, these observations established the relationship between PCSK9 (and, subsequently, lower availability of LDL-Rs) and hypercholesterolemia.
Subsequent work by Cohen and colleagues identified two nonsense, loss-of-function (LOF) PCSK9 mutations prevalent in approximately 2 % of African Americans with low plasma LDL-C levels in the Dallas Heart Study (DHS) [20]. Although rare in European (white) Americans (<0.1 %), these mutations were associated with a 40 % reduction in LDL-C levels, presumably because of increased levels of LDL-Rs. An association of LOF mutation in the PCSK9 gene, low LDL-C level, and decreased CHD was observed during 15 years of follow-up in the Atherosclerosis Risk In Communities (ARIC) study [21]. African American individuals with the nonsense LOF mutations in PCSK9 had 28 % lower LDL-C and a marked 88 % lower incidence in coronary disease compared with individuals who did not have the mutations. A third LOF PCSK9 mutation, a missense mutation, has been shown to be more predominant in white Americans than in African Americans. White Americans with this missense mutation had reduced LDL-C levels and CHD risk (15 and 47 %, respectively) compared with those without the mutation, albeit to less robust levels than the nonsense mutations in the African Americans. This degree of lessened CHD risk is greater than the previously observed magnitude of ASCVD reduction seen with similar levels of LDL-C lowering with statins documented in clinical trials. Therefore, the ARIC findings reflect the potent beneficial effects of lifelong low levels of LDL-C seen with PCSK9 LOF mutations. This profound association of an increase or decrease in CHD risk with PCSK9 GOF or LOF mutations, respectively, provides confirmation of the basic science and the observational data with respect to PCSK9.
Thus far, the LOF PCSK9 mutations linked to increased LDL-R levels have not only been consistently related to low levels of LDL-C but also appear not to be associated with known adverse clinical sequelae. Persons homozygous for LOF PCSK9 mutations appear to live a normal, functional life, free of obvious concomitant diseases or limitations. Participants in the DHS with very low levels of LDL-C were identified and found to have another genetic mutation associated with PCSK9 deficiency [22]. Their lifelong reduction in LDL-C also translated into a profoundly decreased risk for cardiovascular disease (CVD). An individual identified with two mutant copies of PCSK9 gene and LDL-C <20 mg/dL was apparently healthy, giving birth to two children [22, 23]. These findings indicate that lowering PCSK9 (and thus obtaining extremely low LDL-C levels) may be safe, and they emphasize the importance of future studies. Interestingly, in persons with extremely low levels of LDL-C who live in nonindustrialized, hunter-gatherer societies, in patients with genetic hypolipidemia (ie, hypobetalipoproteinemia), and in neonates, typical serum LDL-C levels of 20 to 70 mg/dL are generally well tolerated, with little or no demonstrable ASCVD [24, 25]. In consideration of the fact that new investigational PCSK9 inhibitory agents may lead to very low levels of LDL-C, it is important to confirm the safety of these low LDL-C states.
New 2013 American College of Cardiology/American Heart Association Cholesterol Guidelines and Intensive LDL-C Reduction: Is There a Place for PCSK9 Inhibition?
Based exclusively on the randomized clinical trial evidence, the recent 2013 American College of Cardiology/American Heart Association (ACC/AHA) cholesterol guidelines propose that statin therapy should be the primary and appropriate means to intensively treat elevated LDL-C and CVD risk in patients with hypercholesterolemia [26]. Therefore, the 2013 guidelines recommend the use of statins to consistently reduce LDL-C and decrease CVD.
A potentially limiting factor for the efficacy of statin therapy is an associated increase in PCSK9 levels [27]. The higher levels of PCSK9 in patients receiving statins may limit the degree to which these drugs lower LDL-C. This may partially explain why doubling the doses of conventionally prescribed statins only lowers LDL-C levels by an additional 6 % [28]. PCSK9 inhibition has, therefore, the possibility of removing this barrier and allowing more robust LDL-C lowering in combination with statins. Furthermore, some patients experience limited LDL-C responses to statin therapy, creating another possible area of application for PCSK9 inhibition. Hence, in the future, patients with suboptimal responses to statins and those who are considered statin intolerant may benefit from LDL-reducing therapy with PCSK9 inhibition.
The consistent evidence from a wide range of observational and clinical data demonstrates that lower LDL-C is better for protection against ASCVD. Simply prescribing intensive statin therapy in high-risk individuals, as recommended in the 2013 ACC/AHA guidelines, may not be adequate for predicting effective LDL-C lowering in real-world clinical practice. The guidelines also recommended a ≥50 % reduction in LDL-C levels as an indicator of adequacy of LDL-C–lowering therapy for high-risk patients [26]. A greater LDL-C–lowering effect may be necessary for these patients to obtain sufficient ASCVD event risk reduction. Nonstatins that have been shown in clinical trials to reduce ASCVD events with a good margin of safety are preferred.
In terms of LDL-C lowering, a recent meta-analysis of eight randomized, controlled statin trials among 38,153 patients demonstrated a large interindividual variability in the reductions of LDL-C, non-HDL-C, and apo B achieved with a fixed statin dose [29]. In addition, more than 40 % of patients on high-dose statin therapy did not reach an LDL-C target <70 mg/dL [29]. Further, the meta-analysis showed that patients achieving LDL-C <50 mg/dL had a significantly lower risk for major CV events compared with those who achieved LDL-C between 75 and <100 mg/dL (adjusted hazard ratio [HR], 0.81; 95 % confidence interval [CI], 0.70–0.95). The authors concluded that achieving a very low LDL-C level lowered the risk for major CV events more than achieving moderately low LDL-C levels. Nevertheless, the large meta-analysis by Boekholdt and colleagues is limited in being post hoc observational data. In the recent preliminary report of results from the IMProved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE-IT), patients who received statin and ezetimibe (EZE) achieved mean levels of LDL-C of 53 mg/dL and had improved CV outcomes (ie, 6.4 % relative risk reduction) compared with patients who received statin alone and achieved a mean LDL-C level of 70 mg/dL (http://www.mercknewsroom.com/news-release/prescription-medicine-news/vytorin-ezetimibesimvastatin-significantly-reduced-cardiovas, accessed 14 Dec 2014). These results confirm the direct relationship between LDL-C and CV risk. Continued research, with a focus on intense LDL-C reductions, such as with PCSK9 inhibition, may prove or disprove the “lower is better” hypothesis once and for all [30].
Clinical Trial Evidence for PCSK9 Inhibition: Present Evidence and Future Considerations
Within the last 2 years, new clinical trials have confirmed the robust lipid-lowering effects of PCSK9 inhibitors observed in phase 2 studies. Several pharmaceutical companies have built on the solid foundation of basic science, genetic studies, and epidemiological observations to develop novel drug-development programs. There are several approaches to inhibit the effects of PCSK9 and, consequently, lower LDL-C levels (Table 1). The most prominent agents for which trial data are available have a mode of action based on human monoclonal antibodies (mAbs) to PCSK9 (Fig. 1) and include alirocumab (Regeneron/Sanofi), evolocumab (Amgen), and bococizumab (Pfizer), all in phase 3 trials, and others in early development [31].
The monoclonal antibodies against PCSK9 necessitate subcutaneous (SC) administration. Recent trial results demonstrate substantial reductions of LDL-C, the primary target, as well as reductions of Lp(a) (Tables 2 and 3) [32–51]. Across all studies, the new mAbs have shown a safety profile generally comparable to placebo with the exception of injection-site reactions, which have occurred infrequently but at a higher rate in patients treated with a PCSK9 inhibitor compared with those treated with the study comparator. These injection site reactions are generally mild in severity, with up to 10 % of patients (compared with up to 8 % of placebo-treated patients) discontinuing treatment because of adverse events (AEs) [32–45, 52–54]. Cardiovascular outcome studies of up to approximately 5 years in duration are under way to determine whether PCSK9 inhibition reduces CV event risk.
Alirocumab (REGN727/SAR236553) (Regeneron/Sanofi)
Alirocumab (REGN727/SAR236553) is a PCSK9 inhibitor whose efficacy and safety have been demonstrated in a series of phase 2 and 3 clinical trials [37, 40–42, 50, 52, 55, 56]. The phase 2 trials were 8- or 12-week studies that enrolled a total of 352 patients with hypercholesterolemia or heterozygous familial hypercholesterolemia (HeFH). The reduction from baseline in LDL-C achieved with alirocumab at a dose of 150 mg every 2 weeks was 66.2 to 73.2 % at week 12 [37, 40, 42]. Alirocumab was generally well tolerated, with AEs similar in both the treatment and the placebo groups and mild injection-site reaction being the most common AE reported [37, 40].
Alirocumab, both in combination with other lipid-lowering agents and as monotherapy, is currently being investigated under the auspices of the ODYSSEY clinical trial program, which includes more than 24,000 patients in 14 phase 3 trials (Table 2). The ODYSSEY program encompasses a range of patients, including patients with HeFH, patients with hypercholesterolemia at high CV risk, patients with statin intolerance, and additional patient populations and clinical settings (Table 2). The primary endpoint in these trials is the mean percentage reduction of LDL-C at 24 weeks. Dosing in the majority of the ODYSSEY trials is 75 mg every 2 weeks, with uptitration at 12 weeks to 150 mg every 2 weeks, if necessary [47, 52, 57–61]. However, dosing in ODYSSEY LONG TERM (largest ODYSSEY trial with data available to date) and HIGH FH is 150 mg every 2 weeks throughout the study [51, 62]. All doses of alirocumab are self-administered using a 1-mL prefilled pen.
Results of four clinical trials from the ODYSSEY program have been published to date: ODYSSEY MONO, ODYSSEY COMBO I, ODYSSEY COMBO II, and ODYSSEY LONG TERM [50–52, 56]. ODYSSEY MONO (N = 103) was a randomized, double-blind, active-controlled, parallel-group study to evaluate the efficacy and safety of alirocumab during 24 weeks in patients with primary hypercholesterolemia and moderate CV risk. Patients in the trial were randomized to receive monotherapy with EZE 10 mg, an alternative to statin therapy that inhibits the intestinal absorption of cholesterol, or alirocumab. The majority of patients receiving alirocumab in the trial remained on the initial 75-mg dose and did not require uptitration. The least-squares means (LSM) LDL-C (± SE) reduction from baseline to week 24 was 47.2 % (±3.0 %) with alirocumab versus 15.6 % (±3.1 %) with EZE (P < 0.0001).
Peer-reviewed publications based on the ODYSSEY COMBO I and COMBO II trials evaluated the efficacy and safety of alirocumab in high CV-risk patients with insufficiently controlled hypercholesterolemia who are on maximally tolerated doses of statins [47, 50, 56]. ODYSSEY COMBO I was a 52-week trial in which patients (N = 316) were randomized 2 to 1 to receive alirocumab or placebo [56]. The mean reduction in LDL-C level from baseline to week 24 was 48.2 % in the alirocumab group compared with 2.3 % in the placebo group (P < 0.0001). Most patients in the alirocumab group (75.0 %) at week 24 achieved LDL-C level of <70 mg/dL compared with 9.0 % of patients receiving placebo. The difference in LDL-C–lowering effect between both treatments was sustained at week 52.
Similarly, ODYSSEY COMBO II is an ongoing, 104-week study comparing the efficacy of alirocumab and EZE (N = 720). The primary endpoint analysis published recently showed that at 24 weeks patients in the alirocumab arm achieved a LSM LDL-C reduction (± SE) of 50.6 % (±1.4 %) compared with 20.7 % (±1.4 %) for patients receiving EZE (P < 0.0001) [50]. As in ODYSSEY COMBO I, most patients treated with alirocumab (77.0 %) were able to achieve the target LDL-C level of 70 mg/dL (versus 45.6 % with EZE). The difference in LDL-C–lowering effect between both treatments was maintained throughout the 52-week follow-up.
Longer-term data on the efficacy and safety of alirocumab have recently become available with publication of the ODYSSEY LONG TERM study [51]. This 78-week study compared alirocumab with placebo in patients at high CV risk who were also receiving a maximally tolerated statin dose, with or without other lipid-modifing therapy (N = 2341). The LSM LDL-C reduction from baseline to week 24 was 61.0 % with alirocumab versus an increase of 0.8 % with placebo, and it was consistent for the study duration. The treatment difference between alirocumab and placebo in the LDL-C reduction obtained at week 24 was similar in patients with HeFH and those without it.
Preliminary data from six additional phase 3 ODYSSEY trials of alirocumab in patients with hypercholesterolemia have recently been reported to meet their primary efficacy endpoint of a greater percentage reduction from baseline in LDL-C at 24 weeks compared with placebo or active comparator (LSM change from baseline range of −36.3 to −54.0 %; Table 2) [51, 57, 59, 60, 62].
To better assess the safety profile of alirocumab, a pooled analysis was conducted [63]. The analysis included four phase 2 studies and five phase 3 studies of up to 78 weeks in duration in patients (N = 3752) with hypercholesterolemia who were on stable background statin therapy. Adverse events between the two groups were comparable with the exception of local injection site reaction, which was reported in 7.2 and 5.1 % of patients in the alirocumab and placebo groups, respectively. No significant elevation in liver or muscle enzymes occurred. There were no major differences in the rate of neurologic or skeletal muscle-related treatment-emergent AEs or neurocognitive disorders.
A similar analysis, including 14 phase 2 and 3 trials, was performed to evaluate the safety of reducing LDL-C with alirocumab to very low levels [55]. Of 3340 patients receiving alirocumab, 23.8 % achieved LDL-C <25 mg/dL on at least two consecutive visits and 8.6 % achieved LDL-C <15 mg/dL. Treatment-emergent AEs were generally similar, and no safety signals were observed.
In a post hoc safety analysis of the ODYSSEY LONG TERM trial, the rate of major CV events (a composite endpoint of death from CHD, nonfatal myocardial infarction, ischemic stroke, or hospitalization for unstable angina) was reduced by 48 % in patients receiving alirocumab compared with patients receiving placebo during the 80 weeks of follow-up (HR, 0.52; 95 % CI, 0.31–0.90; P = 0.02) [51]. Long-term CV outcomes of alirocumab treatment are being assessed in ODYSSEY OUTCOMES, which will evaluate the occurrence of above–mentioned major CV events in patients who have recently experienced an acute coronary syndrome [61]. This trial will enroll approximately 18,000 patients with an acute coronary syndrome event 1 to 12 months prior to enrollment and an LDL-C ≥70 mg/dL.
Evolocumab (AMG 145) (Amgen)
Evolocumab (AMG 145) has been shown in multiple recent trials to lower LDL-C by 39 to 75 % (Table 3). Evolocumab is generally well tolerated, with SC administration every 2 or 4 weeks. In the phase 3 studies completed to date, evolocumab doses were either 140 mg every 2 weeks or 420 mg every 4 weeks [32, 35, 39, 44, 49, 64, 65]. Phase 2 evolocumab studies include MENDEL [36], RUTHERFORD [38], GAUSS [45], LAPLACE-TIMI 57 [66, 67], OSLER-1 [34, 49], and TESLA [43]. Phase 3 studies for evolocumab include DESCARTES [32], MENDEL-2 [35], GAUSS-2 [44], LAPLACE-2 [39], OSLER-2 [49], RUTHERFORD-2 [65], and TESLA Part B [64]. These were 12-week randomized, controlled phase 2 and 3 clinical trials of evolocumab as monotherapy or in combination, with poorly controlled hyperlipidemia, statin intolerance, homozygous familial hypercholesterolemia, or HeFH. Overall, efficacy and safety were consistently confirmed.
Importantly, in GAUSS-2, 8 % of patients who were statin intolerant due to muscle-related AEs showed myalgia in the evolocumab arm compared with 18 % in the EZE arm [44]. Moreover, evolocumab treatment led to a 53 to 56 % decrease from baseline in LDL-C level (compared with an 18 % decrease from baseline in the EZE arm). The GAUSS-2 trial is important in demonstrating that robust LDL-C reduction efficacy is coupled with a low incidence of muscle-related side effects, suggesting that PCSK9 inhibition is a promising therapy for the large unmet clinical need in high-risk patients with elevated cholesterol who cannot or will not take statins. Study discontinuations among PCSK9 inhibitor–treated patients were similar to those among patients receiving EZE (8 and 13 %, respectively).
The long-term, randomized, double-blind, placebo-controlled phase 3 trial DESCARTES (N = 901) has confirmed the persistent lipid-lowering effects of evolocumab [32] during a 1-year period. The DESCARTES trial included patients with a wide range of risk for CVD who were receiving background lipid-lowering therapy. In the DESCARTES study, the efficacy finding of LDL-C reduction of 50.1 % at 52 weeks for evolocumab-treated patients (P < 0.001) was similar to the 52.3 % reduction in LDL-C levels at week 52 (P < 0.0001) in an open-label study of long-term evaluation of evolocumab against LDL-C (OSLER-1) [34].
Evolocumab has demonstrated effective and safe LDL-C lowering in a range of patient populations. A pooled analysis focusing on elderly patients enrolled in phase 2 and 3 trials and their open-label extension studies showed a 58.4 to 62.9 % LDL-C reduction at the mean of weeks 10 and 12 for patients ≥65 years and 59.9 to 68.6 % LDL-C reduction for patients ≥75 years [68]. Most documented AEs were mild, and there were no noteworthy differences between patients in the evolocumab and control arms in either age group. No differences in myalgia or neurocognitive events were observed.
A pooled analysis of 12 phase 2 and 3 studies and two extension studies continuing the parent studies showed a similar rate of AEs and serious AEs (including myalgia) between the evolocumab and control arms in both parent studies and year 1 of the extension studies [69]. Injection site reactions were reported by 3.3 % of patients receiving evolocumab and 3.0 % of patients in the control group. No PCSK9 neutralizing antibodies were detected. Increases in creatine kinase and liver enzyme levels were rare and similar in patients receiving evolocumab and in those in the control group.
The rate of CV events was analyzed at 1 year in the open-label extension OSLER-1 and OSLER-2 studies [49]. An exploratory composite analysis published recently showed that patients receiving evolocumab had a significantly lower rate of all CV events than did patients receiving standard-of-care therapy, with Kaplan-Meier estimates of 0.95 and 2.18 %, respectively (HR, 0.47; 95 % CI, 0.28–0.78; P = 0.003). Four other ongoing studies of evolocumab have the potential to provide long-term safety and efficacy data, including the FOURIER (Further Cardiovascular Outcomes Research with PCSK9 Inhibition in Subjects with Elevated Risk) study, which will assess whether treatment with evolocumab compared with placebo reduces recurrent CV events in approximately 22,500 patients with CVD (Table 3).
Bococizumab (RN316) (Pfizer)
A third PCSK9 inhibitor, the fully humanized antibody (ie, antibody originally from a mouse engineered to primarily contain human antibody sequences [15]) bococizumab, is currently in phase 3 development with statin-treated patients with hypercholesterolemia and to date has limited published data available for review. As noted in a recent report by Ballantyne and colleagues, a 24-week, phase 2, randomized, double-blind, placebo-controlled trial investigated the efficacy and safety of bococizumab in 354 patients [70]. The dosing regimen included 50, 100, or 150 mg of bococizumab every 2 weeks, or 200 or 300 mg of bococizumab every 4 weeks, with a possible downtitration if LDL-C level was reduced to at least 25 mg/dL. Bococizumab demonstrated a significant reduction in LDL-C from baseline at week 12 of 19.5 to 52.0 % compared with an increase of 0.6 to 3.3 % with placebo (P < 0.001 for all comparisons with placebo). The safety profile was similar for bococizumab and placebo, with nasopharyngitis and upper respiratory tract infections as the most frequently reported AEs.
Evidence Linking Lp(a) to Cardiovascular Disease Risk: Effect of PCSK9 Inhibition
An LDL particle linked to the plasminogen-like glycoprotein apolipoprotein A, Lp(a) shows a consistent and independent positive association with CVD risk in FH [71]. Observational studies have suggested that a level of Lp(a) > ~50 mg/dL, the 80th percentile for most populations, is an independent, positive CVD risk factor [10, 72]. A large Mendelian randomization study demonstrated a genetically determined doubling of Lp(a) and increased risk for CVD (HR, 1.22; 95 % CI, 1.09–1.37) [73]. The effect of increased CVD risk with elevated Lp(a) may be as robust or greater in persons of African descent, including African Americans since they tend to have two- to four-fold higher Lp(a) levels than European Americans. These higher Lp(a) levels are positively associated with total cholesterol, LDL-C, apo B levels, and CVD risk [74, 75]. Long-term data are now available from a cohort study, which included African Americans, demonstrating that increased Lp(a) levels are associated with increased CVD risk in African Americans [76]. Because of the lack of clinical trial results demonstrating that lowering Lp(a) reduces CVD event risk, the 2013 ACC/AHA guidelines do not identify Lp(a) as a primary target for lipid-lowering therapy [26].
The European Atherosclerosis Society (EAS) Consensus Panel position paper on Lp(a) has recommended targeting elevated circulating plasma levels of Lp(a) (>50 mg/dL) [10]. Although no pharmacotherapy has been proven effective for CVD outcomes, EAS considers evaluating and possibly treating Lp(a) as a priority for reducing CV risk, after lowering of LDL-C.
An additional possible advantage of PCSK9 inhibition is a potential reduction of Lp(a) levels (Tables 2 and 3). Pooled results of phase 2 studies of alirocumab showed that 8- and 12-week treatments led to an Lp(a) reduction of 30.3 % compared with baseline (Table 3) [77]. Treatment with alirocumab for 24 weeks led to up to a 29.3 % reduction from baseline in Lp(a) levels in phase 3 studies (Table 3). Similarly, a pooled analysis of data from 1359 patients in four phase 2 trials assessed the effects of evolocumab on Lp(a) levels, the relationship between Lp(a) and lowering of LDL-C and apo B, and the influence of background statin therapy [78]. Evolocumab treatment for 12 weeks resulted in significant (P < 0.001) mean dose-related reductions in Lp(a), which significantly correlated with percentage reductions in LDL-C and apo B, and this trend was greater than in patients taking statins. In phase 3 studies, evolocumab led to up to a 38.6 % reduction in Lp(a) levels from baseline to week 24 (Table 3). The level of Lp(a) in patients treated with bococizumab was reduced at week 12 by 0 to 10.7 % compared with an increase of 0 to 3.5 % in patients randomized to placebo [70]. The exact mechanism by which PCSK9 inhibitors lower Lp(a) is unclear, but this may be another benefit of these novel agents.
Although it appears that inhibition of PCSK9 may result in significant dose-related reductions in Lp(a), the effect of Lp(a) reduction on CV outcomes is unclear [77]. Unlike statins, which do not have a significant effect on Lp(a), niacin at high doses has been demonstrated to lower Lp(a). Nevertheless, results of the HPS2-THRIVE study did not demonstrate this long-utilized agent to be of additional benefit for CVD outcomes [79]. In consideration of Lp(a) as an additional risk factor for ASCVD, if future studies confirm CVD outcomes benefit with PCSK9 inhibitors, the role of Lp(a) will need additional scrutiny.
Nonantibody PCSK9 Inhibition
Additional nonantibody approaches to PCSK9 inhibition, though currently in the early stages, are being investigated by smaller programs. One agent, ALN-PCS (Alnylam Pharmaceuticals), inhibits PCSK9 synthesis by RNA interference (RNAi). Early results suggest that inhibition of PCSK9 synthesis by RNAi may provide a potentially safe mechanism to reduce LDL-C concentration after a single intravenous dose in healthy volunteers with raised cholesterol [80]. Another nonantibody approach, BMS-962476, is an adnectin inhibitor of PCSK9. Adnectins are therapeutic proteins with high-affinity target binding, similar to antibodies. The first-in-human study of the adnectin inhibitor of PCSK9 demonstrated rapidly reduced free plasma PCSK9 and LDL-C concentrations and showed that adnectin-mediated inhibition was well tolerated [81].
Other preliminary reports from non-mAb agents include preclinical small molecule programs for (1) oral inhibitors of PCSK9, allosteric ligands of PCSK9 that disrupt normal protein folding to inhibit LDL-R binding, (2) small molecules that block the autocatalytic cleavage of PCSK9 to prevent secretion from the cell, and (3) small molecules that block the interaction between PCSK9 and LDL-Rs [31].
Summary and Conclusions
Contemporary lipid management, according to the most recent evidence-based guidelines, is based on LDL-C reduction with statins. However, even with appropriate statin treatment, there may be suboptimal cholesterol management or insufficient LDL-C reduction. A significant number of treated patients are unable to tolerate statins or statin dose escalation, which may result in nonadherence. Patients nonadherent due to intolerance or lack of effect, especially with significant residual risk for major CVD events, could benefit from alternative treatment options. PCSK9 inhibition as a therapeutic approach is rapidly being shown as a potentially important clinical approach to LDL-C lowering. Based on innovative basic research, genetic findings, and observational data, PCSK9 inhibition may conceivably progress from a research tool to a novel therapy with the ability to improve clinical outcomes. Moreover, recent data suggest that PCSK9 inhibition reduces circulating Lp(a) levels. This finding may be of interest to both researchers and clinicians.
The final pathway to widespread clinical use, however, will be based on the results of ongoing long-term safety and efficacy outcomes trials for the PCSK9 inhibitor class. Recent studies with PCSK9 demonstrate rapid translation from basic science and genetic research to lowering LDL-C and, potentially, reducing an important secondary target, Lp(a), which may in the future contribute to protection against CV events.
References
Anderson KM, Castelli WP, Levy D. Cholesterol and mortality. 30 years of follow-up from the Framingham study. J Am Med Assoc. 1987;257:2176–80.
Stamler J, Stamler R, Neaton JD, et al. Low risk-factor profile and long-term cardiovascular and noncardiovascular mortality and life expectancy: findings for 5 large cohorts of young adult and middle-aged men and women. JAMA. 1999;282:2012–8.
Kavey R-EW, Daniels SR, Lauer RM, Atkins DL, Hayman LL, Taubert K. American Heart Association guidelines for primary prevention of atherosclerotic cardiovascular disease beginning in childhood. Circulation. 2003;107:1562–6.
Pagidipati NJ, Gaziano TA. Estimating deaths from cardiovascular disease: a review of global methodologies of mortality measurement. Circulation. 2013;127:749–56.
LaRosa JC. Reduction of serum LDL-C levels: a relationship to clinical benefits. Am J Cardiovasc Drugs. 2003;3:271–81.
Forrester JS, Bairey-Merz CN, Kaul S. The aggressive low density lipoprotein lowering controversy. J Am Coll Cardiol. 2000;36:1419–25.
Cholesterol Treatment Trialists (CTT) Collaborators. The effects of lowering LDL cholesterol with statin therapy in people at low risk of vascular disease: meta-analysis of individual data from 27 randomised trials. Lancet. 2012;380:581–90.
Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol. 2009;29:431–8.
Willeit P, Kiechl S, Kronenberg F, et al. Discrimination and net reclassification of cardiovascular risk with lipoprotein(a): prospective 15-year outcomes in the Bruneck study. J Am Coll Cardiol. 2014;64:851–60.
Nordestgaard BG, Chapman MJ, Ray K, for European Atherosclerosis Society Consensus Panel, et al. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31:2844–53.
Rader DJ, Kastelein JJP. Lomitapide and mipomersen: two first-in-class drugs for reducing low-density lipoprotein cholesterol in patients with homozygous familial hypercholesterolemia. Circulation. 2014;129:1022–32.
Horton JD, Cohen JC, Hobbs HH. Molecular biology of PCSK9: its role in LDL metabolism. Trends Biochem Sci. 2007;32:71–7.
Seidah NG, Benjannet S, Wickham L, et al. The secretory proprotein convertase neural apoptosis-regulated convertase 1 (NARC-1): liver regeneration and neuronal differentiation. Proc Natl Acad Sci U S A. 2003;100:928–33.
Seidah NG, Awan Z, Chrétien M, Mbikay M. PCSK9: a key modulator of cardiovascular health. Circ Res. 2014;114:1022–36.
Catapano AL, Papadopoulos N. The safety of therapeutic monoclonal antibodies: implications for cardiovascular disease and targeting the PCSK9 pathway. Atherosclerosis. 2013;228:18–28.
Abifadel M, Varret M, Rabès J-P, et al. Mutations in PCSK9 cause autosomal dominant hypercholesterolemia. Nat Genet. 2003;34:154–6.
Abifadel M, Rabès J-P, Devillers M, et al. Mutations and polymorphisms in the proprotein convertase subtilisin kexin 9 (PCSK9) gene in cholesterol metabolism and disease. Hum Mutat. 2009;30:520–9.
Humphries SE, Whittall RA, Hubbart CS, for Simon Broome Familial Hyperlipidaemia Register Group and Scientific Steering Committee, et al. Genetic causes of familial hypercholesterolaemia in patients in the UK: relation to plasma lipid levels and coronary heart disease risk. J Med Genet. 2006;43:943–9.
Naoumova RP, Tosi I, Patel D, et al. Severe hypercholesterolemia in four British families with the D374Y mutation in the PCSK9 gene: long-term follow-up and treatment response. Arterioscler Thromb Vasc Biol. 2005;25:2654–60.
Cohen J, Pertsemlidis A, Kotowski IK, Graham R, Garcia CK, Hobbs HH. Low LDL cholesterol in individuals of African descent resulting from frequent nonsense mutations in PCSK9. Nat Genet. 2005;37:161–5.
Cohen JC, Boerwinkle E, Mosley Jr TH, Hobbs HH. Sequence variations in PCSK9, low LDL, and protection against coronary heart disease. N Engl J Med. 2006;354:1264–72.
Cohen JC. Emerging LDL, therapies: using human genetics to discover new therapeutic targets for plasma lipids. J Clin Lipidol. 2013;7:S1–5.
Zhao Z, Tuakli-Wosornu Y, Lagace TA, et al. Molecular characterization of loss-of-function mutations in PCSK9 and identification of a compound heterozygote. Am J Hum Genet. 2006;79:514–23.
European Association for Cardiovascular Prevention & Rehabilitation. ESC/EAS Guidelines for the management of dyslipidaemias: the Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Eur Heart J. 2011;32:1769–818.
O’Keefe Jr JH, Cordain L, Harris WH, Moe RM, Vogel R. Optimal low-density lipoprotein is 50 to 70 mg/dl: lower is better and physiologically normal. J Am Coll Cardiol. 2004;43:2142–6.
Stone NJ, Robinson JG, Lichtenstein AH, et al. 2013 ACC/AHA guideline on the treatment of blood cholesterol to reduce atherosclerotic cardiovascular risk in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines. Circulation. 2014;129:S1–S45.
Dubuc G, Chamberland A, Wassef H, et al. Statins upregulate PCSK9, the gene encoding the proprotein convertase neural apoptosis-regulated convertase-1 implicated in familial hypercholesterolemia. Arterioscler Thromb Vasc Biol. 2004;24:1454–9.
ATP III. Final report. VI: Drug therapy. Circulation. 2002;106:3303–25.
Boekholdt SM, Hovingh GK, Mora S, et al. Very low levels of atherogenic lipoproteins and the risk for cardiovascular events: a meta-analysis of statin trials. J Am Coll Cardiol. 2014;64:485–94.
Ben-Yehuda O, DeMaria AN. LDL-cholesterol targets after the ACC/AHA 2013 guidelines: evidence that lower is better? J Am Coll Cardiol. 2014;64:495–7.
Dragan S, Serban M-C, Banach M. Proprotein convertase subtilisin/kexin 9 inhibitors: an emerging lipid-lowering therapy? J Cardiovasc Pharmacol Ther. 2015;20:157–68.
Blom DJ, Hala T, Bolognese M, for DESCARTES Investigators, et al. A 52-week placebo-controlled trial of evolocumab in hyperlipidemia. N Engl J Med. 2014;370:1809–19.
Giugliano RP, Desai NR, Kohli P, for LAPLACE-TIMI 57 Investigators, et al. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 in combination with a statin in patients with hypercholesterolaemia (LAPLACE-TIMI 57): a randomised, placebo-controlled, dose-ranging, phase 2 study. Lancet. 2012;380:2007–17.
Koren MJ, Giugliano RP, Raal FJ, for OSLER Investigators, et al. Efficacy and safety of longer-term administration of evolocumab (AMG 145) in patients with hypercholesterolemia: 52-week results from the Open-Label Study of Long-Term Evaluation Against LDL-C (OSLER) randomized trial. Circulation. 2014;129:234–43.
Koren MJ, Lundqvist P, Bolognese M, for MENDEL-2 Investigators, et al. Anti-PCSK9 monotherapy for hypercholesterolemia: the MENDEL-2 randomized, controlled phase III clinical trial of evolocumab. J Am Coll Cardiol. 2014;63:2531–40.
Koren MJ, Scott R, Kim JB, et al. Efficacy, safety, and tolerability of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 as monotherapy in patients with hypercholesterolaemia (MENDEL): a randomised, double-blind, placebo-controlled, phase 2 study. Lancet. 2012;380:1995–2006.
McKenney JM, Koren MJ, Kereiakes DJ, Hanotin C, Ferrand A-C, Stein EA. Safety and efficacy of a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease, SAR236553/REGN727, in patients with primary hypercholesterolemia receiving ongoing stable atorvastatin therapy. J Am Coll Cardiol. 2012;59:2344–53.
Raal F, Scott R, Somaratne R, et al. Low-density lipoprotein cholesterol-lowering effects of AMG 145, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease in patients with heterozygous familial hypercholesterolemia: the Reduction of LDL-C with PCSK9 Inhibition in Heterozygous Familial Hypercholesterolemia Disorder (RUTHERFORD) randomized trial. Circulation. 2012;126:2408–17.
Robinson JG, Nedergaard BS, Rogers WJ, for LAPLACE-2 Investigators, et al. Effect of evolocumab or ezetimibe added to moderate- or high-intensity statin therapy on LDL-C lowering in patients with hypercholesterolemia: the LAPLACE-2 randomized clinical trial. JAMA. 2014;311:1870–82.
Roth EM, McKenney JM, Hanotin C, Asset G, Stein EA. Atorvastatin with or without an antibody to PCSK9 in primary hypercholesterolemia. N Engl J Med. 2012;367:1891–900.
Stein EA, Bergeron J, Gaudet D, et al. One year open-label treatment with alirocumab 150 mg every two weeks in heterozygous familial hypercholesterolemic patients [abstract]. J Am Coll Cardiol. 2014;63:A1371.
Stein EA, Gipe D, Bergeron J, et al. Effect of a monoclonal antibody to PCSK9, REGN727/SAR236553, to reduce low-density lipoprotein cholesterol in patients with heterozygous familial hypercholesterolaemia on stable statin dose with or without ezetimibe therapy: a phase 2 randomised controlled trial. Lancet. 2012;380:29–36.
Stein EA, Honarpour N, Wasserman SM, Xu F, Scott R, Raal FJ. Effect of the proprotein convertase subtilisin/kexin 9 monoclonal antibody, AMG 145, in homozygous familial hypercholesterolemia. Circulation. 2013;128:2113–20.
Stroes E, Colquhoun D, Sullivan D, for GAUSS-2 Investigators, et al. Anti-PCSK9 antibody effectively lowers cholesterol in patients with statin intolerance: the GAUSS-2 randomized, placebo-controlled phase 3 clinical trial of evolocumab. J Am Coll Cardiol. 2014;63:2541–8.
Sullivan D, Olsson AG, Scott R, et al. Effect of a monoclonal antibody to PCSK9 on low-density lipoprotein cholesterol levels in statin-intolerant patients: the GAUSS randomized trial. JAMA. 2012;308:2497–506.
Moriarty PM, Jacobson TA, Bruckert E, et al. Efficacy and safety of alirocumab, a monoclonal antibody to PCSK9, in statin-intolerant patients: Design and rationale of ODYSSEY ALTERNATIVE, a randomized Phase 3 trial. J Clin Lipidol. 2014;8:554–61.
Colhoun HM, Robinson JG, Farnier M, et al. Efficacy and safety of alirocumab, a fully human PCSK9 monoclonal antibody, in high cardiovascular risk patients with poorly controlled hypercholesterolemia on maximally tolerated doses of statins: rationale and design of the ODYSSEY COMBO I and II trials. BMC Cardiovasc Disord. 2014;14:121.
Robinson JG, Colhoun HM, Bays HE, et al. Efficacy and safety of alirocumab as add-on therapy in high-cardiovascular-risk patients with hypercholesterolemia not adequately controlled with atorvastatin (20 or 40 mg) or rosuvastatin (10 or 20 mg): design and rationale of the ODYSSEY OPTIONS studies. Clin Cardiol. 2014;37:597–604.
Sabatine MS, Giugliano RP, Wiviott SD, for Open-Label Study of Long-Term Evaluation against LDL Cholesterol (OSLER) Investigators, et al. Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1500–9.
Cannon CP, Cariou B, Blom D, for the ODYSSEY COMBO II Investigators, et al. Efficacy and safety of alirocumab in high cardiovascular risk patients with inadequately controlled hypercholesterolaemia on maximally tolerated doses of statins: the ODYSSEY COMBO II randomized controlled trial. Eur Heart J. 2015;36(19):1186–94.
Robinson JG, Farnier M, Krempf M, for ODYSSEY LONG TERM Investigators, et al. Efficacy and safety of alirocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1489–99.
Roth EM, Taskinen M-R, Ginsberg HN, et al. Monotherapy with the PCSK9 inhibitor alirocumab versus ezetimibe in patients with hypercholesterolemia: results of a 24 week, double-blind, randomized phase 3 trial. Int J Cardiol. 2014;176:55–61.
Farnier M. PCSK9: from discovery to therapeutic applications. Arch Cardiovasc Dis. 2014;107:58–66.
Sahebkar A, Watts GF. New LDL-cholesterol lowering therapies: pharmacology, clinical trials, and relevance to acute coronary syndromes. Clin Ther. 2013;35:1082–98.
Robinson J, Farnier M, Chaudhari U, et al. Adverse events in patients with low-density lipoprotein cholesterol levels <25 or <15 mg/dL on at least two consecutive visits in fourteen randomized, controlled, clinical trials of alirocumab [abstract]. J Am Coll Cardiol. 2015;65:A1350.
Kereiakes DJ, Robinson JG, Cannon CP, et al. Efficacy and safety of the proprotein convertase subtilisin/kexin type 9 inhibitor alirocumab among high cardiovascular risk patients on maximally tolerated statin therapy: the ODYSSEY COMBO I study. Am Heart J. 2015;169(6):906–15.
Bays H, Farnier M, Gaudet D, et al. Efficacy and safety of combining alirocumab with atorvastatin or rosuvastatin versus statin intensification or adding ezetimibe in high cardiovascular risk patients: ODYSSEY OPTIONS I and II [abstract]. Circulation. 2014;130:2118–9.
Kastelein JJP, Robinson JG, Farnier M, et al. Efficacy and safety of alirocumab in patients with heterozygous familial hypercholesterolemia not adequately controlled with current lipid-lowering therapy: design and rationale of the ODYSSEY FH studies. Cardiovasc Drugs Ther. 2014;28:281–9.
Kereiakes DJ, Robinson JG, Cannon CP, et al. Efficacy and safety of alirocumab in high cardiovascular risk patients with suboptimally controlled hypercholesterolemia on maximally tolerated doses of statins: the ODYSSEY COMBO I study [abstract]. Circulation. 2014;130:2119–20.
Moriarty PM, Thompson PD, Cannon CP, et al. ODYSSEY ALTERNATIVE: efficacy and safety of the proprotein convertase subtilisin/kexin type 9 monoclonal antibody, alirocumab, versus ezetimibe, in patients with statin intolerance as defined by a placebo run-in and statin rechallenge arm [abstract]. Circulation. 2014;130:2108–9.
Schwartz GG, Bessac L, Berdan LG, et al. Effect of alirocumab, a monoclonal antibody to PCSK9, on long-term cardiovascular outcomes following acute coronary syndromes: rationale and design of the ODYSSEY Outcomes trial. Am Heart J. 2014;168:682–9.
Ginsberg HN, Rader DJ, Raal FJ, et al. ODYSSEY HIGH FH: efficacy and safety of alirocumab in patients with severe heterozygous familial hypercholesterolemia [abstract]. Circulation. 2014;130:2119.
Jones PH, Bays H, Chaudhari U, et al. Pooled safety and adverse events in nine randomized, placebo-controlled, phase 2 and 3 clinical trials of alirocumab [abstract]. J Am Coll Cardiol. 2015;65:A1363.
Raal FJ, Honarpour N, Blom DJ, for TESLA Investigators, et al. Inhibition of PCSK9 with evolocumab in homozygous familial hypercholesterolaemia (TESLA Part B): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;385:341–50.
Raal FJ, Stein EA, Dufour R, for RUTHEFORD-2 Investigators, et al. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD-2): a randomised, double-blind, placebo-controlled trial. Lancet. 2015;385:331–40.
Desai NR, Giugliano RP, Zhou J, et al. AMG 145, a monoclonal antibody against PCSK9, facilitates achievement of national cholesterol education program–adult treatment panel III low-density lipoprotein cholesterol goals among high-risk patients: an analysis from the LAPLACE-TIMI 57 trial (LDL-C Assessment with PCSK9 monoclonaL Antibody Inhibition Combined With Statin thErapy–Thrombolysis In Myocardial infarction 57). J Am Coll Cardiol. 2014;63:430–3.
Kohli P, Desai NR, Giugliano RP, et al. Design and rationale of the LAPLACE-TIMI 57 trial: a phase II, double-blind, placebo-controlled study of the efficacy and tolerability of a monoclonal antibody inhibitor of PCSK9 in subjects with hypercholesterolemia on background statin therapy. Clin Cardiol. 2012;35:385–91.
Koren M, Rosenson R, Khan B, et al. LDL cholesterol reduction in elderly patients with the pcsk9 monoclonal antibody evolocumab (AMG 145): a pooled analysis of 1779 patients in phase 2, 3 and open label extension studies [abstract]. J Am Coll Cardiol. 2015;65:A1366.
Toth PP, Sattar N, Genest J, et al. A comprehensive safety analysis of 6026 patients from phase 2 and 3 short and long term clinical trials with evolocumab (AMG 145) [abstract]. J Am Coll Cardiol. 2015;65:A1351.
Ballantyne CM, Neutel J, Cropp A, et al. Results of bococizumab, a monoclonal antibody against proprotein convertase subtilisin/kexin type 9, from a randomized, placebo-controlled, dose-ranging study in statin-treated subjects with hypercholesterolemia. Am J Cardiol. 2015;115:1212–21.
Alonso R, Andres E, Mata N, for SAFEHEART Investigators, et al. Lipoprotein(a) levels in familial hypercholesterolemia: an important predictor of cardiovascular disease independent of the type of LDL receptor mutation. J Am Coll Cardiol. 2014;63:1982–9.
Emerging Risk Factors Collaboration. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302:412–23.
Kamstrup PR, Tybjærg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301:2331–9.
Deo RC, Wilson JG, Xing C, et al. Single-nucleotide polymorphisms in LPA explain most of the ancestry-specific variation in Lp(a) levels in African Americans. PLoS One. 2011;6:e14581.
Enkhmaa B, Anuurad E, Zhang W, Berglund L. Significant associations between lipoprotein(a) and corrected apolipoprotein B-100 levels in African-Americans. Atherosclerosis. 2014;235:223–9.
Virani SS, Brautbar A, Davis BC, et al. Associations between lipoprotein(a) levels and cardiovascular outcomes in black and white subjects: the Atherosclerosis Risk in Communities (ARIC) Study. Circulation. 2012;125:241–9.
Gaudet D, Kereiakes DJ, McKenney JM, et al. Effect of alirocumab, a monoclonal proprotein convertase subtilisin/kexin 9 antibody, on lipoprotein(a) concentrations (a pooled analysis of 150 mg every two weeks dosing from phase 2 trials). Am J Cardiol. 2014;114:711–5.
Raal FJ, Giugliano RP, Sabatine MS, et al. Reduction in lipoprotein(a) with PCSK9 monoclonal antibody evolocumab (AMG 145): a pooled analysis of more than 1,300 patients in 4 phase II trials. J Am Coll Cardiol. 2014;63:1278–88.
HPS2-THRIVE Collaborative Group. Effects of extended-release niacin with laropiprant in high-risk patients. N Engl J Med. 2014;371:203–12.
Fitzgerald K, Frank-Kamenetsky M, Shulga-Morskaya S, et al. Effect of an RNA interference drug on the synthesis of proprotein convertase subtilisin/kexin type 9 (PCSK9) and the concentration of serum LDL cholesterol in healthy volunteers: a randomised, single-blind, placebo-controlled, phase 1 trial. Lancet. 2014;383:60–8.
Stein EA, Kasichayanula S, Turner T, et al. LDL cholesterol reduction with BMS-962476, an adnectin inhibitor of PCSK9: results of a single ascending dose study [abstract]. J Am Coll Cardiol. 2014;63:A1372.
Acknowledgments
Editorial support for this manuscript was provided by MicroMass Communications, Inc, with funding from Regeneron Pharmaceuticals, Inc, Tarrytown, NY, and Sanofi US, Bridgewater, NJ.
Sources of support
Support for this educational article was provided by Regeneron Pharmaceuticals, Inc, and Sanofi US.
Conflict of interest
KCF is a consultant for Amgen, AstraZeneca, Boehringer Ingelheim, Eli Lilly and Company, and Sanofi US. SAN has nothing to disclose.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Ferdinand, K.C., Nasser, S.A. PCSK9 Inhibition: Discovery, Current Evidence, and Potential Effects on LDL-C and Lp(a). Cardiovasc Drugs Ther 29, 295–308 (2015). https://doi.org/10.1007/s10557-015-6588-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s10557-015-6588-3