
Abstract
Purpose
The purpose of this study was to describe the skeletal manifestations of primary hyperoxaluria type 1 (PH1), the most common of the primary hyperoxalurias.
Methods
We clinically and radiographically reviewed 12 consecutive patients diagnosed with PH1, aged between 2 and 17 years. All patients had evidence of some type of renal involvement, 4 of whom were at end-stage renal disease (ESRD) and were under dialysis.
Results
The main symptom was skeletal pain and was present only in the 4 severely involved patients and appeared during the second year of dialysis. The 2 most severely involved patients had evidence of pathological fractures. Radiological signs were present in patients with or without symptoms. These radiological signs were of two distinct types: those almost specific of oxalosis, such as dense and radiolucent metaphyseal bands and vertebral osteocondensations, which are found mainly in the severely involved individuals, and those less specific, such as signs of renal osteodystrophy, which are also found in less severely involved patients. Interestingly, our study revealed the presence of spondylolysis in 25% of cases. This latter finding is unique and has not previously been reported in the literature.
Conclusions
The skeletal manifestations of PH1 include specific and less specific radiological signs, with some patients being asymptomatic, and others presenting with bone pain and pathological fractures, as well as spondylolysis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Primary hyperoxaluria (PH) results from endogenous overproduction of oxalate as opposed to secondary hyperoxaluria, which is due to excessive dietary intake or increased intestinal absorption of oxalate [1–11]. Primary hyperoxaluria type 1 (PH1) is an inherited disease due to an inborn error of glyoxylate metabolism localized to the hepatocyte peroxisome, resulting in greatly increased urinary excretion of oxalate and glycolate and eventually to renal failure and end-stage renal disease (ESRD) [1, 2]. With decrease in renal function, the highly insoluble CaOx crystals are deposited in extra-renal tissue, defining oxalosis [3]. After having observed skeletal manifestations not described elsewhere in the literature, our objective was to comprehensively describe the clinical and radiological skeletal manifestations of the disease, based on a case series.
Methods
We conducted a transversal descriptive study of 12 consecutive patients diagnosed with PH1 (Table 1). All of these patients had PH1, as documented by an elevated urinary oxalate concentration and high oxalate over creatinine ratio. Their age ranged from 2 to 17 years. There were 7 girls and 5 boys. All had renal involvement, 4 of whom had ESRD and were undergoing either haemodialysis (3 patients) or peritoneal dialysis (the youngest patient, a 2-year-old boy). All patients had symptoms of urinary lithiasis, and 11 of the 12 had radiological evidence of stones in their urinary tract. Of the patients, 5 had radiological signs evoking nephrocalcinosis. Of the ESRD patients, 1 had received a kidney transplant 2 years earlier, which became non-functional. None of the patients had received a combined liver-kidney transplantation.
All the patients were subjected to a thorough clinical examination and complete skeletal radiographic survey consisting of upper extremities, lower extremities, pelvis and spine X-rays as well as a kidney-ureter-bladder (KUB) radiograph. Bone density was not measured.
Results
Of the 12 patients, 4 had skeletal symptoms—mainly bone pain. These patients were those having reached the ESRD stage. Skeletal symptoms appeared between 1 and 2 years after initiation of dialysis. In the more severely involved patients (one on haemodialysis and one on peritoneal dialysis), bone pain was partly associated with pathological fractures.
Radiographically, bone lesions were visible in all 4 ESRD patients as well as in 3 of the 8 non-symptomatic patients whose renal function was less severely impaired. Some of the radiological signs encountered were almost pathognomonic of oxalosis, i.e., dense metaphyseal bands (DMBs), lucent metaphyseal bands (LMBs) and vertebral osteocondensations. These signs were only seen in the four patients with ESRD.
The most consistent finding seen in all four patients was the presence of condensations on the metaphyseal side of long bone growth plates. The width of the DMBs was positively correlated with the duration of ESRD. The distance between the DMB and the neighboring growth plate was positively correlated with the duration of dialysis (Figs. 1, 2). Moreover, less sharply demarcated metaphyseal condensations were present in two other patients with moderately altered renal function.

Anteroposterior (AP) radiograph of the wrist of a 10-year-old patient (patient 2) showing an early dense metaphyseal band (DMB) on the metaphyseal aspect of the distal radial physis

Anteroposterior (AP) radiograph of both hands of an 8-year-old patient undergoing dialysis (patient 1) showing late dense metaphyseal bands (DMBs; arrows). Note their width and their distance from the growth plates, compared with that shown in Fig. 1. Also, note the fractures of the distal aspect of the third, fourth and fifth metacarpals (arrow heads)
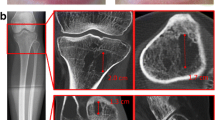
LMBs were also found only in ESRD patients and were typically seen between the physis and the adjacent DMB in long bones. They tended to appear in the same region as the DMB, which became more distant from the growth plate because of longitudinal bone growth (Fig. 3). Here again, this feature was sometimes present as ill-defined metaphyseal radiolucencies; whereas, in other cases, the radiolucent area clearly took the shape of a LMB. In two severely involved patients, we observed the presence of a radiolucent ring around the ossification center of small bones such as the carpal and tarsal bones, and around the patellae (Fig. 4). This radiolucent ring corresponds to the same lesion as the metaphyseal radiolucencies of long bones, since it occurs in the region of maximal new bone formation.

Standing dorsoplantar radiograph of both feet in a 15-year-old patient (patient 3) showing lucent metaphyseal bands (LMBs) and dense metaphyseal bands (DMBs) in the proximal aspect of the first metatarsals

a Anteroposterior (AP) radiograph of the wrist of patient 1 showing lucent rings surrounding the scaphoid and the trapezium. b Lateral radiograph of the left ankle of patient 3 showing lucent rings around the talus and navicular
The third radiographic characteristic of bone oxalosis encountered was the presence of vertebral condensations involving at first the superior and inferior vertebral endplates creating a rugger-jersey spine appearance (Fig. 5), before extending to the rest of the vertebral body and sometimes realizing the typical image of bone-within-the-bone (Fig. 6). This finding was present in the three patients on haemodialysis.

Anteroposterior (AP; a) and lateral (b) radiographs showing the sclerotic superior and inferior end plates realizing a rugger-jersey spine appearance in a 10-year-old patient (patient 2). Also note the characteristic image of nephrocalcinosis

Anteroposterior (AP; a) and lateral (b) radiographs of the lumbar spine in a 15-year-old patient (patient 3) showing the characteristic bone-within-a-bone appearance. Note the spondylolysis (arrow) with a grade-1 slip of L5 over S1 on the lateral view
Other less specific findings were also noted in the four patients with ESRD as well as two others with less severe renal impairment. These consisted of subperiosteal resorption, osteolysis of the distal phalanges, periosteal appositions and osteopenia (Fig. 7).

a Anteroposterior (AP) radiograph of the hand showing subperiosteal resorptions (arrow head) and osteolysis of the distal phalanx of the index finger (arrow) in a 15-year-old patient under dialysis (patient 3). b AP radiograph of the left upper limb showing periosteal appositions (arrows) on the medial and lateral aspects of the proximal humeral metaphysis and diaphysis in the same patient
Pathological fractures were only noted in the two most severely affected patients, both of whom had lost their walking ability (Figs. 2, 8). These fractures were localized in the region of the DMBs. One patient had a spontaneous right femoral neck fracture which appeared after the treatment of a left femoral neck fracture by open reduction and internal fixation. She also sustained spontaneous fractures of both proximal humeral metaphyses and Salter-Harris 2 fractures of the distal metacarpals. The other patient, the last patient of the series, was a 2.5-year-old ambulatory boy with ESRD, who had been on peritoneal dialysis for 3 months and had become non-ambulatory from the beginning. X-rays revealed the presence of a neglected left subtrochanteric stress fracture. Unfortunately, the child’s condition was disastrous and he died soon after the fracture was discovered.

Anteroposterior (AP) radiograph of the pelvis of the severely involved patient 1, showing bilateral femoral neck fractures
Of the 12 patients, 3 showed evidence of spondylolysis, one of whom had a grade 1–2 spondylolisthesis according to Meyerding (Fig. 6b). Of these patients, 2 were still at the very early stages of renal dysfunction; none was symptomatic.
Discussion
There are more and more articles in the literature that relate to skeletal manifestations of PH [12–34], since the prevalence of these manifestations is increasing in conjunction with the longer survival associated with newer treatments for this condition [12, 13]. Indeed, before the introduction of dialysis, patients with PH1 died from uremia at a young age before skeletal changes had developed. It is necessary to point out that the only potentially curative treatment for this condition is a combined kidney-liver transplantation [1], and all other treatment modalities such as renal transplantation, dialysis (whether haemodialysis or peritoneal dialysis) or pyridoxine supplementation do not address the cause of the disease. Dialysis in particular has been shown to be insufficient to prevent deposition of calcium-oxalate crystals in bone.
To our knowledge, this is one of the largest paediatric series studying both clinical and radiological aspects of skeletal lesions in PH1.
Our results concerning the specific and non-specific manifestations are similar to those described elsewhere in the literature [21, 25]. Progression of oxalosis seems to be most striking in areas of rapid growth at the extremities of long bones. Sclerotic and lucent bands develop and both seem to represent different stages of the disease. The DMBs of oxalosis are of greater density, of more homogeneous texture and develop more rapidly than the dense bands of renal osteodystrophy. This is due to superimposition of crystal density on the already increased bone density of secondary hyperparathyroidism [13, 15, 31]. Over time, the inflammatory reaction induced by the oxalate crystals causes bone resorption, which is especially evident in the previously uniform zones of DMBs [17]. Fractures also may occur spontaneously in these regions of weakness. The translucent rims noted around the small bones, such as the vertebrae, the tarsal or carpal bones, reflect the slower growth rate in these regions [33].
Concerning pathological fractures, it is interesting to point out that even if radiological signs of PH1 tend to gradually disappear after combined liver-kidney transplantation, the risk of developing a pathological fracture later on remains [26]. Of our severely affected children, 2 presented pathological fractures that occurred in regions of metaphyseal lucencies. The femoral neck seems to be a zone of predilection for such fractures [15, 21, 26, 31, 33, 34]. Internal fixation of those lower limb fractures interfering with weight bearing transformed the quality of life of these children who had become bedridden. This is in concert with other articles stating that a child with PH1 and bone pain should be regarded as having a fracture until proved otherwise and early internal fixation of hip fractures is mandatory [26, 31].
Literature review has shown that the non-specific signs found in PH1 patients are the result of CaOx deposition, renal osteodystrophy and secondary hyperparathyroidism. Indeed, the bone resorption observed in severely involved bone seems to be due to a combination of hyperparathyroidism and oxalate crystal-induced histiocytic inflammatory response [14, 15, 21]. Some have advocated that the subperiosteal cortical defects initially are related to hyperparathyroidism, but later they are too deep, their contour is too smooth and sclerotic, and they exhibit a slight homogeneous radiodensity [31, 32], all of which distinguish them from the typical features encountered in hyperparathyroidism. This may also be the case of the sclerotic zones observed in the vertebral end plates (rugger-jersey spine appearance), which is frequently observed in hyperparathyroidism.
To our knowledge, no other article mentions spondylolysis, which was present in 3 of our 12 patients, a percentage far beyond the incidence of spondylolysis in the general paediatric population. A thorough search of the literature failed to retrieve any article describing such an association between any of the hyperoxalurias and spondylolysis. This spondylolysis fits into type 5 (pathological spondylolysis) of the Wiltse classification. It may be due to a pathological fracture of the pars interarticularis which may be an area of CaOx crystal deposition, as occurs in long bones. However, it is clear that further studies are necessary to achieve a more comprehensive understanding of this problem.
References
Leumann EP, Hoppe B (2001) The primary hyperoxalurias. J Am Soc Nephrol 12:1986–1993
Maldonado I, Prasad V, Reginato AJ (2002) Oxalate crystal deposition disease. Curr Rheumatol Rep 4:257–264
Danpure CJ (2001) Primary hyperoxaluria. In: Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Voelstein B (eds). The metabolic and molecular bases of inherited disease. McGraw-Hill, New York, pp 3323–3367
Cochat P (1999) Primary hyperoxaluria type 1. Kidney Int 55:2533–2547
Cochat P, Rolland MO, Bozon D, Dumontel C, Divry P (1994) Molecular pathology of type 1 primary hyperoxaluria. Nephrologie 15(6):375–380 (Article in French)
Danpure CJ (1994) Molecular and cell biology of primary hyperoxaluria type 1. Clin Invest 72:725–727
Danpure CJ (1998) The molecular basis of alanine: gloxylate aminotransférase mistargeting: the most common cause of primary hyperoxaluria type 1. J Nephrol 11:8–12
Danpure CJ, Jennings PR, Fryer P, Perdue PE, Allsop J (1994) Primary hyperoxaluria type 1: Genotypic and phenotypic heterogeneity. J Inherit Metab Dis 17:487–499
Leumann E, Hoppe B (1997) Urolithiasis in childhood. In: Proesmans W (ed) Therapeutic strategies in children with renal disease. Baillière’s Clinical Paediatrics 5, London, pp 653–674
Leumann E, Hoppe B (1999) What is new about primary hyperoxaluria? Nephrol Dial Transplant 14:2556–2558
Milliner DS, Wilson DM, Smith LH (1998) Clinical expression and long-term outcomes of primary hyperoxaluria types 1 and 2. J Nephrol 11(Suppl 1):56–59
Anderson DE, Davidson JK, Catto ME (1983) Case report 227: primary hyperoxaluria (oxalosis). Skeletal Radiol 9:266–271
Benhamou CL, Bardin T, Tourliere D, Voisin L, Audran M, Edouard C, Lafage MH, Sebert JL, de Vernejoul MC, Wendling D et al (1991) Bone involvement in primary oxalosis. Study of 20 cases. Rev Rhum Mal Osteoartic 58:763–769
Brancaccio D, Poggi A, Ciccarelli C, Bellini F, Galmozzi C, Poletti I, Maggiore Q (1981) Bone changes in end-stage oxalosis. AJR Am J Roentgenol 136:935–939
Breed A, Chesney R, Friedman A, Gilbert E, Langer L, Lattoraca R (1981) Oxalosis-induced bone disease: a complication of transplantation and prolonged survival in primary hyperoxaluria. J Bone Joint Surg Am 63:310–316
Canavese C, Salomone M, Massara C, Portigliatti Barbos M, Cadario A, Pavan I, Marangella M, Petrarulo M, Rotolo U (1990) Primary oxalosis mimicking hyperparathyroidism diagnosed after long-term hemodialysis. Am J Nephrol 10:344–349
Day DL, Scheinman JI, Mahan J (1986) Radiological aspects of primary hyperoxaluria. AJR Am J Roentgenol 146:395–401
Desmond P, Hennessy O (1993) Skeletal abnormalities in primary oxalosis. Australas Radiol 37:83–85
Elmstahl B, Rausing A (1997) A case of hyperoxaluria: Radiological aspects. Acta Radiol 38:1031–1034
Fisher D, Hiller N, Drukker A (1995) Oxalosis of bone: report of four cases and a new radiological staging. Pediatr Radiol 25:293–295
Gherardi G, Poggi A, Sisca S, Calderaro V, Bonucci E (1980) Bone oxalosis and renal osteodystrophy. Arch Pathol Lab Med 104:105–111
Hoffman GS, Schumacher HR, Paul H, Cherian V, Reed R, Ramsay AG, Franck WA (1982) Calcium oxalate microcrystalline-associated arthritis in end-stage renal disease. Ann Intern Med 97:36–42
Julian BA, Faugere MC, Malluche HH (1987) Oxalosis in bone causing a radiographical mimicry of renal osteodystrophy. Am J Kidney Dis 9:436–440
Kalifa G, Dossans B, Gagnadoux MF, Sauvegrain J (1979) Radiological aspects of oxalosis. J Radiol Electrol Med Nucl 60:45–49 (Article in French)
Kamoun A, Hammou A, Chaouachi S, Bellagha I, Lakhoua R (1995) Radiological signs of type I primary hyperoxaluria. Ann Radiol (Paris) 38:440–446 (Article in French)
Levadoux M, Picon G, Gadea J, Delarue A, Jouve JL, Bollini G (1999) Iterative fractures in type I primary hyperoxaluria. Report of 2 cases. Rev Chir Orthop Reparatrice Appar Mot 85:75–80 (Article in French)
Lagier R, Revell P, Schoenboerner A (1982) Calcium oxalate deposition in growing bone: anatomical and radiological study in a case of primary oxalosis. Metab Bone Dis Rel Res 4:49–59
Martijn A, Thijn CJ (1982) Radiologic findings in primary hyperoxaluria. Skeletal Radiol 8:21–24
Reginato AJ, Ferreiro Seoane JL, Barbazan Alvarez C, Mitja Piferrer J, Vidal Meijon L, Pascual Turon R, Vasconez F, Rivera ER, Clayburne G, Rothfuss S (1986) Arthropathy and cutaneous calcinosis in hemodialysis oxalosis. Arthritis Rheum 29:1387–1396
Ring E, Wendler H, Ratschek M, Zobel G (1989) Bone disease of primary hyperoxaluria in infancy. Pediatr Radiol 20:131–133
Schnitzler CM, Kok JA, Jacobs DW, Thomson PD, Milne FJ, Mesquita JM, King PC, Fabian VA (1991) Skeletal manifestations of primary oxalosis. Pediatr Nephrol 5:193–199
Vichi GF, Bongini U, Seracini D, Lavoratti GC (1995) Progression of bone lesions in a child with primary hyperoxaluria type 1: evaluation by roentgenology an MRI. Pediatr Radiol 25(Suppl 1):S102–S104
Wiggelinkhuizen J, Fisher RM (1982) Oxalosis of bone. Pediatr Radiol 12:307–309
Yalcinkaya F, Tumer N, Onder S, Kuzu I (1998) An 8-year-old boy with renal failure, nephrolithiasis and bone pain. Eur J Pediatr 157:255–256
Author information
Authors and Affiliations
Corresponding author
About this article
Cite this article
El Hage, S., Ghanem, I., Baradhi, A. et al. Skeletal features of primary hyperoxaluria type 1, revisited. J Child Orthop 2, 205–210 (2008). https://doi.org/10.1007/s11832-008-0082-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11832-008-0082-4