
Abstract
The emerging role of interleukin-17 as a hallmark proinflammatory cytokine of the adaptive immune system produced by a new T helper cell subset termed “Th17” has received considerable attention. In this review we will focus on recent information regarding IL-17 and its relevance in autoimmune and chronic inflammatory diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Originally termed cytotoxic T lymphocyte antigen 8, interleukin (IL)-17 is a 17 kDa type 1 transmembrane protein isolated initially from a rodent CD4 T cell cDNA library [1]. It represents the prototype of a recently identified family of cytokines that comprises six members (IL-17A to IL-17F) and five receptors (IL-17RA to IL-17RE) [1, 2]. The most closely related members of the family are IL-17A and IL-17F that bind to the same receptors (IL-17RA and IL-17RC) [1]. IL-17 was initially described in 1995/96 as a proinflammatory cytokine, and is produced by several cell types including CD4+ T lymphocytes, CD8+ T lymphocytes, CD3+ CD4−CD8− T cells, γδ-T cells, natural killer cells and neutrophils [3]. The discovery of Th17 cells came from murine models of autoimmunity such as experimental autoimmune encephalomyelitis (EAE), collagen-induced arthritis (CIA) and inflammatory bowel diseases, which have been historically thought to be mainly due to Th-1 responses. The CD4+ T lymphocyte effector subset termed “Th17” has been considered a remarkable discovery that was named after its signature cytokine, IL-17 [4]. Th17 cells are considered as a distinct T helper lymphocyte subset because: (a) they arise from naïve T cells when primed in the presence of specific factors; (b) exhibit a particular cytokine production profile; and (c) their differentiation is controlled by exclusive transcription factors. Development of Th17 cells can be divided into three stages: differentiation, driven by transforming growth factor-β1(TGF-β), IL-1β and IL-6, autocrine proliferation triggered by IL-21, and amplification maintained by IL-23 [5, 6].
Human Th17 cells originate in response to IL-1β and IL-23 from a subset of CD4+ T cell precursors, detectable in the thymus and umbilical cord blood (UCB), expressing the CD161 antigen, a lectin-like receptor that is the equivalent of murine NK1.1 [7]. Th17 cells constitutively express a unique transcription factor, the retinoic acid receptor-related orphan receptor C (RORC), which induces transcription of the IL-17 gene in naïve helper T cells, and is required for the development of IL-17 producing cells in the presence of interleukin-6 and TGF-β. Activation of RORC also causes expression of the IL-23 receptor whose engagement by IL-23 enhances the expression of IL-17 and IL-22. Finally, Th17 cells express high levels of the CC chemokine receptor 6 (CCR6) and theβ2 chain of the IL-12 receptor [7].
Although it is widely accepted that murine Th17 cells originate from naïve CD4+ T lymphocytes in the presence of IL-6 and TGF-β, and that their development is then stabilized or amplified by IL-23 and IL-21, several studies have denied the role of TGF-β in human Th17 cell differentiation [8, 9]. These differences between humans and mice were attributed either to the difficulty in ensuring a truly naïve T cell population in humans, or to a blocking activity of TGF-β on the amplification of Th1 cells [8]. More recently, three independent studies show, in contrast to the earlier observations, that the differentiation of human Th17 cells also requires, in addition to IL-1β and IL-6, the activity of TGF-β suggesting that in previous studies the role of this cytokine had been underestimated because CD4+ T cells were cultured in media added with human or bovine serum that usually contains TGF-β [10, 12–14]. However, the results of these studies are somehow contradictory. First, Manel et al. [8, 10] found that TGF-β, IL-1β, IL-6 and IL-21 (or IL-23) are necessary and sufficient to induce IL-17 expression in naïve UCB human CD4+ T lymphocytes, and that TGF-β upregulates RORC expression but simultaneously inhibits its ability to induce IL-17 expression. Second, Volpe et al. [8, 11] suggest that TGF-β, IL-23, IL-1β and IL-6 are all essential for human Th-17 cell development, but differentially modulate the cytokines produced by Th17 lymphocytes, and that the absence of TGF-β favors a shift from a Th17 profile to a Th-1-like profile. Finally, Yang et al. [10, 12] demonstrate that TGF-β and IL-21 promote the differentiation of human naïve T CD4+ T lymphocytes in Th17 cells accompanied by the expression of RORC.
Th17 cells and their effector cytokines (IL-17A, IL-17F, IL-21 and IL-22) mediate host defensive mechanisms to various infections, especially those caused by extracellular bacteria and fungi [13]. In fact, IL-17A-deficient mice show enhanced susceptibility to experimental K. pneumoniae and Toxoplasma gondii infections, but not to intracellular bacterial infections (i.e. Mycobacterium tuberculosis and Listeria monocytogenes) [13]. The role of IL-17A and IL-17F in fungal infections remains controversial. It has been recently shown that IL-17A negatively regulates Th1 responses to Aspergillus fumigatus and Candida albicans favoring the growth of these pathogens [13]. By contrast, Fenoglio et al. [14] demonstrate that in early HIV-1-infected patients peripheral blood Vdelta1 T lymphocytes, bearing the CD27 surface marker of memory T cells, proliferate in vitro in response to Candida albicans and co-express cytoplasmic interferon (IFN)-γ and IL-17.
Of note, IL-17 and IL-22 can synergistically or additively increase antimicrobial proteins in skin keratinocytes, and induce the expression of several host-defence genes (i.e. human beta defensin-2) in bronchial epithelial cells [15]. This antimicrobial-peptide production augments the epithelial-barrier function, and is critical for mucosal host defence [13]. Recent studies show that CD4+ T cells in the intestinal mucosa comprise significant numbers of Th17 lymphocytes [16]. A hypothetical model suggests that discrete subsets of intestinal bacteria produce toll-like receptor ligands, which activate antigen presenting cells inducing Th17 development, and release ATP that stimulates dendritic cells to synthesize IL-6 and TGF-β thus promoting Th17 differentiation [16].
The possibility that polarized Th17 cells can be switched in vitro and in vivo to Th17/Th1 and then to Th1 suggests the “plasticity” of this T cell subset, and supports the concept of a shared ancestry between Th17 and Th1 cells both expressing CD161 [17]. Of note, the expression of Th17 genes (RORC2, CCR6 and IL-23) in Th1 synovial cells from inflamed joints, and the overlap in TCR sequences between Th17 and Th1 lymphocytes suggest that Th17 to Th1 conversion may be a real phenomenon. Finally, Cosmi et al. [18] demonstrate the existence of human Th17/Th2 lymphocytes, originating from memory CCR6+ CD161+ CD4+ T cells when exposed to IL-4, which can produce IL-4, IL-5, IL-9 and IL-13 in addition to IL-17A, IL-8 and IL-22. These cells are able to induce the production of IgE confirming that Th17/Th2 may express the most important functional properties of both Th17 and Th2 lymphocytes [18].
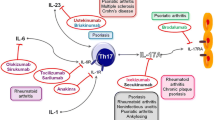
The concept of Th17 cells was conceived within the setting of experimental autoimmune diseases, and these cells have been intimately associated, since their first description, with autoimmune responses. In fact, in addition to its potent proinflammatory capacity, IL-17 exerts its effects through several mechanisms including the recruitment of monocytes and neutrophils by increasing the local production of chemokines (IL-8, monocyte chemoattractant protein-1, growth-related oncogene protein-α), the facilitation of T cell infiltration and activation by stimulating the expression of intercellular adhesion molecule-1, and the amplification of the immune response by inducing the production of IL-6, prostaglandin E2, granulocyte–macrophage colony-stimulating factor and granulocyte colony-stimulating factor [1, 19]. In addition, IL-17 synergizes with other cytokines, such as IL-1β, tumor necrosis factor (TNF)-α and IFN-γ and its receptor IL-17RA is expressed broadly and mediates IL-17 effects through a number of immune and non-immune cells (particularly endothelial and epithelial cells) [6]. However, there is now general agreement that both Th17 and Th1 cells are involved in autoimmune diseases. In particular, Th17 cells mediate the inflammatory response, whereas Th1 cells determine the tissue damage. In experimental autoimmune uveitis (EAU), autoimmunity to the retina can be both Th17 and Th1 driven [20]. In fact, IL-23 plays an important role in the development and intensity of disease through the generation of Th17 cells, whereas IL-12 promotes the generation of IFN-γ-producing Th1 cells that cause retinal damage. Of note, each effector phenotype by itself is sufficient to induce pathology in the absence of the reciprocal hallmark cytokine. Similarly, IL-23 and IL-12 have a significant role in the induction and maintenance of EAE as shown by the ability to transfer the disease through tolerance-induced myelin-specific T cells after antigenic challenge in the presence of these cytokines [21]. This finding also suggests that in this animal model of autoimmunity, the development of Th17 and Th1 lymphocytosis capable of engaging distinct proinflammatory pathways characterized by neutrophilic and macrophage infiltrates, respectively. Finally, Bending et al. [22] and Martin-Orozco et al. [23] report that Th17 and Th1 cells are involved in the pathogenesis of type 1 diabetes. In fact, induction of diabetes in NOD/SCID mice via adoptive transfer of Th1 cells from BDC2.5 transgenic mice is prevented by treatment of recipient mice with a neutralizing IFN-γ-specific antibody, suggesting a major role of Th1 lymphocytes in the induction of the disease. Nevertheless, transfer of highly purified Th17 cells from BDC2.5 transgenic mice also causes diabetes in NOD/SCID recipients with similar rates of onset as in the transfer of Th1 cells. However, treatment of NOD/SCID mice with neutralizing IL-17-specific antibody does not prevent the development of the disease. Therefore, it has been suggested that islet-reactive Th17 lymphocytes are able to promote pancreatic inflammation, but only induce type 1 diabetes after conversion into IFN-γ producing cells thus confirming the plasticity of the Th17 population. The critical points of the pathophysiology of Th-17 cells are summarized in the Fig. 1.

Potential role of Th-17 cells in autoimmune and chronic inflammatory diseases
Based on the current knowledge, it appears that Th17 cells are responsible for many of the inflammatory and autoimmune responses once attributed to Th1 lymphocytes. This review focuses on the key role of IL-17 in the pathogenesis of autoimmune and chronic inflammatory diseases.
Rheumatoid arthritis
Rheumatoid arthritis (RA) is characterized by the chronic inflammation of the synovium membrane of the joints. Resulting cell interactions induce proinflammatory cytokine production that in turn activates the release of proteases leading to bone and cartilage destruction. Concordant results using mouse and human models of RA show that IL-17 is involved in the proinflammatory responses associated with joint inflammation, cartilage damage and bone erosion [24]. The link between IL-17 and RA was reinforced when it was shown that pieces of RA synovium could produce bioactive IL-17, and that the synovial fluid from RA patients contains IL-17 that promotes osteoclastogenesis by inducing the expression of the receptor activator of NF-κB Ligand (RANK-L) on mesenchymal cells [24–26]. Furthermore, a strong correlation between IL-15 concentration and IL-17 level in the synovial fluid is reported in RA patients [26]. Moreover, a subset of infiltrating T cells in RA synovium expresses IL-17. RA synovium pieces induce a massive production of IL-6 by RA synoviocytes, and anti-IL-17 monoclonal antibody significantly decreases IL-6 production [24]. In addition to synovium inflammation, IL-17 induces bone resorption [24, 27] and contributes to joint prosthesis loosening and periodontal disease [24, 28]. In a clinical study of RA patients, the expression of IL-17 in synovium biopsies is associated with an increase in both activity and severity of the disease [20, 24–30]. It is also proposed that IL-17 may activate the alternative complement pathway proteins C3 and factor B, both of which are upregulated in RA synovial tissue, through synergy with TNF-α. Abnormalities in the activation of the alternative complement pathway have also been implicated in the pathogenesis of autoimmune arthritis [31].
IL-17 may intensify local inflammation by promoting angiogenesis, and recruiting innate immune cells into the joints. Indeed, IL-17 increases the production and expression of vascular endothelial growth factor (VEGF) in RA fibroblast-like synovial cells in vitro thus favoring neutrophils, macrophages and T lymphocytes recruitment into the joints [31]. Moreover, RA synovium-derived T cells express the lymphocyte adhesion molecules LFA-1 and CD2, which are the ligands of synovial cell ICAM-1 and LFA-3 molecules, respectively, and constitutively express IL-17, while the surrounding synovial macrophages and fibroblasts produce IL-15, a known stimulus for IL-17 [31]. Furthermore, IL-23, which is a differentiating cytokine for Th17 cells, plays a key role in the pathogenesis of joint inflammation and in the progression of the disease [31, 32]. Indeed, Murphy et al. [32] have demonstrated that IL-23/p19-deficient mice are resistant to the development of the autoimmune inflammation of joints, and are unable to generate IL-17 producing CD4+ Th17 lymphocytes. Additionally, IL-17 induces in synoviocytes the expression of cyclooxygenase-2 (COX-2), a stress response molecule contributing to the high levels of prostaglandin E2 (PGE2) observed during inflammation. Of note, PGE2 favors the expansion of the Th17 lineage by shifting dendritic cells from IL-12 to IL-23 phenotype [30]. By contrast, experimental models of arthritis demonstrate that COX-2 deficiency attenuates acute and chronic inflammation [25]. Finally, IL-27, which is the most recently described member of the IL-12 family, is suggested to be involved in the early initiation of the Th1 responses [33]. IL-27 binds to a receptor composed of WSX-1/TCCR and gp130, the latter of which serves as a common signal transduction receptor for IL-6-related family members. IL-27 inhibits the IL-6 plus transforming growth factor (TGF)-β-mediated differentiation of Th17 lymphocytes and mice defective for IL-27 receptor WSX-1 show higher levels of circulating Th17 cells. Therefore, IL-27 generally exerts anti-inflammatory activity and may be regarded as a suppressor of autoimmunity [33].
Systemic lupus erythematosus
Systemic lupus erythematosus (SLE) has been classically considered a complex autoimmune disease in which T cell-driven autoimmune response against ubiquitously expressed autoantigens results in clinically and pathologically diverse manifestations [1, 34]. Although the presence of a large array of autoantibodies is perhaps the most typical characteristic of SLE patients, target organ infiltration and chronic inflammation are essential pathogenic features that result in end-organ damage in most SLE clinical manifestations such as nephritis, vasculitis and discoid lupus [1, 35]. Recent evidence indicates that IL-17 plays a role in the pathogenesis of SLE. SLE patients have higher serum levels of IL-17 and IL-23 than healthy controls [1, 36].
Moreover, the frequency of IL-17-producing T lymphocytes is increased in the peripheral blood of SLE patients, and plasma IL-17 levels show a positive correlation with SLE disease activity [1, 36]. Recent work demonstrates that a significant fraction of the IL-17 produced in SLE patients derives from double-negative (DN) TCR-αβ+CD4−CD8− T lymphocytes [37]. Scarce in healthy individuals, DNT cells are expanded in the peripheral blood of SLE patients, and produce IL-17 and IFN-γ. Moreover, DN T lymphocytes and IL-17-producing T cells are also found in kidney biopsies from patients with lupus nephritis. The finding of DN T lymphocytes within a T cell infiltrate confirms their capacity to accumulate in inflamed tissue, and strongly suggests that they play a pathogenic role in the local inflammatory response favoring the amplification and the perpetuation of the damage in the organs targeted by SLE [1, 37]. Lastly, IL-17 also induces the activation of B lymphocytes increasing autoantibodies production [1]. Zhang et al. find that T cells from lupus-prone animals express high levels of IL-17A, and that IL-17 A+ cells can be identified in the kidneys of mice with active nephritis [38]. These IL-17A+ cells are overwhelmingly CD4−CD8−, a finding that is in keeping with the human data. Moreover, as the mice age and the disease worsen, lymphocytes progressively express higher levels of IL-17A. These observations provide a link between IL-17A and immunopathology in murine lupus. This adds SLE to a list of experimental autoimmune diseases (e.g., experimental encephalomyelitis, experimental arthritis) where Th17 lymphocytes are found in the areas of tissue damage [25, 30, 38]. The lymphocytes isolated from lupus-prone animals progressively express higher levels of IL-23 receptor mRNA as their disease becomes more severe. Even more importantly, IL-23 induces proliferation of DN T lymphocytes in vitro. Therefore, IL-23 seems to act as a “trophic/inducing” cytokine for a specific subset of T lymphocytes that do not bear the CD4 or CD8 molecules and produce IL-17 instead of the classic IL-2 and IFN-γ. Moreover, IL-23 acts as a differentiating factor for activated but not naïve CD4+ T cells [38]. Finally, IL-23 also promotes an autoimmune humoral response with autoantibodies deposition in the kidneys and complement activation resulting in nephritis. One possible mechanism that would explain this phenomenon is that IL-23-induced Th17 lymphocytes (primarily DN T lymphocytes) provide excessive help to B lymphocytes, favoring IgG deposition in the kidneys, rather than attracting leukocytes in the site of inflammation [38].
Psoriasis
Psoriasis is a polygenic chronic inflammatory disease with a significant proportion of patients suffering from additional joint involvement, which may finally lead to joint destruction and significant functional impairment. Interestingly, evidence is accumulating in recent years that psoriasis may be a multisystem disease involving even coronary arteries and the heart [25, 39, 40]. Epidermal thickening (acanthosis) of the skin is a hallmark of psoriasis. Skin biopsies from patients with psoriasis show a high expression of IL-17 as well as of IL-23 and IL-22. Nevertheless, the respective contribution of these cytokines in the pathogenesis of the disease remains unclear, even if it has been demonstrated that IL-22 mediates IL-23-induced acanthosis [24, 25]. The positive results from a clinical trial with an anti-IL-17 antibody obtained in patients with psoriasis [24] seem to confirm a major role for IL-17 in the development of psoriasis, but do not exclude an indirect effect mediated by other cytokines like IL-22 and IL-23. Moreover, in skin biopsies obtained from patients with psoriasis and treated with a TNF-α inhibitor, a decreased expression of IL-23, IL-22 and IL-17 is observed, suggesting possible interactions of these cytokines with TNF-α [24].
Multiple sclerosis
Multiple sclerosis (MS) is a chronic inflammatory demyelinating disease of the central nervous system generally affecting the white matter, which develops in genetically predisposed individuals. It is well known that IFN-γ plays an important role in MS [25], however, recent research shows that monocyte-derived dendritic cells obtained from patients with MS secrete higher amounts of IL-23 than healthy controls, suggesting that the IL-23/Th17 pathway may also play a role in the development of MS [41]. Using in situ hybridization, Matusevicius et al. demonstrate higher numbers of IL-17 mRNA-positive mononuclear cells in the peripheral blood of 40% of patients with MSas compared with healthy subjects [41]. Interestingly, patients with disease exacerbation have 3.5-times more IL-17 mRNA-positive mononuclear cells than patients in remission [41]. Furthermore, patients with MS have a significant number of IL-17 mRNA-positive mononuclear cells in the cerebrospinal fluid as compared to patients suffering from other non-inflammatory neurological diseases, suggesting an enrichment or accumulation of Th17 lymphocytes at the sites of inflammation in MS [41]. Finally, CD4+ T cells isolated from patients with MS and stimulated in vitro with an anti-CD3 antibody, produce significantly more IL-17 than T cells from healthy controls, whereas secrete comparable amounts of IFN-γ [41].
Crohn’s disease
Crohn’s disease and ulcerative colitis are the two main chronic inflammatory bowel diseases. Crohn’s disease affects specific locations of the digestive tract that differ from those affected in ulcerative colitis. In both diseases, local inflammation in the mucosa leads to the destruction of the lamina, with complications such as perforations and internal or external fistulas. Inflammatory joint manifestations different from those seen in RA may be associated. Although their exact etiology is still not completely understood, it has been proposed that their pathogenesis is characterized by an exaggerated immune response in genetically susceptible individuals [42]. As in RA and psoriasis, the contribution of TNF-α is demonstrated by the positive effect of TNF-α inhibition in a significant proportion of patients with chronic inflammatory bowel diseases [24]. Studies performed with endoscopic bowel biopsies show that IL-23, IL-17 and IL-12 are over expressed in mucosal lesions of Crohn’s disease [43]. Studies in autoimmune animal models, traditionally recognized as Th1-disease models, revealed that tissue inflammation does not develop in mice deficient in the IL-23p19 subunit, while inflammation is seen in IL-12p35 deficient mice, suggesting that these models of autoimmunity are linked to Th17 and not primarily to Th1 responses [44]. Moreover, CD40-induced colitis depends on IL-23p19 secretion, while IL-12p35 secretion controls wasting disease and serum cytokine production, but not mucosal immunopathology [44]. Novel data also indicate that IL-23 restrains regulatory T cells (Treg) activity to drive T lymphocyte-dependent colitis. The frequency of naïve T cell-derived forkhead box P3 (FOXP3)+ cells in the colon increases in the absence of IL-23, indicating a role for IL-23 in controlling Treg induction. FOXP3-deficient T cells induce colitis when transferred into recipients lacking IL-23p19, showing that IL-23 is not essential for intestinal inflammation in the absence of FOXP3 [44, 45]. These data suggest that the over-riding immunosuppressive activity is an important function of IL-23 in the intestine. An increased number of T lymphocytes expressing retinoid-related orphan receptor-γt (RORγt), the master transcription factor for Th17 cells, in the lamina propria of patients with Crohn’s disease is also demonstrated [44]. However, a number of investigators identify a subset of Th17 lymphocytes that may coproduce the Th1 cytokine IFN-γ [7, 44]. This finding is particularly prominent at sites of inflammation in active Crohn’s disease, suggesting that IFN-γ may not always downregulate IL-17A production, and may contribute to the pathogenetic and proinflammatory functions of Th17 cells. Indeed, increased expressions of IL-17A and IL-17F mRNA are found in the intestinal mucosa of patients with Crohn’s disease [44]. Two independent studies also demonstrate Th17 cells in the peripheral blood and in the gut from patients with Crohn’s disease. These cells are characterized by the expression of RORγt, IL-23R and CCR6, whereas they lack CXCR3, a chemokine receptor that is characteristic of Th1 cells [7, 44]. Moreover, Annunziato et al. demonstrate IL-17A-producing T cells in the gut, including T lymphocyte populations that also express both IL-17A and IFN-γ named “Th17/Th1” cells [7]. Finally, very recent findings implicate CD161 as a novel surface marker for human Th17 lymphocytes, and prove the exclusive origin of these cells from a common CD161+ CD4+ T cell progenitor [7, 44]. Therefore, the interactions between Th1 and Th17 lymphocytes and the role of IFN-γ on Th17 cells may be more complex than previously assumed, and require further analysis to delineate the specific contributions of these cell lines to the development of Crohn’s disease.
Allergic rhinitis and asthma
Allergic rhinitis and asthma are characterized by an inflammatory reaction associated with increased production of Th-2-type cytokines, such as IL-4 and IL-13. Indeed, peripheral blood mononuclear cells of allergic rhinitis patients predominantly produce IL-4 with respect to IFN-γ. However, Th-1-type cytokines may also be involved in inflammatory events arising in allergic rhinitis by producing IFN-γ [46]. In fact, serum IL-17 levels seem to be significantly related to some clinical and inflammatory parameters in patients with persistent moderate-to-severe allergic rhinitis evaluated during the pollen season. Moreover, serum IL-17 levels correlate well with clinical severity as documented by symptom score and drug consumption. In addition, serum IL-17 levels correlate with allergic inflammation as documented by the relationship found with the number of peripheral blood eosinophils. [46]. Finally, Th-17 cells may be implicated in chronic events of allergic reaction that endure over time until allergen exposure occurs [46]. Therefore, IL-17A may be proposed as a new biomarker of disease progression and allergy severity. Ex vivo analysis of peripheral CD3+ CD4+ T lymphocytes clearly shows that allergic patients have significantly more elevated percentages of CD4+ IL-17+ and CD4+ IFN-γ+IL17+ T cells than normal controls, further underlying the possible role of Th17 cells in allergic inflammation during pollen exposure. Furthermore, allergen stimulus is able to induce an expansion of both CD4+ IL-17+ and CD4+ IFN-γ+IL17+ allergen-specific T clones at a higher frequency in allergic patients than in normal controls and CD8+ IL-17+ and CD8+ IFN-γ+IL17+ T cells also occur more frequently in allergic patients than in normal subjects [47]. As previously reported, Cosmi et al. [18] postulate that Th17/Th2 lymphocytes originate from memory CCR6+ CD161+ CD4+ T cells when exposed to IL-4, suggesting that an IL-4 rich microenvironment favors the shifting of Th17 cells to Th17/Th2 cells. The demonstration in vitro and ex vivo of CD4+ T lymphocytes able to produce both Th17 and Th2-related cytokines, together with their increase in the circulation of patients with asthma, raises the important question of the pathophysiological role of this novel subset in allergic disorders. Lajoie et al. [48] demonstrate a critical role for complement-mediated regulation of the IL-23/Th17 axis in severe asthma. Indeed, as the gene encoding C5 has been identified as a susceptibility gene for asthma, these authors explore the possibility that its alterations may underlie excessive Th17 production in susceptible strains of mice. They prove a direct link among C5aR signaling, IL-17A production and severe airway hyperresponsiveness (AHR), as the enhancement of AHR severity noted in mice after C5aR blockade is completely reversed by concurrent IL-17A blockade. The ability of C5a to stimulate IL-10 production probably explains its ability to suppress IL-12p70 and IL-23p19, and, thus, Th1 and Th17 responses, respectively, supporting the hypothesis that C5a regulates the maintenance of tolerance at the mucosal surface [48]. In contrast, polymorphisms in the gene encoding C5 are associated with protection from asthma in humans [48]. The activation of C3 after the exposure to allergens favors the shift toward the activation of the IL-23/Th17 axis. Finally, once produced, IL-17A alone or synergistically with IL-13, initiates an amplification loop by directly inducing more C3 production by pulmonary cells, enhancing IL-23 production, and thereby perpetuates the response. Therefore, this aberrant Th-17 response occurs as a result of a shift from C5a-driven tolerance toward C3a-driven Th-17 responses at the airway surface [48]. Finally, IL-17 expression by both T cells and eosinophils is increased in the airways of asthmatic patients, and is required during initial allergic sensitization, although it is yet uncertain whether it is correlated with neutrophilic inflammation and the development of airflow obstruction [48]. Collectively, these results confirm that Th17 lymphocytes may be involved in the immune response to causal allergens, and could contribute to the worsening of the inflammatory disease in patients with allergic rhinitis and asthma.
The implications of Th-17 cells in the reported autoimmune and chronic inflammatory diseases are summarized in Table 1.
Conclusions and future directions
The complex series of processes involved in the protection against immune responses toward self-antigens is known as immunological tolerance. Mechanisms operating at the level of primary lymphoid organs (bone marrow and thymus) and at the level of secondary lymphoid organs (spleen, lymph nodes and mucosal lymphoid tissue) have been described. Failure or breakdown of mechanisms responsible for the development or maintenance of immunological tolerance, result in immune responses against autoantigens, and thus, in the development of autoimmune diseases [49]. Moreover, in the majority of autoimmune diseases, both genetic and environmental factors together contribute to induce the breakdown of tolerance against self-antigens. Infections are certainly critical in triggering autoimmune responses through different mechanisms including those inducing Th1 or Th17 cell responses against self-peptides cross-reactive with microbial peptides (epitope mimicry) [49]. The identification of IL-17 producing T lymphocytes as a distinct subset of proinflammatory helper T cells has broken down old paradigms concerning the role of Th1 lymphocytes in autoimmune disorders. Growing evidence indicates that IL-17 plays a key role in various steps of RA, SLE and other autoimmune and chronic inflammatory diseases development, and is associated not only with T cell-mediated tissue injury, but also with the production of pathogenic autoantibodies. Although a growing list of diseases has been associated with IL-17, the final demonstration of its contribution to disease pathogenesis is still missing. Therefore, it appears crucial to determine the details and resolve the inconsistencies surrounding the involvement of the Th17 network in autoimmune diseases in order to design specific therapies that not only reduce the degree of inflammation, but also delay the progression of the disease. Cytokine targeting with various inhibitors including monoclonal antibodies against TNF-α and IL-6 receptor as well as with TNF-α soluble receptor and IL-1 receptor antagonist is efficacious in several immune mediated diseases. Analogous tools are now available to target the IL-17 pathway including monoclonal antibodies against IL-17 and IL-17R, and active research is looking for small molecules able to control RORγt, the transcription factor characteristic of the Th17 pathway.
References
Nalbandian A, Crispín JC, Tsokos GC (2009) Interleukin-17 and systemic lupus erythematosus: current concepts. Clin Exp Immunol 157(2):209–215
Weaver CT, Hatton RD, Mangan PR, Harrington LE (2007) IL-17 family cytokines and the expanding diversity of effector T cell lineages. Annu Rev Immunol 25:821–852
Korn T, Oukka M, Kuchroo VK, Bettelli E (2007) Th17 cells: effector cells with inflammatory properties. Semin Immunol 19:362–371
Korn T, Bettelli E, Oukka M, Kuchroo VK (2009) IL-17 and Th17 cells. Annu Rev Immunol 27:485–517
Zenewicz LA, Antov A, Flavell RA (2009) CD4 T-cell differentiation and inflammatory bowel disease. Trends Mol Med 15(5):199–207
Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL (2003) Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem 278:1910–1914
Cosmi L, De Palma R, Santarlasci V, Maggi L, Capone M, Frosali F et al (2008) Human interleukin 17-producing cells originate from a CD161+ CD4+ T cell precursor. J Exp Med 205(8):1903–1916
Annunziato F, Romagnani S (2009) Do studies in humans better depict Th17 cells? Blood 114(11):2213–2219
Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F (2007) Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol l8(9):942–949
Manel N, Unutmaz D, Littman DR (2008) The differentiation of human Th17 cells requires transforming growth factor-beta and induction of the nuclear receptor ROR gamma t. Nat Immunol 9(6):641–649
Volpe E, Servant N, Zollinger R, Bogiatzi SI, Hupé P, Barillot E et al (2008) A critical function for transforming growth factor-beta, interleukin-23 and proinflammatory cytokines in driving and modulating human Th17 responses. Nat Immunol 9(6):650–657
Yang L, Anderson DE, Baecher-Allan C, Hastings WD, Bettelli E, Oukka M et al (2008) IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature 454(7202):350–352
Ouyang W, Kolls JK, Zheng Y (2008) The biological functions of T helper 17 cell effector cytokines in inflammation. Immunity 28(4):454–467
Fenoglio D, Poggi A, Catellani S, Battaglia F, Ferrera A, Setti M et al (2009) Vdelta1 T lymphocytes producing IFN-gamma and IL-17 are expanded in HIV-1-infected patients and respond to Candida albicans. Blood 113(26):6611–6618
Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M et al (2006) Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med 203:2271–2279
Honda K, Takeda K (2009) Regulatory mechanisms of immune responses to intestinal bacteria. Mucosal Immunol 2(3):187–196
Nistala K, Adams S, Cambrook H, Ursu S, Olivito B, de Jager W et al (2010) Th17 plasticity in human autoimmune arthritis is driven by the inflammatory environment. Proc Natl Acad Sci USA 107(33):14751–14756
Cosmi L, Maggi L, Santarlasci V, Capone M, Cardilicchia E, Frosali F et al (2010) Identification of a novel subset of human circulating memory CD4(+) T cells that produce both IL-17A and IL-4. J Allergy Clin Immunol 125(1):222–230
Schwarzenberger P, Huang W, Ye P, Oliver P, Manuel M, Zhang Z et al (2000) Requirement of endogenous stem cell factor and granulocyte-colony-stimulating factor for IL-17-mediated granulopoiesis. J Immunol 164:4783–4789
Luger D, Silver PB, Tang J, Cua D, Chen Z, Iwakura Y et al (2008) Either a Th17 or a Th1 effector response can drive autoimmunity: conditions of disease induction affect dominant effector category. J Exp Med 205(4):799–810
Kroenke MA, Carlson TJ, Andjelkovic AV, Segal BM (2008) IL-12- and IL-23-modulated T cells induce distinct types of EAE based on histology, CNS chemokine profile, and response to cytokine inhibition. J Exp Med 205(7):1535–1541
Bending D, De La Peña H, Veldhoen M, Phillips JM, Uyttenhove C, Stockinger B et al (2009) Highly purified Th17 cells from BDC2.5NOD mice convert into Th1-like cells in NOD/SCID recipient mice. J Clin Investig 119:565–572
Martin-Orozco N, Chung Y, Chang SH, Wang YH, Dong C (2009) Th17 cells promote pancreatic inflammation but only induce diabetes efficiently in lymphopenic hosts after conversion into Th1 cells. Eur J Immunol 39(1):216–224
Miossec P (2009) IL-17 and Th17 cells in human inflammatory diseases. Microbes Infect 11(5):625–630
Kunz M, Ibrahim SM (2009) Cytokines and cytokine profiles in human autoimmune diseases and animal models of autoimmunity. Mediators Inflamm 979258, pp 20
Ziolkowska M, Koc A, Luszczykiewicz G, Ksiezopolska-Pietrzak K, Klimczak E, Chwalinska-Sadowska H et al (2000) High levels of IL-17 in rheumatoid arthritis patients: IL-15 triggers in vitro IL-17 production via cyclosporin A-sensitive mechanism. J Immunol 164(5):2832–2838
Sato K, Suematsu A, Okamoto K, Yamaguchi A, Morishita Y, Kadono Y et al (2006) Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J Exp Med 203:2673–2682
Andersson MK, Lundberg P, Ohlin A, Perry MJ, Lie A, Stark A et al (2007) Effects on osteoclast and osteoblast activities in cultured mouse calvarial bones by synovial fluids from patients with a loose joint prosthesis and from osteoarthritis patients. Arthritis Res Ther 9:R18
Kirkham BW, Lassere MN, Edmonds JP, Juhasz KM, Bird PA, Lee CS et al (2006) Synovial membrane cytokine expression is predictive of joint damage progression in rheumatoid arthritis: a two year prospective study (the DAMAGE study cohort). Arthritis Rheum 54:1122–1131
Peck A, Mellins ED (2009) Breaking old paradigms: Th17 cells in autoimmune arthritis. Clin Immunol 132(3):295–304
Wiekowski MT, Leach MW, Evans EW, Sullivan L, Chen SC, Vassileva G et al (2001) Ubiquitous transgenic expression of the IL-23 subunit p19 induces multiorgan inflammation, runting, infertility, and premature death. J Immunol 166:7563–7570
Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA et al (2003) Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med 198:1951–1957
Crispin JC, Kyttaris VC, Juang YT, Tsokos GC (2008) How signaling and gene transcription aberrations dictate the systemic lupus erythematosus T cell phenotype. Trends Immunol 29:110–115
Cohen RA, Bayliss G, Crispin JC, Kane-Wanger GF, Van Beek CA, Kyttaris VC et al (2008) T cells and in situ cryoglobulin deposition in the pathogenesis of lupus nephritis. Clin Immunol 128(1):1–7
Wong CK, Lit LC, Tam LS, Li EK, Wong PT, Lam CW (2008) Hyperproduction of IL-23 and IL-17 in patients with systemic lupus erythematosus: implications for Th17-mediated inflammation in auto-immunity. Clin Immunol 127:385–393
Crispín JC, Oukka M, Bayliss G, Cohen RA, Van Beek CA, Stillman IE et al (2008) Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J Immunol 181(12):8761–8766
Zhang Z, Kyttaris VC, Tsokos GC (2009) The role of IL-23/IL-17 axis in lupus nephritis. J Immunol 183(5):3160316–3160319
Ludwig RJ, Herzog C, Rostock A, Ochsendorf FR, Zollner TM, Thaci D et al (2007) Psoriasis: a possible risk factor for development of coronary artery calcification. Br J Dermatol 156(2):271–276
Ortonne JP (2008) Psoriasis, metabolic syndrome and its components. Ann Dermatol Venereol 135(Suppl 4):S235–S242
Vaknin-Dembinsky A, Balashov K, Weiner HL (2006) IL-23 is increased in dendritic cells in multiple sclerosis and downregulation of IL-23 by antisense oligos increases dendritic cell IL-10 production. J Immunol 176(12):7768–7774
Bennett JL, Stüve O (2009) Update on inflammation, neurodegeneration, and immunoregulation in multiple sclerosis: therapeutic implications. Clin Neuropharmacol 32(3):121–132
Brand S (2009) Crohn’s disease: Th1, Th17 or both? The change of a paradigm: new immunological and genetic insights implicate Th17 cells in the pathogenesis of Crohn’s disease. Gut 58(8):1152–1167
Yagi Y, Andoh A, Inatomi O, Tsujikawa T, Fujiyama Y (2007) Inflammatory responses induced by interleukin-17 family members in human colonic subepithelial myofibroblasts. J Gastroenterol 42:746–753
Izcue A, Hue S, Buonocore S, Arancibia-Cárcamo CV, Ahern PP, Iwakura Y et al (2008) Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity 28(4):559–570
Dambacher J, Beigel F, Zitzmann K, De Toni EN, Göke B, Diepolder HM et al (2009) The role of the novel Th17 cytokine IL-26 in intestinal inflammation. Gut 58(9):1207–1217
Ciprandi G, Filaci G, Battaglia F, Fenoglio D (2010) Peripheral Th-17 cells in allergic rhinitis: new evidence. Int Immunopharmacol 10(2):226–229
Robinson DS (2009) Regulatory T cells and asthma. Clin Exp Allergy 39(9):1314–1323
Lajoie S, Lewkowich IP, Suzuki Y, Clark JR, Sproles AA, Dienger K et al (2010) Complement-mediated regulation of the IL-17A axis is a central genetic determinant of the severity of experimental allergic asthma. Nat Immunol 11(10):928–935
Romagnani S (2006) Immunological tolerance and autoimmunity. Intern Emerg Med 1(3):187–196
Conflict of interest
None.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Murdaca, G., Colombo, B.M. & Puppo, F. The role of Th17 lymphocytes in the autoimmune and chronic inflammatory diseases. Intern Emerg Med 6, 487–495 (2011). https://doi.org/10.1007/s11739-011-0517-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11739-011-0517-7