
Abstract
CD4+ T helper cells are classical but constantly reinterpreted T-cell subset, playing critical roles in a diverse range of inflammatory responses or diseases. Depending on the cytokines they release and the immune responses they mediate, CD4+ T cells are classically divided into two major cell populations: Th1 and Th2 cells. However, recent studies challenged this Th1/Th2 paradigm by discovering several T-helper cell subsets with specific differentiation program and functions, including Th17 cells, Treg cells, and Tfh cells. In this chapter, we summarize the current understanding and recent progresses on the Th17 lineage differentiation and its effector impacts on variety of inflammatory responses or disease pathogenesis.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
5.1 Introduction
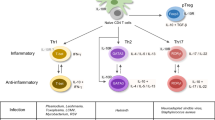
Upon various pathogens or injuries challenging, our body initiates both innate and adaptive inflammatory responses to protect us from infection or insult. CD4+ T helper cells are critical components for appropriate establishment of adaptive immune responses. After engagement of T-cell receptor (TCR) and co-stimulatory molecules, naive CD4+ T cells differentiate into different effector T helper cells under the control of distinct cytokines produced by particular pathogens or injuries activated antigen presenting cells (APCs). Over 25 years, Mosmann and Coffman’s Th1/Th2 paradigm in CD4+ T helper cells development shapes our view of the landscape of adaptive immunity [189]. Although this paradigm helps us a lot for understanding many aspects of adaptive immunity, some intriguing phenomena cannot be explained until the discovery of a third T helper cells lineage: the Th17 subset, which produces Interleukin-17 (IL-17, also called IL-17A) as its signature cytokine [129] (Fig. 5.1). The function of this newly emerged effector T helper cell subset appears to be distinct from those of Th1 and Th2 subsets and even reinforces certain roles of adaptive immunity system such as host defense and tissue repair responses, which cannot be fully achieved by the Th1/Th2 paradigm [129]. However, Th17 cells are also strong inducers of local tissue inflammation. Persistent and uncontrolled inflammation triggered by Th17 cells becomes a major stimulator in the pathogenesis of many human chronic diseases, which include autoimmune diseases and cancer [129]. The cytokines and the key transcription factors for its differentiation, maintenance, and expansion have been identified. The signaling pathways mediated by IL-17 and the responses mediated by its effector cytokines have also been elucidated.

The differentiation of Th17 cells. Naïve T cells can differentiate into three subsets of effector T helper cells under the control of distinct sets of cytokines. After TCR ligation, IL-12 and IL-4 promote Th1 and Th2 differentiation respectively, while the Th17 differentiation is controlled by TGF-β, IL-6, IL-23, IL-1β, and self-secreted IL-21. The differentiation of Th17 cells is inhibited by IFN-γ or IL-4. T-bet (also named Tbx21), GATA3, or RORγt (also known as Rorc) represents the linage-specific transcription factors for Th1, Th2, or Th17, respectively
In this chapter, we review the current understanding of the regulation of Th17 differentiation by the key cytokines and transcription factors and recent progresses of Th17 cell fate commitment and plasticity in both mouse and human. We also discuss the interplay of Th17 cells with other effector T cells such as Th1 and Treg cells. More importantly, Th17 cells produce characterized cytokines like IL-17A, IL-17F, and IL-22. All those unique cytokines mediate Th17-driven inflammatory responses in both physiological and pathogenic conditions. The signaling pathways and the efforts mediated by those cytokines are also addressed here.
5.2 The Discovery of Th17 Cells
Autoimmune diseases such as multiple sclerosis (MS) and rheumatoid arthritis (RA) are chronic inflammatory diseases. Experimental autoimmune encephalomyelitis (EAE) and collagen-induced arthritis (CIA) mouse models are the most common tools to study those human autoimmune disorders [19, 257]. From a long time, those diseases were associated with uncontrolled self-reactive Th1 responses, since the IFN-γ level was highly correlated with the pathogenesis of EAE and CIA. Blockage of the key Th1 differentiation cytokine IL-12 with antibodies eliminated the progression of EAE and CIA [148, 172]. Rodent genetic evidences demonstrated that both Th1 favorite transcription factors Tbx21- and Stat4-deficient mice were protected from EAE [17, 35]. All the above evidences indicate that the IFN-γ-producing self-reactive Th1 cells are responsive for the induction of those autoimmune responses. However, this Th1-dominant presumption was intrigued by some experimental contradictions: either the Ifng- or its receptor deficient mice were unexpectedly noticed to be susceptible to EAE, rather than resistant to EAE [132, 274]. Similar phenotypes were also observed in both Il12p35 (one subunit of IL-12)-and Il12rb2-deficient mice in EAE model [78, 318]. This paradox raises a question that whether there are unperceived cell populations required for the pathogenesis of EAE. In 2000, IL-12p40 was found to form a novel cytokine IL-23 with a newly identified subunit IL-23p19 [210]. Thus, IL-12p40 is a common subunit for both IL-12 and IL-23, but IL-12p35 and IL-23p19 are the unique subunit for IL-12 and IL-23, respectively. By comparing Il12p35-deficient mice with Il23p19-deficient mice in EAE or CIA model, researchers found that IL-23 rather than IL-12 was critical for the induction in both models [142, 44, 192]. These results also well explained the phenotype paradox between IL-12 antibodies blockage and Il12p35- or its receptor deficient mice in EAE, since blockage strategies targeted IL-12p40 subunit, which also affect the function of IL-23. Following studies showed that IL-23 was crucial for the development of IL-17-producing T helper cells, which was then named as Th17 cells [87, 215].
5.3 Th17 Cell Differentiation
Th17 cells are characterized by production of effector cytokines IL-17 as well as IL-17F, IL-21, and IL-22. Comparing with Th1 and Th2 lineages, Th17 lineage is a more plastic population in its fate determination and its differentiation is controlled by a distinct set of cytokines: after TCR activation and co-simulation, TGF-β and IL-6, which activate transcription factors Smads and STAT3 respectively, induce the expression of Th17 polarized transcription factor retinoid-related orphan receptor (ROR) γt to initialize Th17 differentiation from naive T cells. IL-21, the cytokine produced by Th17 cells, further promotes this process in a positive feedback manner. After upregulation of IL-23R by the above cytokines in Th17 cells, IL-23 binds to its receptor to drive the terminal differentiation of Th17 cells for their fully function achievement. Other Th17 cell commitment promoting or regulatory cytokines and transcription factors are also identified in recent years, and we will also discuss them in detail as below (Fig. 5.2).

The regulation of Th17 cell differentiation. Upon TCR activation, Th17 cells can be induced in presence of cytokines IL-6, IL-23, and IL-21 that activate STAT3. Activated STAT3 binds to the promoter regions and activates transcription of RORγt and RORα. Transcription factors BATF and IRF4 play a central role in RORγt mediated Th17 cell differentiation. Together with STAT3, RORγt, and RORα activate the expression of Th17 effector cytokines IL-17A, IL-17F, as well as IL-21 and IL-22. IL-6 mediated STAT3 activation also increases the expression of IL-23R, thus promoting the polarizing of Th17 cells. STAT3 activation also induces the expression of HIF1α to inhibit Foxp3 and promotes Th17 differentiation. IL-21 secreted by early Th17 cells functions as a self-amplified autocrine cytokine through IL-21 receptor. IL-1β promotes Th17 polarization by activation of MAPK and Akt/mTOR pathway. IL-1β also induces IRF4 to promote IL-21 secretion. Th17 differentiation is also promoted by activation of aryl hydrocarbon receptors (AHR). TGF-β signals through Smads to limit expression of genes encoding T-bet, Gata3 and other Th1 and Th2-related factors, thus enhancing the Th17 differentiation. TGF-β signaling cooperates with retinoic acid (RA) and IL-2-induced STAT5 activation to promote Foxp3 induction and Treg differentiation. Both Foxp3 and RORγt form complexes with Runx1 and regulate each other reciprocally. IL-12 and IFN-γ, which activate STAT4 and STAT1, respectively, promote Th1 differentiation by induction of T-bet and IFN-γ and then inhibit Th17 polarization. IL-27 also activates STAT1 to upregulate T-bet expression and thus inhibit Th17 development. Similarly, IL-4 signaling through STAT6 to induce GATA3 to suppress Th17 polarization and promotes Th2 differentiation
5.3.1 Positive Regulation of Th17 Differentiation by Cytokines
5.3.1.1 TGF-β
TGF-β is a pleiotropic cytokine in T-cell functions and its contribution in Th17 cell differentiation remains contradictory in different conditions [151]. In vivo evidences shown that Tgfb1-transgenic mice resulted in enhanced generation of Th17 cells and aggressive EAE phenotype when immunized with MOG35–55 in CFA [16]. Dominant negative Tgfbr2-transgenic mice did not develop the pathological signs of EAE and have no Th17 cells in the spinal cords [282]. T-cell specific Tgfb1-deficient mice also did not develop any clinical sign of EAE, and the number of Th17 cells was greatly reduced in those mice [153]. Although Treg derived TGF-β was shown to induce Th17 differentiation in vitro [281], but it was not required for Th17 differentiation in vivo, since mice with Foxp3-Cre-mediated disruption of TGF-β had similar Th17 cells and comparable EAE pathogenesis when compared with wild-type controls [84]. However, Ox40-Cre-mediated deletion of TGF-β in both activated CD4+ T cells and Tregs resulted in reduced Th17 cell frequencies and ameliorated EAE clinic signs [84], indicating that the production of TGF-β is critical for the pathogenic effector T cells during the organ specific autoimmune diseases. This study also found that, among the activated CD4+ T cells, the Th17 cells themselves produced considerable TGF-β to further promote their functions in an autocrine manner [84].
However, how TGF-β contributes to Th17 cell differentiation remains unclear. TGF-β was reported as an inhibitory cytokine for both Th1 and Th2 cell differentiation by suppressing the transcription factors T-bet and GATA-3, respectively [77, 199]. It was likely that TGF-β promote Th17 differentiation in an indirect way by inhibiting the fate determination of other effector T-cell subsets such as Th1 and Th2 cells. The studies have also shown that TGF-β enhanced Th17 responses by suppressing the transcription factors Eomes and Gfi1, which are critical for Th1 and Th2 related cytokines production [108, 331]. Indeed, comparing with IL-6 which activates STAT3 pathway, TGF-β induced much less expression of RORγt in T cells [327]. The IL-6 plus TGF-β produced stronger expression of RORγt than IL-6 alone [327], it is still unclear how TGF-β mechanistically contributes to Th17 cell differentiation. One study found that Th17 cells generated by IL-23, IL-6, and IL-1β rather than those induced by TGF-β and IL-6 caused the pathogenesis of EAE in a transfer model [74]. It seems that Th17 cells generated by TGF-β and IL-6 were not pathogenic, but they are regulatory Th17 cells which produced high amount of IL-10 [74, 180]. Thus, TGF-β in low concentrations could efficiently initiate the Th17 cell differentiation in cooperated with IL-6, but under this condition, its signaling was insufficient to generate inflammatory Th17 responses [281]. In the contrast, the high concentration of TGF-β favored the generation of induced FOXP3+ regulatory T cells (iTregs) rather than Th17 cells by inhibiting the expression of IL-23R and disrupting the function of RORγt [328], and high concentration TGF-β also inhibited IL-22 expression to generate IL-17+IL-22– cells [324]. The plastic interplay between Th17 cells and iTregs will be discussed further in the section below.
5.3.1.2 IL-6
IL-6 is also a multiple functional cytokine in immune system and is produced by many immune and stromal cell types [279]. IL-6 binds to its receptor complex IL-6Rα and gp130 to activate STAT1 and STAT3 for downstream biological activities [94]. Its roles in autoimmune diseases have been studied. Several lines of clinic evidences revealed that the patients with MS or RA exhibit higher levels of IL-6 in their cerebrospinal fluid or synovial fluids than normal controls [97, 243]. Previous studies found that Il6-deficient mice are protective from CIA and EAE [5, 206, 235] and anti-IL-6 receptor antibodies blockage in mice also suppressed the progression of both CIA and EAE diseases [65, 243]. More importantly, in human, the therapy by Tocilizumab, a humanized anti-IL-6 receptor antibody, had become a novel therapeutic strategy to prevent many autoimmune diseases including RA [200]. The resistance phenotype of Il6-deficient mice in the models of EAE and CIA has been poorly explored until different groups identified that IL-6 as a critical differentiation factor for Th17 cell generation. As described above, IL-6 was a strong suppressor of the TGF-β-driven induction of Foxp3 in T cells, instead, IL-6 plus TGF-β induced a unique transcriptional program resulting in the differentiation of Th17 cells [16]. At this condition, TGF-β also induced the expression of IL-6Rα and gp130 indicating that TGF-β is crucial to maintain the responsiveness of T cells to IL-6 [298]. IL-6 then binds to IL-6Rα and gp130 to recruit and phosphorylate the transcription factors STAT3. Deficiency of STAT3 in T cells impaired the induction of RORγt and RORα and consequentially abrogates the generation of Th17 cells [306, 307]. Accordingly, CD4-Cre-mediated STAT3 depletion in T cells protected mice from EAE [88]. Thus, IL-6 is a key switch factor that favors Th17 cell differentiation while suppressing the generation of Tregs.
5.3.1.3 IL-21
IL-21 was firstly described in 2000 by showing that this cytokine has a role in the proliferation of natural killer (NK) cell, B cell, and T-cell populations in different conditions [216]. IL-21 is a member of IL-2 family of cytokines and signals though the common γ chain of this family and IL-21R [159]. Although it can be produced by several activated CD+ T cells, IL-21was found to be highly produced in T follicular helper (Tfh) cells and Th17 cells [128, 165, 203, 327]. IL-6 served as a strong inducer of IL-21 [263] though the transcription factor STAT3 but not RORγt [327]. It was found that IL-21 plus TGF-β can also generate Th17 cells in vitro as IL-6 plus TGF-β [128, 203, 327], while the relative contribution of IL-6 and IL-21 to Th17 cell differentiation in vivo was still controversial. Whereas Th17 can secrete IL-21, it is likely to keep a similar role as IFN-γ and IL-4 in Th1 and Th2 cell development, IL-21 is also a positive feedback amplification factor for Th17 cells. Although IL-21 was induced by IL-6 in Th17 cells, IL-6 plus TGF-β induced Th17 cell differentiation was independent of IL-21 signaling and both Il21- and Il21r-deficient mice shown similar susceptibility to the control mice in EAE model [42, 249]. Those data suggest that IL-6, rather than IL-21, plays a dominant role in Th17 differentiation in inflammatory conditions where IL-6 is massively produced. While in the absence of inflammation, IL-21 might contribute to the maintaining of precursor pool of Th17 cells, as the memory Th17 cell frequency was reduced in Il21r-deficient mice [128]. Thus, it is likely that IL-21 helps to maintain and amplify the pool of Th17 precursors when the level of IL-6 is relative low while under inflammatory conditions when IL-6 is highly produced, IL-21 is dispensable for the differentiation of Th17 cells.
5.3.1.4 IL-23
As discussed above, IL-23 was described as a novel heterodimer cytokine which is composed by IL-23p19 and IL-12p40 subunits in 2000 [210]. Later, researchers found that Il23p19-deficient mice, rather than Il12p35-deficient mice were protective from the induction of EAE and harbored very few IL-17-producing cells in CNS [44, 142]. The capability of IL-23 in promoting IL-17 production in activated T cells led to a notion that IL-23 was strongly connected with the generation of Th17 cells [2]. Since IL-23R is not expressed on naïve T cells, it is likely that IL-23 is dispensable for the de novo generation of Th17 cells. After the discovery of initiation factors for Th17 cell differentiation (IL-6, IL-21, and TGF-β), it became clear that IL-23 was not required for the de novo differentiation of Th17 cells but critical for the maintaining and expansion of differentiated Th17 cells. At the present of IL-6, IL-21, and TGF-β, naïve T cells began to differentiate into Th17 cells under the control of transcription factors STAT3 and RORγt, which were critical for the expression of IL-23R in those cells [174, 203, 327]. More recently, one study found that increased sodium chloride concentrations markedly boost the induction of both murine and human Th17 cells and this induction was dependent on NFAT5 and SGK1 [125]. The kinase SGK1 is critical for regulating the expression of IL-23R and thus stabilizing the Th17 cell fate [296]. Increased salt concentration induced its expression, in turn to promote IL-23R expression and enhance Th17 cell-mediated autoimmune responses [296]. Beside to Th17 cells, IL-23 signaling was also crucial for the production of IL-17 or IL-22 in many innate immune cells [45]. Several genome-wide association studies showed the associations of Il23r gene SNPs with Crohn’s disease and psoriasis in human [23, 58, 160].
5.3.1.5 IL-1β
The cytokine IL-1β has a broad range of influence on infectious diseases as well as autoimmune disorders [52]. More recently, studies showed that both IL-1β and IL-18 have a role in promoting IL-17 production from Th17 cells [140, 264]. Both cytokines synergized with IL-23 to enhance IL-17 secretion from TCR stimulated T cells [140, 264]. As IL-23, mice lacking IL-1β signaling was resistant to both EAE and CIA induction [169, 264). Following studies showed that IL-1β signaling was required for the early stage of Th17 differentiation by converting Foxp3+ T cells into Th17 cells [36]. After polarization, IL-1β also favored Th17 cells to maintain their own fate [36]. Mechanically, IL-1β signaling promoted Th17 cell function by induction of transcription factor RORγt and IRF4 [36]. The processing of functional IL-1β requires two signals; conversely, inactive pro-IL-1β is produced by TLR signaling in innate immune cells (Signal 1) and then is cleaved by the caspase-1 to become mature and active cytokines (Signal 2) [275]. Caspase1- or other inflammasome components such as Asc- and Nlrp3-deficient mice were all found to be resistant to EAE induction, indicating that they may function though processing of IL-1β [67, 79, 109, 116, 244]. A Nlrp3 gene mutation, which hyper activated inflammasome, promoted a Th17-dominant responses though uncontrolled production of IL-1β [183]. The inflammasome agonists such as uric acid crystal and extracellular ATP were all reported to promoted Th17 differentiation though inflammasome-derived IL-1β [9, 40].
5.3.1.6 TNFα
TNFα is another important inflammatory cytokine with diverse function in immune system [14]. It was found that both Il1b- and Tnf-deficient mice were resistant to spontaneous arthritis in SGK mice [90]. In CIA model of DBA/1 mice, antibodies blockage of either IL-1β or TNFα had therapeutical effects on joint pathology [120]. Tnf-deficient mice were also protected from EAE induction [111]. Although neither of these cytokines, alone or together, was sufficient for initiation step of Th17 differentiation, TNFα as well as IL-1β was found to amplify Th17 differentiation in vitro [281]. Together with IL-1β, the DC cell-derived TNFα promoted IL-6 plus TGF-β directed Th17 differentiation [196, 281]. TNFα or IL-1β can also indirectly promote Th17 development by induction of IL-6. It is likely that cytokines such as inflammatory environments derived IL-1β and TNFα contribute to Th17 differentiation by generating an inflammatory niche which favors its differentiation. As IL-17 is a strong inducer of both TNFα and IL-1β, it is possible for Th17 cells to interact with local or infiltrated cells to set up a positive feedback loop in such an inflammatory niche.
5.3.1.7 IL-17C
IL-17C was primitively described as a novel IL-17 family cytokine, which shared similar proinflammatory effects with IL-17 [105, 150, 302]. Recently, we and others identified IL-17RE, an orphan receptor of IL-17 receptor family, as the functional receptor for IL-17C [28, 224, 253]. IL-17RE was found to be highly expressed on intestinal epithelial cells and critical for IL-17C-mediated mucosal immunity to pathogen infection or colitis [224, 227, 253] (discussed below). While surprisingly, comparing to other CD4+ T cells, Th17 cells also harbored high level of IL-17RE [28], indicating that IL-17C may also contribute to autoimmune response by targeting Th17 cells. The expression of IL-17RE was induced by IL-6 plus TGF-β and was fully upregulated at the present of IL-23 in T cells [28]. Deficient of its ligand, IL-17C, protected mice from EAE induction [28]. IL-17C bound to its receptor IL-17RE in Th17 cells and induced the expression of IκBζ, a nuclear IkappaB family member, to promote the production of IL-17 and Th17 cell response [28].
5.3.2 Positive Transcription Factors
5.3.2.1 RORγt and RORα
The T-bet, GATA3, and Foxp3 represent the lineage-specific transcription factors for Th1, Th2, and Treg cells, respectively. RORγt (also named as Rorc), a splicing variant of RORγ expressed in T cells [93, 181], was found to be the linage-specific transcription factor for Th17 cells [114]. However, in Rorc-deficient mice, the frequencies of Th17 cells were not absent but only reduced, indicating that other transcription factors play redundant role in controlling Th17 fate determination. Another ROR family member, RORα, was subsequently identified as a coordinator factor with RORγt to promote Th17-cell differentiation [307]. The deficiency of both RORγt and RORα completely abolished the generation of Th17 cells both in vitro and in vivo [307]. Both RORγt and RORα were synergistically induced by IL-6 or IL-21 plus low amounts of TGF-β [102, 307]. However, the mechanisms by which RORγt and RORα regulate IL-17 production have not yet been fully elucidated.
5.3.2.2 STAT3
Both IL-6 and IL-21 activate STAT3 in T cells, indicating a master role of STAT3 in Th17 cell programming. T-cell specific deletion of STAT3 impaired Th17 differentiation [306, 307], while retroviral overexpression of a constitutively active STAT3 in T-cells can enhance IL-17 production [179]. The STAT3 directly controlled expression of RORγt, RORα, and IRF4, which were transcriptional factors required for Th17 differentiation [59]. More importantly, STAT3 also bound directly to the promoters of IL-17A, IL-17F, IL-21, IL-6R, and IL-23R, indicating a direct control of STAT3 on the Th17 differentiation [31, 59, 203]. Patients with Hyper IgE syndrome (HIES) harbored a dominant negative form of STAT3 which led to a serious impairment of Th17 generation in those patients [49, 99, 166, 185, 186, 226]. The mutation of STAT3 and subsequent impairment of IL-17 production may explain the inefficient cleanup of bacterial and fungal infections in those patients.
5.3.2.3 IRF4
IRF4, a transcription factor which has been shown to be critical for the differentiation of the Th1 and Th2 cells [162, 225], was also found to be essential for the development of Th17 cells. Irf4-deficient mice were resistant to EAE induction and its deficient-T cells were unable to induce the expression of RORγt and RORα and consequently could not be differentiated into Th17 cells when those cells were treated with TGF-β plus IL-6 or IL-21 [21, 102], while restitution of RORγt and RORα in those cells could partially compensate for the reducing of IL-17 production [102]. In addition, the expression of Foxp3 was increased in Irf4-deficient T cells, suggesting the interplay of the Foxp3 and RORγt balance in Treg /Th17-cell differentiation [21]. IRF-4 was negatively regulated by a binding protein named IBP (IRF-4-binding protein), which is shown to play a regulatory role in Th17-cell differentiation [29]. IBP prevented IRF-4 from binding to the transcriptional elements of Il17 and Il21 genes and mice lacking IBP developed arthritis-like syndrome and enhanced Th17 responses [29].
5.3.3 Negative Regulation of Th17 Differentiation
5.3.3.1 STAT5
STAT5 is an essential downstream transcription factor of IL-2 signaling, which is critical for the survival of T cells. Genetic abolishment or antibody neutralization of IL-2 promoted differentiation of the Th17 cells in vivo [144]. This inhibition effect of IL-2 on Th17 differentiation was independently of Foxp3 and RORγt [308]. Whereas STAT3 is a critical positive regulator of RORγt and Th17 development, disruption of STAT5 led to increased Th17 cell development, probably due to the loss of IL-2-mediated inhibitory effect [144]. This suppressive effect of STAT5 on the differentiation of Th17 might be due to its competition with STAT3 to bind to the same locus sites encoding Il17 [308].
5.3.3.2 Gfi1
Gfi1 was highly expressed in Th1 and Th2 cells and its expression was controlled by IFN-γ/STAT1 and IL-4/STAT6 pathways. TGF-β, which is critical for either Th17 or Tregs differentiation, suppressed Gfi1 expression [107]. Over-expression of Gfi1 strongly repressed IL-17A expression in both human EL4 T-cell line and primary T cells. This suppressive effect was mainly due to the blockage of RORγt recruitment to the promoter region of IL-17A. In contrast, Gfi1-deficient T cells produced more IL-17A than wild-type T cells under Th17 favored differentiation conditions [107]. However, the influences of Gfi1 on iTregs were not as obvious as on Th17 cells [107].
5.3.3.3 LXR
Liver X receptors (LXRs) are nuclear receptors which are originally involved in cholesterol homeostasis and are activated by endogenous oxysterols or oxidized cholesterol derivatives [323]. Recently, LXR was found as a negative regulator for Th17 differentiation. LXR suppressed Th17 responses by promoting the transcription factors Srebp-1 to bind to the Il17 promoter region, and thus interfering with the AhR-mediated Il17 transcription [46]. Over-expression of LXR inhibited mouse Th17 differentiation and Lxr-deficiency mice were more susceptive to EAE induction than wild-type mice [46].
5.3.3.4 TCF1
T-cell factor 1 (TCF-1) is a transcription factor to activate canonical Wnt pathway and is crucial for normal T cell development [254]. TCF1 was showed to regulate differentiation and maintenance of memory CD8+ cells [329]. Recently, it was also found that TCF1 suppressed IL-17 expression by directly binding to the regulatory region of Il17 and thereby repressing its transcription [312]. Induction of IL-17 in TCF1 depletion T cells was coupled with up-regulation of RORγt and STAT3 in those cells [168]. Moreover, Tcf1-deficient Th17 cells expressed increased levels of IL-7Rα, which potentially enhanced Th17 cell survival [312]. Accordingly, Tcf1-deficient mice showed exacerbated EAE phenotype [168, 312]. TCF1 might also repress Th17 responses by induction of Eomes [329].
5.3.3.5 Eomes
Cytotoxic activities of T cells were noticed under controlling of T box transcription factors including T-bet and Eomes through up-regulation of perforin, FasL and granzyme B [63, 223]. Eomes interacted with GATA3 to prevent its binding to IL-5 promoter in memory Th2 cells or functioned together with T-bet to mediate generation of IFN-γ producing CD8+ cells [62, 76, 207]. In Th17 cells, ectopic expression of Eomes inhibited Th17 cell differentiation through directly binding to the promoter region of Rorc and Il17. Depletion of Eomes expression could substitute for TGF-β in induction of Th17 cell differentiation [108].
5.3.3.6 NFIL3
NFIL3, also called E4BP4, is a transcription factor with basic leucine zipper structure which regulates multiple immune responses [171]. Recently, it was found that this transcription factor suppressed Th17 cell differentiation though directly binding and inhibiting the Rorc promoter [313]. More interestingly, NFIL3 was negatively regulated by the transcription factor REV-ERBα, which is critical for circadian clock pathways [187]. Consequently, Th17 lineage frequency varied diurnally and was altered in Rev-erbα-deficient mice [313]. Disruption of light cycle elevated intestinal Th17 cell level and increased susceptibility to intestinal inflammatory disorders [313].
5.3.4 Negative Regulation of mTOR in Th17 Differentiation
The mTOR signaling is critical for regulating organism growth and homeostasis [143]. Recently, mTOR signaling is found to represent a crucial regulator of T-cell differentiation [95]. The mTOR signaling pathways have two distinct complexes: mTORC1 and mTORC2. The conditional depletion of Rictor, a key component of mTORC2, impaired the differentiation of Th1, Th2, and Tregs but not Th17 cells [146], while mTORC1 signaling was found to selectively regulate Th17 differentiation [51]. Recently, it was found that IL-1β signaling induced the activation of JNK and mTOR kinases in Th17 cells. IL-1β-induced Th17 proliferation was impaired in Mtor-deficient Th17 cells, indicating the critical role of mTOR activation in Th17 development [82]. Meanwhile, the study also found a negative regulator of IL-1β and TLR signaling, named SIGIRR, was induced during Th17 cell development and SIGIRR inhibited Th17 differentiation and expansion by suppression of IL-1β signaling. Comparing to wild-type Th17 cells, Sigirr-deficient Th17 cells shown increased IL-1β-induced JNK and mTOR activation [82]. IL-1β-induced mTOR activation was dependent on AKT, which was constitutively suppressed by GSK3α medicated phosphorylation. Thus, GSK3α severed as a brake to prevent over-expansion of Th17 cells. Upon IL-1β stimulation, a kinase IKKi was found to phosphorylate GSK3α and in turn to release the negative regulation of GSK3α on mTOR activation in Th17 cells [81].
5.3.5 MicroRNA in Th17 Differentiation
MicroRNAs (miRNAs) are about 19–24 nucleotide (nt) single-stranded RNA molecules that post-transcriptionally regulate the expression of genes [333]. In mammals, it was suggested that nearly 85% of microRNA-mediated decay of mRNA [83]. More than 600 different microRNAs were expressed in T cells and many of them were critical regulators for the differentiation stage and activation status of different T-cell populations [124]. As helper T-cell subsets are critical for autoimmune pathogenesis and proper host defense responses, the regulation of lineage commitment by microRNAs are studied to a greater extent. T-cell specific deletion of Dicer, a critical enzyme for microRNA biogenesis resulted in reduced T cells proportion in the thymus or periphery lymph tissues [39, 191]. Recent studies also showed that specific miRNAs are involved in regulation of certain types of T help cell development. T-cell deficiency of miR-155 led the activated CD4+ T cells to favorite a Th2 bias under neutral conditions in vitro [230, 270]. It is noticed that miR-155 inhibited the function of Th2 transcriptional factor c-Maf to promote the differentiation of Th1 cells [230]. However, the Th1 differentiation was negatively regulated by miR-29. MiR-29 was reported to suppress Th1 differentiation by targeting the Th2 favored transcription factor T-bet and Eomes or the cytokine IFN-γ [167, 247, 255]. Beside to Th1 and Th2 cells, the microRNAs which regulate Th17 cells function have also been identified. A recent study showed that miR-326 targeted Est-1, a negative regulator of Th17 differentiation, to promote Th17 cell expansion and Th17 related autoimmune pathogenesis [56]. Likewise, miR-301a promoted Th17 differentiation by inhibiting PIAS3, which negatively regulated IL-6-IL-23-STAT3 cascades [195]. MiR-155 also enhanced STAT signaling through suppression of SOCS1 and this might explained the depressed Th17 differentiation and ameliorated EAE phenotype in Mir155-deficient mice [194, 310]. More recently, miR-21 was showed to promote Th17 differentiation by disrupting Smad-7, a negative regulator for TGF-β signaling. Like MiR-155, Mir21-deficient mice also protected from EAE induction [75]. Meanwhile, microRNA let-7a was recently found to be a negative regulator of Th17 differentiation by suppressing IL-6 secretion in a mouse model of Con A-induced hepatitis [320].
5.4 Interplay Between Th17 Cells and Other T Helper Cells
5.4.1 Interplay Between Th17 and Th1, Th2 Cells
The effector T-helper cell subsets was noticed as distinct and terminal differentiated lineages by studies showing that, once naive T cells have been programmed into Th1 or Th2 cells, these polarities could not be reversed even when new polarizing conditions were reintroduced [193]. The discoveries of specific master transcription factors, T-bet for Th1 while GATA3 for Th2 differentiation, had also improved this lineage model. Furthermore, several studies showed that Th1 and Th2 antagonized each other to favor their own stable terminally differentiated phenotype [53, 198, 266]. As the crosstalk between Th1 and Th2 cells, cross regulation may also exist between Th17 and Th1 or Th2 cells. Either IFN-γ or IL-4 inhibited Th17 differentiation and IL-17 induction [87]. In addition, the IL-12 family cytokine IL-27, which induced T-bet and IL-12Rβ2 expression to promote Th1 responses inhibited Th17 cell differentiation through activation of STAT1 [13, 258]. Loss of T-bet in T cells strongly promoted IL-17 expression in Th17 cells [178, 309]. As discussed above, IL-4 could induce Gfi-1 to favor optimal Th2 cell differentiation [332] and suppressed Th17 or iTreg cells differentiation [331]. Thus, both Th1 and Th2 favored signaling are inhibitors for Th17 development, while the effects of Th17 derived cytokines on Th1 and Th2 differentiation are still need to be addressed.
Th17 cells are distinct T-helper cell subset and are not derived from the Th1 lineage, since those cells lacking T-bet or Stat4 also expressed RORγt and developed into Th17 cells under Th17 polarizing condition [114]. However, under both homeostatic and inflammatory conditions, IFN-γ+IL-17+ cells can be easily detected, suggesting that intricacy is existed between the Th1 and Th17 cell differentiation. In vitro polarizing Th17 cells could lose both IL-17 and IL-17F expression when IL-6 and TGF-β stimulation was withdrawn [149]. T cells cultured in Th17 polarizing conditions still failed to keep IL-17 and IL-17F expression and could be converted to Th1 or Th2 cells when those cells were stimulated with IL-12 or IL-4, respectively [147, 149]. In vitro polarized Th17 cells failed to keep their IL-17-producing memory when transferred into mice and many of them converted into IFN-γ-producing Th1 cells under a colitis condition [147]. Similar phenomenon was also noticed when Th17 cells were introduced into NOD-SCID mice [15]. In contrast, in vivo memory Th17 cells with CD4+CD62Llow marker appeared more resistant to conversion [149], suggesting in vivo generated Th17 cells are a stable and distinct lineage of T helper cells.
As described above, Th17 cells polarized by TGF-β and IL-6, but without IL-23, were shown to express IL-10 [180]. Since Th17 cells are abundant in the lamina propria of intestine even in steady state, this containment is critical to control over-activated Th17 cells in gut. Upon infection or injury, these Th17 cells can easily become inflammatory Th17 cells when other Th17 polarizing cytokines are released by the local environment, and these cells can also rapidly convert into Th1-like cells for intracellular pathogens cleanup. As Th17-mediated inflammatory responses also cause serious tissue destruction, these pathogenic Th17 cells may be appropriately and timely re-shifted toward protective IL-10-producing Th17 cells when dangerous signals are removed.
5.4.2 Interplay Between Th17 and Treg Cells
TGF-β signaling was essential for formation of immune tolerance, in part due to its capability for induction of peripheral iTreg cells and for maintenance of thymus derived nTreg cells [30, 152, 175]. However, at lower concentration, TGF-β was also critical for induction of proinflammatory Th17 cells [16, 174, 281]. Mice deficient in TGF-β failed to develop ethier Foxp3+ Treg or Th17 cells and had uncontrolled autoimmune disorders which was caused by aggressive Th1 cells responses [152, 282], indicating a close relationship between Treg and Th17 cell development program in against Th1 differentiation. After TCR ligation, TGF-β could induce both Foxp3 and RORγt expression in CD4+ T cells [328]. These Foxp3+ RORγt+ T cells were found both in murine and human and noticed as a transitional state for Treg and Th17 cells [284, 328]. In small intestine, the Foxp3+ RORγt+ cells produced less IL-17 than Foxp3–RORγt+ cells [328]. In agreeing with this, Foxp3 abolition led to a notable increase in IL-17 production without affecting RORγt expression [70], indicating that Foxp3 may antagonize RORγt function to favor Treg development. Indeed, Foxp3 suppressed Th17 responses by directly interacting with both RORγt and RORα [57, 307, 328]. Foxp3–RORγt+ T cells induced by TGF-β do not produce much IL-17 and have the dual potential to develop into either Treg or Th17 cells depending on the different inflammatory environment. As described, a proinflammatory environment (IL-6, IL-23, IL-1, and IL-21) plus low concentration or TGF-β inhibited Foxp3 function while further enhanced RORγt expression in favor of Th17 differentiation. In contrast, high concentration TGF-β further firmed Foxp3 expression and thus promoted Treg differentiation rather than Th17 cells [328]. This Treg favored shift was further enhanced by IL-2 and retinoic acid (RA) signaling, both of which were suppressors for RORγt but enhancers for Foxp3 expression [41, 144, 190, 261]. As mentioned before, several transcription factors have been noticed to modulate the Foxp3 and RORγt balance during CD4+ T-cell differentiation. Runx1 was found to cooperate with RORγt to promote Th17 cell differentiation [317]. Additionally, in Treg cells, Runx1 and Foxp3 were also indicated to from a complex which was required for inhibition of IL-2 and IFN-γ expression and Treg suppressive activity [209]. Thus, Runx1 is required for governing potential plasticity in Foxp3+ RORγt+ T cells. IRF4 is critical for Th17 cell differentiation and its absence led to increased Foxp3 expression, reduced RORγt expression, and thus loss of IL-17 secretion [21]. Stat3, a transcription factor that was commonly activated by IL-6, IL-21, or IL-23, was also crucial for Th17 cell differentiation by binding to the promoter regions of Il17 and Il17f [31]. These proinflammatory cytokines were found to inhibit TGF-β-induced Foxp3 expression in a Stat3-dependent manner [306, 327].
5.4.3 Epigenetic Regulation Between Th17 and Other T Helper Cells
Epigenetic regulation is critical for controlling gene expression through changing chromatin structure, histone or DNA modifications, and small noncoding RNAs expression and T helper cells are also under control of these regulatory strategies [8, 184, 290]. Recent studies by using chromatin immunoprecipitation on gene array chips (ChIP-chip) and high-throughput sequencing (ChIP-Seq) found several histone modification and DNA methylation changes accompanying with CD4+ T-cell differentiation. Trimethylation of histone H3 lysine 4 (H3K4me3) was a permissive mark, while H3 lysine 27 (H3K27me3) was a silence mark to be found at the promoters and the enhancers of different subset specific genes [289]. In Th1 cells, H3K4me3 mark was found at the Ifng locus while H3K27me3 mark was found at the Il4 and Il17 loci. In Th2 cells, however, Il4 locus was modified with H3K4me3 while Ifng and Il17 loci were modified with H3K27me3. Notably, the gene Tbx21 and Gata3 showed a bivalent status in Th2 and Th1 cells respectively, indicating cell lineage interconversion may occur between these two types of cells [133, 289]. However, Rorc locus was suppressed in both Th1 and Th2 cells, indicating that these two types of cells can hardly reprogrammed into Th17 cells. Consistent with the high plasticity of Th17 cells, the gene Tbx21 and Gata3 all showed a bivalent status in Th17 cells, indicating that Th17 cells can easily transmit into Th1- or Th2-like cells under appropriate conditions [133]. Tbx21, Gata3, or Rorc sites were all found to be bivalent modified in Treg cells, suggesting that, like Th17 subset, Tregs are also highly dynamic cell population. However, nTreg cells derived from thymus and iTreg cells derived from peripheral showed different reprogramming propensity. In iTreg cells, the Il17 locus was silenced by H3K27me3 modification whereas H3K4me3 was found at the Rorc locus, agreeing with the notion that TGF-β induced RORγt expression in T cells, but the differentiation of Th17 cells was inhibited by Foxp3 [289, 328]. However, both H3K4me3 and H3K27me3 bivalently existed at the Rorc locus in nTreg cells, allowing for the generation of Foxp3+RORγt+ cells [289]. When nTreg cells were subjected to Th1- or Th17-polarizing culture conditions, a remarkable number of IFN-γ+Foxp3+ or IL-17+Foxp3+ cells appeared without affecting the expression of Foxp3 [289, 300, 305]. However, TGF-β-induced Treg cells easily lost Foxp3 expression and gained IL-17 expression when these cells were cultured in a Th17 cell-favoring condition [305].
5.5 Human Th17 Differentiation
Sooner after the discovery of cytokines and transcription factors that promote mouse Th17 cell differentiation, efforts were made to induce the differentiation of human Th17 cells. As mouse Th17 cells, human Th17 cells also highly expressed the master transcription factors RORC2, the human homologue to RORγ t [7, 291], and forced expression of RORC2 in cord blood derived T helper cells induced IL-17A, IL-17F, and IL-26, but not IL-22 expression [173]. The functional roles of other Th17-related transcription factors in human Th17 cell development are not fully understood. It was reported that transduction of RORA in human cord blood cells led to increased IL-17 expression [173], indicating that this human homologue to RORα also cooperates with RORC2 to induce Th17 cells as in mice. STAT3 was also found to be important for the development of human Th17 cells. Patients with hyper-IgE sydrome carried dominant negative mutations in STAT3 gene and T cells obtained from these patients fail to develop into Th17 cells in vitro due to the lacking of IL-6-induced STAT3 activation and subsequently RORC2 expression [99, 166, 186]. AHR was also expressed in human Th17 cells [280], but it is still not clear whether it contributes to these cells development.
The cytokines required for the differentiation of human Th17 cells appeared similar to mouse Th17 cells, including IL-6, IL-23, IL-1, and IL-21, however, there were some debates on the necessity of TGF-β in human Th17 differentiation [129]. Several studies found that TGF-β was not required for human Th17 cells differentiation [1, 291]. But one study argued that these cells used above were not totally equal to naïve T cells in mouse. Other studies reported that the human naive cord blood T cells were dependent on TGF-β for their differentiation into Th17 cells. TGF-β was required for induction of RORC, but excess TGF-β inhibited its expression and function [173, 283, 303].
Given the complexity of TGF-β for human Th17 differentiation, the requirement of TGF-β in mouse Th17 differentiation has been re-checked. Tbx21- and Stat6-deficient T cells could differentiate into Th17 cell when stimulated with IL-6 alone, even in the absence of TGF-β [48], indicating that TGF-β indirectly regulated IL-17 production by inhibition of factors that required for other cell fates [145]. In addition, the IL-23R was induced in the absence of TGF-β, and IL-23 addition could further induce this receptor expression. In addition, the combination of IL-6, IL-23, and IL-1 was sufficient to induce IL-17 production in a TGF-β independent way [74]. Consistently, Th17 cells were also found in the gut of Tgfb1-deficient mice [74, 221].
5.6 Th17 Effective Cytokines
5.6.1 IL-17A and IL-17F
5.6.1.1 IL-17 Family Cytokines
Th17 cells secret characteristic cytokines IL-17A (also called IL-17) and IL-17F. IL-17 family also contains other four cytokines IL-17B, IL-17C, IL-17D, and IL-17E (also named IL-25), which are homologous to the fundamental member IL-17A by bioinformatics analysis. The IL-17 family of receptors, which contains IL-17RA, IL-17RB, IL-17RC, IL-17RD, and IL-17RE, are signal transmembrane receptors with unique structure architecture among the members. All receptor members harbor two fibronectin III-like domains in their extracellular region and a SEF/IL-17R (SEFIR) domain in their intracellular part [68]. IL-17RA (also called IL-17R) has been long known as the receptor of IL-17. However, recent findings indicate that IL-17RA may sever as a common receptor for all IL-17 family cytokines. Other receptors like IL-17RB, IL-17RC, or IL-17RE have been discovered as specific receptors for IL-17E, IL-17A, as well as IL-17F or IL-17C, respectively. IL-17RD (also called Sef), a negative regulator of FGF signaling, has recently been identified to have a role in IL-17A signaling [182, 231] (Fig. 5.3).

The IL-17 family cytokines and their receptors. IL-17 family cytokines contain six members: IL-17A to IL17F, while the receptor family has five members: IL-17RA to IL-17RE. The most studied members, IL-17A and IL-17F, form homo- or hetero-dimer to bind IL-17RA and IL-17RC receptor complex and activate downstream signaling for host defense, autoimmune diseases and other inflammation responses. Although IL-17B was found as a ligand of IL-17RB, the in vivo functional evidences for this pair are still largely unclear. IL-17C binds to IL-17RA and IL-17RE receptor complex to trigger downstream signaling for host defense and autoimmune diseases. IL-17E associated with IL-17RA and IL-17RB receptor complex to mediate Th2 responses. Neither the receptor for IL-17D nor the legend for IL-17RD has been found. The adaptor protein Act1 has been considered as a key adaptor in IL-17A, IL-17F, IL-17C, as well as IL-17E mediated signaling
In murine, IL-17F shares the highest homology in amino acid sequence to IL-17 (about 50%), while IL-17B, IL-17C, IL-17D, and IL-17E share 16–30% sequence identity to IL-17 [115]. The high similarity of IL-17 and IL-17F may be responsible for the similar pro-inflammatory outcomes they mediate. IL-17 and IL-17F form homo/hetero-dimer to bind to their receptors, IL-17RA and IL-17RC, and therefore activate NF-κB, MAPKs, and C/EBPs signaling pathways which consequently upregulate the expression of proinflammatory genes [336]. IL-17 also cooperates with other inflammatory cytokines such as TNFα to synergistically induce certain chemokines expression by stabilizing their mRNAs [336]. Recently, we also found IL-17 enhanced NF-κB activation though an alternative way by downregulating the expression of microRNA-23b in residential cells of several autoimmune diseases [335].
As discussed above, IL-17 and IL-17F are considered to be predominantly produced in the Th17 cells. The differentiation of Th17 cells is controlled by a unique set of cytokines (TGF-β, IL-6, IL-1β, IL-23, and IL-21) and transcription factors (RORγt, RORα, STAT3, IRF4, BATF, AHR, Runx1, and IκBξ). However, beside to Th17 cells, a subset of CD8+ T cells named Tc17 cells can also produce those two cytokines. In addition, several innate immune cells have also been found to release IL-17 and IL-17F. These cells, which include γδT cells, iNKT cells, NK cells, LTi cells, and neutrophils, are noticed as major sources for innate IL-17 and IL-17F. Furthermore, nonlymphoid cells such as intestinal Paneth cells and colonic epithelial cells may also have the capability to produce IL-17 or IL-17F [45, 115]. The rapid releasing of IL-17 or IL-17F by those cells may contribute to appropriate immune responses against many pathogens.
IL-17E (also called IL-25) functions distinctly different from IL-17 and IL-17F. IL-17E promotes Th2 cell responses by inducing the expression of IL-4, IL-5, and IL-13. Il25-transgenic mice or mice treated with ectopic IL-17E showed increased releasing of Th2 factors along with eosinophilia [105, 123, 213, 268]. In contrast, due to reduced Th2 response, Il25-deficient mice displayed impaired Th2 responses, thus were more susceptible to parasite infection but were resistant to allergic responses in lung [11, 64, 212, 268, 322]. Either immune cells (macrophages, dendritic cells, mast cells, eosinophils, basophils, and T cells) or nonimmune cells (epithelial cells and Paneth cells) could release IL-17E under certain conditions. As its producing cells, IL-17E can target diverse cell types such as Th2, Th9, NKT, monocytes, macrophages, non-B non-T (NBNT), and epithelial cells [115]. IL-17E is activated downstream signaling through the IL-17RA and IL-17RB receptor complex [228]. The adaptor protein Act1 was also found to mediate IL-17E signaling [38, 121, 265]. Recently, one study showed that intestinal commercial bacteria induced IL-17E production from intestinal epithelia cells and IL-17E in turn inhibited Th17 expansion by suppression of IL-23 production in macrophages [314]. In addition, Il25-deficient mice were susceptible to EAE induction [126], indicating a negative role of IL-17E in controlling Th17 responses.
Although the biological functions of IL-17B and IL-17D remain largely unknown [252], recent findings have begun to uncover the functional roles of IL-17C. We and others demonstrated that IL-17C was the ligand for the orphan receptor IL-17RE and delivered its signal through IL-17RA and IL-17RE complex [28, 224, 253]. IL-17C is specifically induced in epithelial cells and keratinocytes by pathogens or inflammatory cytokines and acted as an autocrine cytokine on those cells. Similar to IL-17, IL-17C activates common signaling pathways including NF-κB and MAPKs cascades [224, 253]. IL-17C also targeted Th17 cells for promoting IL-17 production [28]. In vivo studies from Il17c- or Il17re-deficient mice demonstrated that this pathway was critical for protection against intestinal pathogens as well as the progression of several autoimmune diseases including psoriasis, IBD and MS [28, 224, 253].
5.6.1.2 IL-17A and IL-17F Medicated Signaling
Positive regulator
Soon after it was cloned from activated murine T lymphocyte hybridoma cDNA library [232], the downstream signaling pathways of IL-17A were revealed. Typically, IL-17A activates NF-κB, MAPKs, and C/EBPs cascades to upregulate expression of inflammatory genes [251]. IL-17A also synergizes with TNFα to induce gene expression through stabilization of TNFα-induced mRNAs [251]. Other studies also found that it can activate JAK-PI3K and JAK-STAT pathways, although detailed downstream molecular mechanisms were still poorly understood [101, 234]. Recently, we found that IL-17A downregulated microRNA-23b to promote inflammatory responses in tissue resident cells [335]. IL-17RA was initially noticed as the receptor for Il17a- and Il17ra-deficient fibroblasts have no response to IL-17A stimulation [273, 304, 311]. Later, IL-17RC was found as a receptor component for IL-17A signaling by interacting with IL-17RA as well as IL-17A. Deficiency of IL-17RC in mice impaired IL-17A induced downstream gene expression [304]. Thus, upon IL-17A ligation, IL-17RA binds to IL-17RC to form a heterodimeric receptor complex to initiate intracellular signaling pathways. The receptor proximal signaling mechanisms have been uncovered recently (Fig. 5.4).

The activation of IL-17 signaling. After IL-17A or IL-17F binding to the receptors, Act1 is recruited to the IL-17RA and IL-17RC receptor complex. Act1 then recruits and poly-ubiquitinates TRAF6 for the activation of NF-κB and JNK pathways. IL-17A-induced NF-κB signaling also suppresses the miR-23b expression and therefore releases its inhibition on TAB2 and TAB3 complex to amplify NF-κB activation. Act1 is also critical for IL-17A induced activation of C/EBP pathway. In addition, Act1 also recruits TRAF2 and TRAF5 to mediate mRNA stabilization pathway. Normally SF2 is bound to certain mRNAs for their degradation. Upon IL-17A challenging, Act1/TRAF5/TRAF2 complex recruits SF2 form mRNA and thus protect them from degradation. The phosphorylation of Act1 at Ser-311 by the kinase IKKi is crucial for the interaction of Act1 with TRAF2 or TRAF5. Meanwhile, Act1 also recruits and ubiquitinates an ARE-binding protein HuR through TARF2 and TRAF5. Act1/HuR complex then bind to the SF2 free mRNA to stabilize them. Hsp90 has been shown to be required for IL-17A signaling by promoting the function of Act1
TRAF6 was firstly found as a positive adaptor in IL-17A signaling, and it was essential for IL-17A-induced activation of NF-κB and JNK pathways and expression of downstream genes such as IL-6 [240]. However, the intercellular region of IL-17RA did not contain any predicted TRAF6 binding motif and TRAF6 was found not responsible for IL-17A-induced mRNA stabilization pathway, suggesting that additional adaptors upstream of TRAF6 might exist for both TRAF6 dependent or independent pathways [89].
By bioinformatic searching, all IL-17R family receptors were found to harbor an intracellular SEFs and IL-17Rs (SEFIR) domain. Furthermore, this SEFIR domain structure also was also noticed in a cytosolic protein called Act1 (also known as CIKS) [202]. After IL-17 stimulation, IL-17R recruited Act1 to its intracellular region through SEFIR-SEFIR domain interaction [27, 220]. Act1 then recruited TRAF6 to IL-17R complex. Act1 was not only a simple adaptor for downstream molecules recruitment, but also was noticed as a U-box like E3 ligase, which mediated Lys63-linked ubiquitination of TRAF6 through recruitment of the Ubc13-Uev1A E2 complex [157]. We recently found that IL-17A downregulated the expression of microRNA-23b (miR-23b) through NF-κB pathway and subsequently releasing its suppression effect on TAB2 and TAB3 to further amplify NF-κB activation [335]. Thus, IL-17A can trigger a positive feedback loop through removing the suppression of miR-23b on NF-κB activation. Since the SEFIR domains are required for the interaction between Act1 and IL-17R, a detailed domain mapping study found that SEFIR domains contain a CC’ loop structure and this structure was responsible for the interaction of adaptor and receptor [158]. A mimicking decoy peptide of Act1 CC’ loop inhibited IL-17 induced inflammatory responses [158]. Though the SEFIR domain is important for IL-17A signaling transduction and inflammatory gene production, other studies also found that, beside to SEFIR domain, the C-terminal region and “TIR-like loop” (TILL) motif of IL-17RA, the cytoplasmic tail region of IL-17RC and the N-terminal domain of Act1, were also critical for IL-17A-mediated signaling [98, 170, 208, 248].
Act1 but not TRAF6 was required for IL-17A-mediated mRNA stabilization of KC induced by TNFα [89], indicating that Act1 mediates either TRAF6 dependent or independent pathways for IL-17A signaling. Recently, the inducible kinase IKKi (also called IKKε) was found for the Act1-mediated mRNA stabilization pathway. IKKi was showed to bind to Act1 upon IL-17 stimulation in mouse fibroblasts. Deficiency of IKKi in airway epithelial cells did not influence IL-17 induced NF-κB activation, but largely impaired IL-17-mediated KC mRNA stabilization in those cells [22]. IKKi directly phosphorylated Act1 at residue Ser-311 after IL-17A stimulation and the phosphorylation of this site was critical for IL-17A-mediated mRNA stabilization of KC but not for NF-κB activation [22]. Another study showed that The Ser-311 phosphorylation of Act1 was required for recruiting TRAF2 and TRAF5 to generate an Act1-TRAF2-TRAF5 complex. IL-17A induced formation of Act1-TRAF2/TRAF5 complex further recruit mRNA splicing factor 2 (SF2) to prevent its binding and cleavaging of KC mRNA [262]. Besides, Act1 also recruited and ubiquitinated an ARE-binding protein HuR to stabilize the mRNA which released from SF2 [96]. More importantly, deficiency of HuR in epithelium resulted in impaired IL-17A-mediated inflammation in lung, agreeing with the essential role of Act1 in epithelium of lung [96]. More recently, upon IL-17A exposure, heat shock protein 90 (Hsp90) was found to be associated with Act1 and this interaction was critical for the binding of Act1 with other signaling molecules [285]. Inhibitors of Hsp90 prevented the signaling complex formation and in turn inhibited the activation of downstream signaling pathways [285]. Interestingly, Hsp90 lost a psoriasis associated Act1 mutant D10N [60, 104, 256] and this mutation failed to activate downstream signaling mediated by IL-17A [285]. To date, Act1 cooperates with Hsp90 to generate a receptor proximal anchor platform to assemble two distinct IL-17A-mediated cascades: (1) TRAF6-dependent pathway and (2) IKKi-TRAF2-TRAF5-dependent pathway.
IL-17F shares the same receptor set IL-17RA-IL-17RC with IL-17A for downstream signaling. Surface plasmon resonance (SPR) analysis found that human IL-17RA had a higher binding affinity to human IL-17A while human IL-17RC preferred to bind to human IL-17F [138, 295]. Although the binding affinities of those two cytokines are different, they mediated inflammatory responses completely required either IL-17RA or IL-17RC [98, 100, 273, 295, 304], suggesting their mediated downstream signaling all through the IL-17RA and IL-17RC heterodimer complex. Similar to IL-17A, IL-17F also needed Act1 and TRAF6 for downstream signaling [304] and induced NF-κB, MAPKs, and C/EBP activation in different cell types [122, 304, 330].
Negative regulator
As IL-17A exhibits a broad influence on driving inflammatory responses, its signaling is under strict control to prevent harmful persistent inflammation (Fig. 5.5). One early study found that IL-17A activated the kinases GSK-3β and ERK to phosphorylate C/EBPβ at Thr188 and Thr179 in its regulatory 2 domain, and this dual-phosphorylation in turn inhibited IL-17A induced expression of proinflammatory genes [245].

The regulation of IL-17 signaling. Once IL-17 signaling is activated, several mechanisms are adopted to prevent over activation of the signaling. Upon activation, TRAF3 is recruited to IL-17RA and IL-17RC complex to interfere the formation of IL-17R/Act1/TRAF6 signaling complex, while TRAF4 binds to Act1 to disrupt the Act1/TRAF6 signaling complex formation. Although IKKi is required for IL-17A mediated mRNA stabilization pathway, IKKi and TBK1 also triple-phosphorylate Act1 to disrupt the interaction between Act1 and TRAF6 to suppress activation of NF-κB in a TRAF6-dependent negative feedback manner. IL-17A also activates ERK and GSK3β to phosphorylate C/EBPβ and thus suppresses C/EBP activation. USP25, a deubiquitinase, directly removes both TRAF5 and TRAF6 ubiquitination to inhibit IL-17-induced signaling. Another deubiquitinase A20 specifically removes the ubiquitination of TRAF6 to suppress IL-17-mediated NF-κB activation. With persistent IL-17A exposure, Act1 is phosphorylated and then ubiquitinated and degraded by SCFβ- TrCP E3 ubiquitin ligase complex, therefore avoiding over activation of the signaling
TRAF3 was a critical adaptor for TLRs and RIG-I induced type I interferon production in antiviral responses or was a negative regulator for CD40 or BAFF induced noncanonical NF-κB activation [33, 85, 92, 131, 204, 276, 299]. We recently identified that TRAF3 was an important negative regulator for IL-17A signaling [334]. Forced expression of TRAF3 in cells prevented IL-17A-mediated signaling activation and downstream cytokine production, while silencing of TRAF3 enhanced the activation of NF-κB and MAPKs pathways as well as expression of downstream genes. TRAF3 was found to directly bind to IL-17R for disrupting the interaction complex of IL-17R, Act1, and TRAF6, thus suppressed IL-17 signaling activity. Transgenic TRAF3 in mice inhibited the IL-17 induced inflammatory responses and subsequently controlled EAE progression [100]. Thus, TRAF3 was identified as the first receptor proximal negative regulator of IL-17A signaling.
Similar to TRAF3, TRAF4 was lately identified to negatively control IL-17A signaling. Upon IL-17A treatment, deficiency of TRAF4 in cells led to remarkable exacerbated activation of IL-17A signaling and increased expression of chemokines. Genetic depletion of TRAF4 caused earlier onset of Th17 cell induced EAE pathogenesis. TRAF4 suppressed IL-17A signaling by disruption the binding between Act1 and TRAF6 [316]. Therefore, in an alternative way, TRAF4 also interferes with the formation of positive signaling complex for controlling IL-17A signaling.
Ubiquitin-proteasome pathway mediated Protein degradation is a widely used strategy to desensitized specific signaling by erasing key signaling adaptors [197]. We recently found that Act1 protein level significant decreased upon IL-17A persistent stimulation, without affecting its mRNA level. Mechanically, Act1 was degraded in a Lys48-linked polyubiquitination manner by the SCFβ- T rCP-1/2 E3 ubiquitin ligase complex, which was the same for degradation of the inhibitor of nuclear factorκBα (IκBα), indicating that both negative and positive signaling events of IL-17 is controlled by the same degradation complex [246]. The degradation of Act1 eventually led to dismissing of downstream pathway activation and consequently desensitized cells to IL-17 exposure.
As described, IKKi phosphorylated Act1 at Ser-311 for IL-17A-mediated mRNA stabilization pathway [22]. Recently, we also found that both IKK-related kinases IKKi and TBK1 were recruited to Act1 upon IL-17A stimulation [222]. Both kinases phosphorylated Act1 on three serine sites different from Ser-311 and phosphorylation of those sites intertered with the association of Act1 with TRAF6 and subsequently suppressed IL-17A induced NF-κB activation. Notably, TRAF6 itself was needed for the activation of TBK1 and Act1 [222]. Therefore, in addition to its positive role in IL-17A signaling by phosphoryting Act1 at Ser-311, IKKi also displayed a redundant role with TBK1 though phosphorylation of Act1 on other Serine sites to control the signaling.
Since Act1 is an E3 ligase for TRAF6 to promote the signaling of IL-17A, the deubiquitinating enzyme (DUB) for negative regulation may exist. Recently, the ubiquitin-specific protease USP25 was found as a negative regulator of IL-17A signaling [326]. Overexpression of this deubiquitinating enzyme inhibited IL-17A induced downstream signaling. By contrast, deficiency of USP25 in MEFs or primary lung epithelial cells led to enhanced IL-17A-mediated responses [326]. Genetic depletion of USP25 in mice consequently enhanced IL-17A induced respiratory inflammation and exacerbated EAE induction [326]. USP25 was found to directly remove TRAF5 and TRAF6 ubiquitination induced by IL-17A and subsequently erase the signals for both TRAF6 dependent and independent pathways [326]. More recently, A20, another deubiquitinating enzyme, was identified as a negative regulator of IL-17A induced NF-κB and MAPK activation [69]. Notably, A20 itself was induced by IL-17A exposure and in turn formed a negative feedback control for IL-17A signaling. Deficiency of A20 in cells led to exacerbated IL-17A induced inflammatory gene expression [69]. Mechanically, IL-17R signaling directly recruited A20 to the distal domain of the receptor and therefore erased the ubiquitination of TRAF6 and also the activation of TRAF6 dependent cascades [69].
5.6.2 Th17 Effector Cytokines in Inflammatory Diseases
5.6.2.1 Th17 Effector Cytokines in Host Defense Against Infection
Th17 cells are found to be critical for host defense responses by releasing its effector cytokines IL-17A, IL-17F, and IL-22. The secreted IL-17A, IL-17F, or IL-22 in turn induces expression of host defense molecules in epithelial cells or keratinocytes to protect host from specific pathogens. Deficiency of IL-17A and IL-17F in mice caused increased susceptibility to the infection of extracellular pathogens, including Klebsiella pneumonia, Citrobacter rodentium, and Staphylococcus aureus [10, 110]. It was found that, an intestinal commensal bacterium, segmented filamentous bacteria (SFB), preferentially induced gut Th17 differentiation in steady state. Mice without SFB led to reduced Th17 proportion in intestine and were more susceptible to Citrobacter rodentium infection [113]. However, another study demonstrated that it was Il22-deficient mice but not Il17rc (a specific receptor for IL-17A and IL-17F)-deficient mice were susceptible to the infection [325]. IL-22 is a critical cytokine for mucosal immunity. It could directly promote epithelia proliferation to keep the epithelia barrier integrity during the infection of pathogens. IL-22 also acted alone or in synergy with other cytokines, such as IL-17A, IL-17F, IL-17C, or TNFα, to induce the expression of antimicrobial proteins such as S100A-proteins, β-defensins, or RegIII family proteins in host defense responses [10, 155, 253, 293, 294, 325]. In addition, IL-22 also induced the expression of multiple mucus proteins in globet cells, which are important for mucosal damage protection [260]. Furthermore, as IL-17A and IL-17F, this cytokine also stimulated the production of numerous inflammatory chemokines and cytokines production during infection [10, 155, 293]. Upon Klebsiella pneumoniae infection, neutralizing IL-22 led to remarkable bacterial dissemination in the lung and spleen [10]. Comparing with wild-type mice, the survival rate of infected animals treated with anti-IL-22 decreased significantly [10]. In this study, Th17 derived IL-22 acted in synergy with IL-17 to promote lung epithelia proliferation. The combination of IL-22 with IL-17 also induced several pro-inflammatory cytokines and chemokines or antimicrobial peptides expression in lung epithelial cells [10]. Another study also found that, comparing with wild-type mice, Il22-deficient mice died quickly after Klebsiella pneumoniae infection [301]. As described above, after Citrobacter rodentium infection, deficiency of IL-22 in mice led to increased intestinal damage, bacterial load and mortality [325].
In addition to extracellular pathogens, IL-17A or IL-22 also protected mice from intracellular bacterial and fungal infection. Upon Francisella tularensis infection, either neutralization or genetic depletion of IL-17A increased the bacterial burden in mice [156]. It was found that IL-17A, but not IL-17F or IL-22, was responsible for IL-12 production from dendritic cells and promoted Th1 activity during the infection [156]. IL-22, however, was found to protect Il12p35-deficient mice from liver necrosis upon Salmonella enterica serovar Enteritidis infection. Neutralization of IL-22 in Il12p35-deficient mice led to increased cell necrosis in the Salmonella enterica serovar Enteritidis infected liver, indicating a protective role of IL-22 against this pathogen [239]. Th17 differentiation was strongly induced by an oral opportunistic fungus, Candida albicans [233]. Deficiency of IL-17A, but not IL-17F in mice, remarkably increased the susceptibility to oral candidiasis, indicating that IL-17A is critical against this pathogen-mediated pathogenesis [233]. In addition, in Il17ra-deficient mice, without IL-17A and IL-17F signaling, neutralization of IL-22 in mice increased Candida albicans burden in stomach [50]. Similarly, increased fungal burden in the kidney and stomach was found in Il22-deficient mice when these mice were intravenously infected with Candida albicans [50]. Mice intragastrically infected with C. albicans showed upregulated IL-22 in the stomach while several antimicrobial peptides such as S100A8, S100A9, RegIIIβ, and RegIIIγ were all decreased in Il22-deficient mice in this model [50], indicating that IL-22 protects against pathogen infections through inducing antibacterial genes in the epithelium.
5.6.2.2 Th17 Effector Cytokines in Autoimmune Diseases
Autoimmune diseases, such as MS and RA, have long been noticed as Th1, Th2, or B cells related diseases. Recently growing evidences showed that the Th17 subset and its unique cytokines IL-17A, IL-17F, and IL-22 display board effects on the pathogenesis of MS and RA as well as many other autoimmune diseases including inflammatory bowel disease, psoriasis, systemic lupus erythematosus, and type 1 diabetes mellitus [336].
MS is a central nervous system autoimmune disease characterized with loss of protective myelin sheaths around the brain and spinal cord axons. It was found that the expression of IL-17A as well as its producing T cells was highly elevated in the brain lesion regions of MS patients [161, 277]. Deficiency of IL-23p19, a specific cytokine subunit critical for Th17 cell differentiation, protected mice from EAE [142]. Genetic depletion of either IL-17A or its specific receptor IL-17RC suppressed the induction of EAE in mice [100, 127, 304]. However, it was found that IL-17F was not required for the initiation stage of EAE but the later stage of this disease seems decreased in Il17f-deficient mice [304]. One study identified that Il22ra2 is a MS risk gene [18]. However, another study showed that Il22-deficient mice were still susceptible to EAE induction, suggesting that IL-22 appears not required for EAE [134].
RA is a systemic autoimmune disease which is characterized by symmetric inflammation that causes progressive joint cartilage destruction. Comparing with healthy volunteers and osteoarthritis patients, patients with RA showed remarkable increased IL-17A level in their rheumatoid synovium [130, 337]. In experimental murine models of RA, neutralizing of IL-17A ameliorated, while forced expression of IL-17A aggravated diseases progression [163, 164]. Il17a-deficient mice were protected from either collagen induced arthritis (CIA) or spontaneous autoimmune arthritis developed in Il1rn-deficient mice [110, 196]. The contribution of IL-17F to the arthritis pathogenesis was found to be extremely limited [110]. Increased IL-22 in serum was also found in human patients of RA, and its expression level was shown to correlate with disease activity [47]. In the CIA model, IL-22 level was increased and deficiency of IL-22 protected mice from CIA induction [71]. Blockade of IL-22 in Il1rn-deficient mice significantly reduced the inflammation and bone erosion [176]. However, another study revealed that mice treated with IL-22 prior to the onset of CIA had delayed progression of CIA and neutralization of IL-10 abrogated IL-22 protective effect [236]. These studies indicated that the role of IL-22 in RA still needs to be further characterized.
Inflammatory bowel diseases (IBD) is a chronic autoimmune intestinal inflammatory disease, which is caused by abnormal innate or/and adaptive immune responses. IBD can be classified into two major types, Crohn disease (CD) and ulcerative colitis (UC). IL-17A level was found to be upregulated in either CD or UC patients specimens [66, 259]. The expression of IL-17F was also higher in CD patients compared with UC patients [241]. Genome-wide association study (GWAS) data also identified IL-23R, STAT3, CCR6, as well as Act1, which were either critical for Th17 differentiation or IL-17 signaling, were IBD-associated genes [12, 37, 58, 286]. However, in murine models of IBD, the roles of IL-17A were still controversial. Antibody blockage of IL-17A or genetic depletion of IL-17A in mice caused exacerbated dextran sodium sulfate (DSS) induced colitis, indicating that IL-17A has a protective role of in this model [205, 304]. In contrast, comparing with wild-type mice, Il17a-deficient mice were protected from DSS induced colitis in another study [112]. The inconformity of those studies may largely due to the differences of gut microbiota among different facilities. In a parallel study of IL-17A, IL-17F was identified to enhance the inflammation of intestine as Il17f-deficient mice were protected from colitis induced by DSS [304]. Increased IL-22 expression has also been found in IBD patients [6, 20] and its serum level was correlated with disease severity [237]. The expression of its receptor IL22RA1 was also increased in UC patients tissues [117, 242]. Both antibody blockade of IL-22 function and genetic depletion of IL-22 or STAT3 in mice led to increased severe colitis, body weight loss and histological score, indicating IL-22 has a protective function in this model [218, 260, 315]. By contrast, IL-22 gene delivery reduced local intestine inflammation and induced the expression of mucus-associated proteins from goblet cells [260].
Psoriasis is a chronic skin immune disorder characterized by hyperplasia of dermis which is probably due to dysregulated interaction between immune cells and keratinocytes. Both IL-17A and IL-17F were remarkably upregulated in psoriatic skin biopsies from psoriasis patients [118, 119, 291]. As GWAS studies in IBD, similar approach also identified Act1 and Il23r as psoriasis-associated genes in psoriasis specimens [23, 60, 104, 256]. In mice, skin intradermal delivery of IL-23 could cause a psoriasis-like symptoms and the skin hyperplasia was meliorated in either Il17a- or Il22-deficient mice [229]. IL-17A cooperated with other cytokines like TNFα, IFN-γ, or IL-22 to induce the expression of inflammatory genes and antibacterial molecules in human keratinocytes [4, 34, 86, 155, 269]. IL-22 has also been found as a critical mediator in the psoriatic pathology. Increased serum IL-22 level was found in psoriatic patients and showed correlation with the disease severity [294]. IL-22-transgenic mice had acanthosis and hypogranularity symptoms which resembles psoriasis. These mice were born with stiff and shiny skin and then died quickly after broth [292]. Consistently, IL-23-mediated dermal inflammation was decreased in Il22-deficient mice [324]. Similarly, in an autoimmune psoriasis model, neutralization of IL-22 in wild-type mice showed either no development or very mild development of the disease [278]. Like IL-17A, IL-22 also could synergize with several other cytokines to promote the progression of psoriasis [211].
IL-17A is also found to be involved in the pathogenesis of other autoimmune diseases like SLE and T1DM. It was noticed that IL-17A was highly expressed in both SLE patients-derived double negative (DN) T cells and MRL/lpr mice [43, 321]. In a mouse model of SLE, comparing with Fcgr2b-deficient mice, recent study identified that Il17a and Fcgr2b-double deficient mice were resistance to the development of lupus [219]. IL-17A could activate inflammatory genes and autoantibodies production in peripheral blood mononuclear cells (PBMC) isolated from lupus patients [55]. These studies suggest that IL-17 is critical for the pathogenesis of SLE. Although the function of IL-17A in human T1DM progression remains unclear, in murine model of T1DM, IL-17A was noticed to aggravate autoimmune diabetes progression. Antibody blockage of IL-17A ameliorated autoimmune diabetes pathogenesis in non-obese diabetic (NOD) mice [61]. However, deficiency of IL-17A in NOD mice led to comparable hyperglycemia when compared with control NOD mice [127]. It was also noticed that plasma IL-22 levels decreased and correlated with disease severity in SLE patients [32]. However, the role of IL-22 in lupus still needs to be further studied.
As IL-17A signaling is critical for mutiple autoimmune diseases progression, the usage of blocking antibodies against IL-17 or IL-17RA to treat different autoimmune diseases such as RA or psoriasis has shown promising outcomes in clinical trials [72, 73, 103, 214]. Two humanized IL-17A antibodies, AIN457 and LY2439821, were in phase II trials for RA, MS, and psoriasis therapy and were shown no strong adverse safety concerns [73, 103]. A fully human monoclonal antibody to IL-17RA, Brodalumab, was developed to block IL-17R-mediated signaling and was undergoing in phase II trials for psoriasis and RA [214]. As IL-17RA is a common receptor subunit shared by other members of IL-17 family, the strategy for targeting IL-17RC, which is also critical for IL-17A-mediated signaling, maybe more specific.
5.6.3 Th17 Effector Cytokines in Allergy
Allergic diseases such as allergic asthma and atopic dermatitis (AD) are common chronic inflammatory diseases, which are characterized by infiltration of eosinophil, mast cells, and T cells. It was found that both IL-17A and IL-17F were remarkably increased in bronchial specimens from asthma patients [3, 54, 188]. Enforced expression of both IL-17A and IL-17F led to increased infiltration of neutrophil, but no eosinophil in lung [105, 304]. In OVA-induced airway allergic model, IL-17A was noticed to promote while IL-17F suppressed the allergic responses [110, 196, 304]. However, IL-17F was also showed not required for OVA induced neutrophilia in lung [110]. It was found that host dust mite induced airway inflammation was also dependent on IL-17A [139]. More recently, IL-17A, but not IL-17F, was found to promote airway hyper-responsiveness by directly enhancing the contraction of airway smooth muscle [137]. However, IL-22 has been found to play a protective role in allergic diseases. Increased IL-22 expression was found in both atopic dermatitis (AD) patients and asthmatic patients [201, 217]. In a mouse asthma model, neutralization of IL-22 increased the eosinophil recruitment into the lung [267]. One group showed that IL-22 inhibited antigen-induced airway inflammation by decreasing IL-25 production in lung epithelia cell [267]. Another group showed that IL-22 attenuated allergic responses by inhibiting DC functions [238].
5.6.4 Th17 Effector Cytokines in Cancer
Th17 cells had been implicated to associate with cancer development in human, although its roles in tumor progression are still in controversy between different studies. In hepatocellular carcinoma or colorectal cancer specimens, patients characterizing with high level of IL-17A expression correlated with a poor prognosis [272, 319]. By contrast, patients with ovarian cancer had poor prognosis when the infiltrated IL-17A producing cells numbers and IL-17A expression level were low [135], indicating that the influences of IL-17A on tumorigenesis are largely dependent on the type and stage of tumors.
Several studies by using Il17a-deficient mice implicated that IL-17A mainly has a pro-tumorigenic role among different tumor models. Subcutaneous transplantation of bladder carcinoma cell line MB49 or melanoma cell line B16 in Il17a-deficient mice led to delayed growth rate of those cells [287]. In either spontaneous or chemical-induced colon cancer models, disruption of IL-17A in mice caused reduced intestinal tumor development in those mice [26, 106]. More recently, it was noticed that either Il23p19- or its receptor deficient mice on an Apc-CRC background displayed reduced tumorigenesis in intestine, agreeing with the early studies with Il17a-deficient mice [80]. Enterotoxigenic Bacteroides fragilis (ETBF), an intestinal commensal bacteria, was found to promote intestinal tumorigenesis in Apc min/+ mice through promoting Th17 responses [297]. As described, this hematopoietic IL-23-Th17-IL-17A axis was found to mediate microbiota-driven tumor growth. However, we also found that IL-17C contributed to this process in a different way. Unlike IL-17A, which was produced by Th17 cells, IL-17C was specifically secreted by IECs [250]. Furthermore, IL-23 and IL-17A were shown to be critical for tumor cell growth but not survival during intestinal tumorigenesis [80, 250]. By contrast, IL-17C signaling was critical for IEC survival but not proliferation during the early stage of intestinal tumorigenesis [250]. Although IL-17C has been reported to promote Th17 differentiation [28], we found that the intestinal local resident cells rather than infiltrated hematopoietic cells such as Th17 were critical for IL-17C-mediated tumorigenesis, suggesting that IL-17C-mediated IEC survival instead of Th17 cell differentiation is critical for promoting intestinal tumorigenesis [250]. Both IL-17A and IL-23 were also found to promote other cancers such as cutaneous chemical carcinogenesis, as Il17a-, Il17ra-, or Il23p19-deficient mice developed remarkably decreased skin tumors [91, 141, 288].
As mentioned above, Th17 cells were also correlated with ameliorated prognosis in some studies. Similarly, in some mouse models, those IL-17A producing cells were also found to suppress tumor progression. One study showed that Th17 transplantation inhibited tumor growth through the activation of anti-tumor CD8+ cells [177]. Although there were studies showed that IL-17A enhanced tumor metastasis by showing that deficiency of IL-17A in mice reduced lung metastasis [24, 154, 287], other studies found controversial results by showing that IL-17A ablation led to increased lung metastasis [136, 177]. Thus, it is likely that the pro- or anti-tumor property of IL-17A is mainly dependent on the different tumor types and stages. The role of IL-17F in tumor development remains unclear. Similar to IL-17A, IL-17F was noticed to be critical for intestinal tumorigenesis in Apc min/+ mice [25]. By contrast, IL-17F played a protective role in in chemical induced colon cancer model [271].
5.7 Conclusion and Perspective
The discovery of Th17 cells as a distinct T helper cell subset refreshes our understanding of both adaptive immunity and its related immune disorders. Th17 effector cytokines, IL-17A, IL-17F, and IL-22, show unique proinflammatory properties comparing to the typical cytokines of either Th1 or Th2 subsets. Recent progresses of Th17 differentiation and plasticity also expand our knowledge about T-cell development and complexity between different subpopulations. Recent advances in uncovering the molecular mechanisms of signal transduction of Th17 cytokines IL-17A, IL-17F, and IL-22, coupling with largely expanded understanding of their roles in human inflammatory diseases as well as in the relevant mouse models, provide great opportunities to develop new targets and therapeutic strategies. Further investigation of the programs of Th17 differentiation and the signaling mediated by Th17 effector cytokines will benefit for the understanding and therapy of human inflammatory diseases.
References
Acosta-Rodriguez EV, Napolitani G, Lanzavecchia A, Sallusto F (2007) Interleukins 1beta and 6 but not transforming growth factor-beta are essential for the differentiation of interleukin 17-producing human T helper cells. Nat Immunol 8(9):942–949. doi:10.1038/ni1496
Aggarwal S, Ghilardi N, Xie MH, de Sauvage FJ, Gurney AL (2003) Interleukin-23 promotes a distinct CD4 T cell activation state characterized by the production of interleukin-17. J Biol Chem 278(3):1910–1914. doi:10.1074/jbc.M207577200
Al-Ramli W, Prefontaine D, Chouiali F, Martin JG, Olivenstein R, Lemiere C, Hamid Q (2009) T(H)17-associated cytokines (IL-17A and IL-17F) in severe asthma. J Allergy Clin Immunol 123(5):1185–1187. doi:10.1016/j.jaci.2009.02.024
Albanesi C, Cavani A, Girolomoni G (1999) IL-17 is produced by nickel-specific T lymphocytes and regulates ICAM-1 expression and chemokine production in human keratinocytes: synergistic or antagonist effects with IFN-gamma and TNF-alpha. Journal of immunology 162(1):494–502
Alonzi T, Fattori E, Lazzaro D, Costa P, Probert L, Kollias G, De Benedetti F, Poli V, Ciliberto G (1998) Interleukin 6 is required for the development of collagen-induced arthritis. J Exp Med 187(4):461–468
Andoh A, Zhang Z, Inatomi O, Fujino S, Deguchi Y, Araki Y, Tsujikawa T, Kitoh K, Kim-Mitsuyama S, Takayanagi A, Shimizu N, Fujiyama Y (2005) Interleukin-22, a member of the IL-10 subfamily, induces inflammatory responses in colonic subepithelial myofibroblasts. Gastroenterology 129(3):969–984. doi:10.1053/j.gastro.2005.06.071
Annunziato F, Cosmi L, Santarlasci V, Maggi L, Liotta F, Mazzinghi B, Parente E, Fili L, Ferri S, Frosali F, Giudici F, Romagnani P, Parronchi P, Tonelli F, Maggi E, Romagnani S (2007) Phenotypic and functional features of human Th17 cells. J Exp Med 204(8):1849–1861. doi:10.1084/jem.20070663
Ansel KM, Lee DU, Rao A (2003) An epigenetic view of helper T cell differentiation. Nat Immunol 4(7):616–623. doi:10.1038/ni0703-616
Atarashi K, Nishimura J, Shima T, Umesaki Y, Yamamoto M, Onoue M, Yagita H, Ishii N, Evans R, Honda K, Takeda K (2008) ATP drives lamina propria T(H)17 cell differentiation. Nature 455(7214):808–812. doi:10.1038/nature07240
Aujla SJ, Chan YR, Zheng M, Fei M, Askew DJ, Pociask DA, Reinhart TA, McAllister F, Edeal J, Gaus K, Husain S, Kreindler JL, Dubin PJ, Pilewski JM, Myerburg MM, Mason CA, Iwakura Y, Kolls JK (2008) IL-22 mediates mucosal host defense against gram-negative bacterial pneumonia. Nat Med 14(3):275–281. doi:10.1038/nm1710
Ballantyne SJ, Barlow JL, Jolin HE, Nath P, Williams AS, Chung KF, Sturton G, Wong SH, McKenzie AN (2007) Blocking IL-25 prevents airway hyperresponsiveness in allergic asthma. J Allergy Clin Immunol 120(6):1324–1331. doi:10.1016/j.jaci.2007.07.051
Barrett JC, Hansoul S, Nicolae DL, Cho JH, Duerr RH, Rioux JD, Brant SR, Silverberg MS, Taylor KD, Barmada MM, Bitton A, Dassopoulos T, Datta LW, Green T, Griffiths AM, Kistner EO, Murtha MT, Regueiro MD, Rotter JI, Schumm LP, Steinhart AH, Targan SR, Xavier RJ, Consortium NIG, Libioulle C, Sandor C, Lathrop M, Belaiche J, Dewit O, Gut I, Heath S, Laukens D, Mni M, Rutgeerts P, Van Gossum A, Zelenika D, Franchimont D, Hugot JP, de Vos M, Vermeire S, Louis E, Belgian-French IBDC, Wellcome Trust Case Control C, Cardon LR, Anderson CA, Drummond H, Nimmo E, Ahmad T, Prescott NJ, Onnie CM, Fisher SA, Marchini J, Ghori J, Bumpstead S, Gwilliam R, Tremelling M, Deloukas P, Mansfield J, Jewell D, Satsangi J, Mathew CG, Parkes M, Georges M, Daly MJ (2008) Genome-wide association defines more than 30 distinct susceptibility loci for Crohn’s disease. Nat Genet 40(8):955–962. doi:10.1038/ng.175
Batten M, Li J, Yi S, Kljavin NM, Danilenko DM, Lucas S, Lee J, de Sauvage FJ, Ghilardi N (2006) Interleukin 27 limits autoimmune encephalomyelitis by suppressing the development of interleukin 17-producing T cells. Nat Immunol 7(9):929–936. doi:10.1038/ni1375
Baud V, Karin M (2001) Signal transduction by tumor necrosis factor and its relatives. Trends Cell Biol 11(9):372–377
Bending D, De la Pena H, Veldhoen M, Phillips JM, Uyttenhove C, Stockinger B, Cooke A (2009) Highly purified Th17 cells from BDC2.5NOD mice convert into Th1-like cells in NOD/SCID recipient mice. J Clin Invest 119(3):565–572. doi:10.1172/JCI37865
Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK (2006) Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature 441(7090):235–238. doi:10.1038/nature04753
Bettelli E, Sullivan B, Szabo SJ, Sobel RA, Glimcher LH, Kuchroo VK (2004) Loss of T-bet, but not STAT1, prevents the development of experimental autoimmune encephalomyelitis. J Exp Med 200(1):79–87. doi:10.1084/jem.20031819
Beyeen AD, Adzemovic MZ, Ockinger J, Stridh P, Becanovic K, Laaksonen H, Lassmann H, Harris RA, Hillert J, Alfredsson L, Celius EG, Harbo HF, Kockum I, Jagodic M, Olsson T (2010) IL-22RA2 associates with multiple sclerosis and macrophage effector mechanisms in experimental neuroinflammation. Journal of immunology 185(11):6883–6890. doi:10.4049/jimmunol.1001392
Brand DD, Latham KA, Rosloniec EF (2007) Collagen-induced arthritis. Nat Protoc 2(5):1269–1275. doi:10.1038/nprot.2007.173
Brand S, Beigel F, Olszak T, Zitzmann K, Eichhorst ST, Otte JM, Diepolder H, Marquardt A, Jagla W, Popp A, Leclair S, Herrmann K, Seiderer J, Ochsenkuhn T, Goke B, Auernhammer CJ, Dambacher J (2006) IL-22 is increased in active Crohn’s disease and promotes proinflammatory gene expression and intestinal epithelial cell migration. Am J Physiol Gastrointest Liver Physiol 290(4):G827–838. doi:10.1152/ajpgi.00513.2005
Brustle A, Heink S, Huber M, Rosenplanter C, Stadelmann C, Yu P, Arpaia E, Mak TW, Kamradt T, Lohoff M (2007) The development of inflammatory T(H)-17 cells requires interferon-regulatory factor 4. Nat Immunol 8(9):958–966. doi:10.1038/ni1500
Bulek K, Liu C, Swaidani S, Wang L, Page RC, Gulen MF, Herjan T, Abbadi A, Qian W, Sun D, Lauer M, Hascall V, Misra S, Chance MR, Aronica M, Hamilton T, Li X (2011) The inducible kinase IKKi is required for IL-17-dependent signaling associated with neutrophilia and pulmonary inflammation. Nat Immunol 12(9):844–852. doi:10.1038/ni.2080
Cargill M, Schrodi SJ, Chang M, Garcia VE, Brandon R, Callis KP, Matsunami N, Ardlie KG, Civello D, Catanese JJ, Leong DU, Panko JM, McAllister LB, Hansen CB, Papenfuss J, Prescott SM, White TJ, Leppert MF, Krueger GG, Begovich AB (2007) A large-scale genetic association study confirms IL12B and leads to the identification of IL23R as psoriasis-risk genes. Am J Hum Genet 80(2):273–290. doi:10.1086/511051
Carmi Y, Rinott G, Dotan S, Elkabets M, Rider P, Voronov E, Apte RN (2011) Microenvironment-derived IL-1 and IL-17 interact in the control of lung metastasis. J immunol 186(6):3462–3471. doi:10.4049/jimmunol.1002901
Chae WJ, Bothwell AL (2011) IL-17F deficiency inhibits small intestinal tumorigenesis in ApcMin/+ mice. Biochem Biophys Res Commun 414(1):31–36. doi:10.1016/j.bbrc.2011.09.016
Chae WJ, Gibson TF, Zelterman D, Hao L, Henegariu O, Bothwell AL (2010) Ablation of IL-17A abrogates progression of spontaneous intestinal tumorigenesis. Proc Natl Acad Sci USA 107(12):5540–5544. doi:10.1073/pnas.0912675107
Chang SH, Park H, Dong C (2006) Act1 adaptor protein is an immediate and essential signaling component of interleukin-17 receptor. J Biol Chem 281(47):35603–35607. doi:10.1074/jbc.C600256200
Chang SH, Reynolds JM, Pappu BP, Chen G, Martinez GJ, Dong C (2011) Interleukin-17C promotes Th17 cell responses and autoimmune disease via interleukin-17 receptor E. Immunity 35(4):611–621. doi:10.1016/j.immuni.2011.09.010
Chen Q, Yang W, Gupta S, Biswas P, Smith P, Bhagat G, Pernis AB (2008) IRF-4-binding protein inhibits interleukin-17 and interleukin-21 production by controlling the activity of IRF-4 transcription factor. Immunity 29(6):899–911. doi:10.1016/j.immuni.2008.10.011
Chen W, Jin W, Hardegen N, Lei KJ, Li L, Marinos N, McGrady G, Wahl SM (2003) Conversion of peripheral CD4 + CD25- naive T cells to CD4+ CD25+ regulatory T cells by TGF-beta induction of transcription factor Foxp3. J Exp Med 198(12):1875–1886. doi:10.1084/jem.20030152
Chen Z, Laurence A, Kanno Y, Pacher-Zavisin M, Zhu BM, Tato C, Yoshimura A, Hennighausen L, O’Shea JJ (2006) Selective regulatory function of Socs3 in the formation of IL-17-secreting T cells. Proc Natl Acad Sci USA 103(21):8137–8142. doi:10.1073/pnas.0600666103
Cheng F, Guo Z, Xu H, Yan D, Li Q (2009) Decreased plasma IL22 levels, but not increased IL17 and IL23 levels, correlate with disease activity in patients with systemic lupus erythematosus. Ann Rheum Dis 68(4):604–606. doi:10.1136/ard.2008.097089
Cheng G, Cleary AM, Ye ZS, Hong DI, Lederman S, Baltimore D (1995) Involvement of CRAF1, a relative of TRAF, in CD40 signaling. Science 267(5203):1494–1498
Chiricozzi A, Guttman-Yassky E, Suarez-Farinas M, Nograles KE, Tian S, Cardinale I, Chimenti S, Krueger JG (2011) Integrative responses to IL-17 and TNF-alpha in human keratinocytes account for key inflammatory pathogenic circuits in psoriasis. J Invest Dermatol 131(3):677–687. doi:10.1038/jid.2010.340
Chitnis T, Najafian N, Benou C, Salama AD, Grusby MJ, Sayegh MH, Khoury SJ (2001) Effect of targeted disruption of STAT4 and STAT6 on the induction of experimental autoimmune encephalomyelitis. J Clin Invest 108(5):739–747. doi:10.1172/JCI12563
Chung Y, Chang SH, Martinez GJ, Yang XO, Nurieva R, Kang HS, Ma L, Watowich SS, Jetten AM, Tian Q, Dong C (2009) Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity 30(4):576–587. doi:10.1016/j.immuni.2009.02.007
Ciccacci C, Biancone L, Di Fusco D, Ranieri M, Condino G, Giardina E, Onali S, Lepre T, Pallone F, Novelli G, Borgiani P (2012) TRAF3IP2 gene is associated with cutaneous extraintestinal manifestations in Inflammatory Bowel Disease. J Crohns Colitis. doi:10.1016/j.crohns.2012.02.020
Claudio E, Sonder SU, Saret S, Carvalho G, Ramalingam TR, Wynn TA, Chariot A, Garcia-Perganeda A, Leonardi A, Paun A, Chen A, Ren NY, Wang H, Siebenlist U (2009) The adaptor protein CIKS/Act1 is essential for IL-25-mediated allergic airway inflammation. J immunol 182(3):1617–1630
Cobb BS, Nesterova TB, Thompson E, Hertweck A, O’Connor E, Godwin J, Wilson CB, Brockdorff N, Fisher AG, Smale ST, Merkenschlager M (2005) T cell lineage choice and differentiation in the absence of the RNase III enzyme Dicer. J Exp Med 201(9):1367–1373. doi:10.1084/jem.20050572
Conforti-Andreoni C, Spreafico R, Qian HL, Riteau N, Ryffel B, Ricciardi-Castagnoli P, Mortellaro A (2011) Uric acid-driven Th17 differentiation requires inflammasome-derived IL-1 and IL-18. J immunol 187(11):5842–5850. doi:10.4049/jimmunol.1101408
Coombes JL, Siddiqui KR, Arancibia-Carcamo CV, Hall J, Sun CM, Belkaid Y, Powrie F (2007) A functionally specialized population of mucosal CD103 + DCs induces Foxp3+ regulatory T cells via a TGF-beta and retinoic acid-dependent mechanism. J Exp Med 204(8):1757–1764. doi:10.1084/jem.20070590
Coquet JM, Chakravarti S, Smyth MJ, Godfrey DI (2008) Cutting edge: IL-21 is not essential for Th17 differentiation or experimental autoimmune encephalomyelitis. J immunol 180(11):7097–7101
Crispin JC, Oukka M, Bayliss G, Cohen RA, Van Beek CA, Stillman IE, Kyttaris VC, Juang YT, Tsokos GC (2008) Expanded double negative T cells in patients with systemic lupus erythematosus produce IL-17 and infiltrate the kidneys. J immunol 181(12):8761–8766
Cua DJ, Sherlock J, Chen Y, Murphy CA, Joyce B, Seymour B, Lucian L, To W, Kwan S, Churakova T, Zurawski S, Wiekowski M, Lira SA, Gorman D, Kastelein RA, Sedgwick JD (2003) Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 421(6924):744–748. doi:10.1038/nature01355
Cua DJ, Tato CM (2010) Innate IL-17-producing cells: the sentinels of the immune system. Nat Rev Immunol 10(7):479–489. doi:10.1038/nri2800
Cui G, Qin X, Wu L, Zhang Y, Sheng X, Yu Q, Sheng H, Xi B, Zhang JZ, Zang YQ (2011) Liver X receptor (LXR) mediates negative regulation of mouse and human Th17 differentiation. J Clin Invest 121(2):658–670. doi:10.1172/JCI42974
da Rocha LF, Jr., Duarte AL, Dantas AT, Mariz HA, Pitta Ida R, Galdino SL, Pitta MG (2012) Increased serum interleukin 22 in patients with rheumatoid arthritis and correlation with disease activity. J Rheumatol 39(7):1320–1325. doi:10.3899/jrheum.111027
Das J, Ren G, Zhang L, Roberts AI, Zhao X, Bothwell AL, Van Kaer L, Shi Y, Das G (2009) Transforming growth factor beta is dispensable for the molecular orchestration of Th17 cell differentiation. J Exp Med 206(11):2407–2416. doi:10.1084/jem.20082286
de Beaucoudrey L, Puel A, Filipe-Santos O, Cobat A, Ghandil P, Chrabieh M, Feinberg J, von Bernuth H, Samarina A, Janniere L, Fieschi C, Stephan JL, Boileau C, Lyonnet S, Jondeau G, Cormier-Daire V, Le Merrer M, Hoarau C, Lebranchu Y, Lortholary O, Chandesris MO, Tron F, Gambineri E, Bianchi L, Rodriguez-Gallego C, Zitnik SE, Vasconcelos J, Guedes M, Vitor AB, Marodi L, Chapel H, Reid B, Roifman C, Nadal D, Reichenbach J, Caragol I, Garty BZ, Dogu F, Camcioglu Y, Gulle S, Sanal O, Fischer A, Abel L, Stockinger B, Picard C, Casanova JL (2008) Mutations in STAT3 and IL12RB1 impair the development of human IL-17-producing T cells. J Exp Med 205(7):1543–1550. doi:10.1084/jem.20080321
De Luca A, Zelante T, D’Angelo C, Zagarella S, Fallarino F, Spreca A, Iannitti RG, Bonifazi P, Renauld JC, Bistoni F, Puccetti P, Romani L (2010) IL-22 defines a novel immune pathway of antifungal resistance. Mucosal Immunol 3(4):361–373. doi:10.1038/mi.2010.22
Delgoffe GM, Pollizzi KN, Waickman AT, Heikamp E, Meyers DJ, Horton MR, Xiao B, Worley PF, Powell JD (2011) The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat Immunol 12(4):295–303. doi:10.1038/ni.2005
Dinarello CA (2009) Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 27:519–550. doi:10.1146/annurev.immunol.021908.132612
Djuretic IM, Levanon D, Negreanu V, Groner Y, Rao A, Ansel KM (2007) Transcription factors T-bet and Runx3 cooperate to activate Ifng and silence Il4 in T helper type 1 cells. Nat Immunol 8(2):145–153. doi:10.1038/ni1424
Doe C, Bafadhel M, Siddiqui S, Desai D, Mistry V, Rugman P, McCormick M, Woods J, May R, Sleeman MA, Anderson IK, Brightling CE (2010) Expression of the T helper 17-associated cytokines IL-17A and IL-17F in asthma and COPD. Chest 138(5):1140–1147. doi:10.1378/chest.09-3058
Dong G, Ye R, Shi W, Liu S, Wang T, Yang X, Yang N, Yu X (2003) IL-17 induces autoantibody overproduction and peripheral blood mononuclear cell overexpression of IL-6 in lupus nephritis patients. Chin Med J 116(4):543–548
Du C, Liu C, Kang J, Zhao G, Ye Z, Huang S, Li Z, Wu Z, Pei G (2009) MicroRNA miR-326 regulates TH-17 differentiation and is associated with the pathogenesis of multiple sclerosis. Nat Immunol 10(12):1252–1259. doi:10.1038/ni.1798
Du J, Huang C, Zhou B, Ziegler SF (2008) Isoform-specific inhibition of ROR alpha-mediated transcriptional activation by human FOXP3. J Immunol 180(7):4785–4792
Duerr RH, Taylor KD, Brant SR, Rioux JD, Silverberg MS, Daly MJ, Steinhart AH, Abraham C, Regueiro M, Griffiths A, Dassopoulos T, Bitton A, Yang H, Targan S, Datta LW, Kistner EO, Schumm LP, Lee AT, Gregersen PK, Barmada MM, Rotter JI, Nicolae DL, Cho JH (2006) A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 314(5804):1461–1463. doi:10.1126/science.1135245
Durant L, Watford WT, Ramos HL, Laurence A, Vahedi G, Wei L, Takahashi H, Sun HW, Kanno Y, Powrie F, O’Shea JJ (2010) Diverse targets of the transcription factor STAT3 contribute to T cell pathogenicity and homeostasis. Immunity 32(5):605–615. doi:10.1016/j.immuni.2010.05.003
Ellinghaus E, Ellinghaus D, Stuart PE, Nair RP, Debrus S, Raelson JV, Belouchi M, Fournier H, Reinhard C, Ding J, Li Y, Tejasvi T, Gudjonsson J, Stoll SW, Voorhees JJ, Lambert S, Weidinger S, Eberlein B, Kunz M, Rahman P, Gladman DD, Gieger C, Wichmann HE, Karlsen TH, Mayr G, Albrecht M, Kabelitz D, Mrowietz U, Abecasis GR, Elder JT, Schreiber S, Weichenthal M, Franke A (2010) Genome-wide association study identifies a psoriasis susceptibility locus at TRAF3IP2. Nat Genet 42(11):991–995. doi:10.1038/ng.689
Emamaullee JA, Davis J, Merani S, Toso C, Elliott JF, Thiesen A, Shapiro AM (2009) Inhibition of Th17 cells regulates autoimmune diabetes in NOD mice. Diabetes 58(6):1302–1311. doi:10.2337/db08-1113
Endo Y, Iwamura C, Kuwahara M, Suzuki A, Sugaya K, Tumes DJ, Tokoyoda K, Hosokawa H, Yamashita M, Nakayama T (2011) Eomesodermin controls interleukin-5 production in memory T helper 2 cells through inhibition of activity of the transcription factor GATA3. Immunity 35(5):733–745. doi:10.1016/j.immuni.2011.08.017
Eshima K, Chiba S, Suzuki H, Kokubo K, Kobayashi H, Iizuka M, Iwabuchi K, Shinohara N (2012) Ectopic expression of a T-box transcription factor, eomesodermin, renders CD4(+) Th cells cytotoxic by activating both perforin- and FasL-pathways. Immunol Lett 144(1–2):7–15. doi:10.1016/j.imlet.2012.02.013
Fallon PG, Ballantyne SJ, Mangan NE, Barlow JL, Dasvarma A, Hewett DR, McIlgorm A, Jolin HE, McKenzie AN (2006) Identification of an interleukin (IL)-25-dependent cell population that provides IL-4, IL-5, and IL-13 at the onset of helminth expulsion. J Exp Med 203(4):1105–1116. doi:10.1084/jem.20051615
Fujimoto M, Serada S, Mihara M, Uchiyama Y, Yoshida H, Koike N, Ohsugi Y, Nishikawa T, Ripley B, Kimura A, Kishimoto T, Naka T (2008) Interleukin-6 blockade suppresses autoimmune arthritis in mice by the inhibition of inflammatory Th17 responses. Arthritis Rheum 58(12):3710–3719. doi:10.1002/art.24126
Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, Bamba T, Fujiyama Y (2003) Increased expression of interleukin 17 in inflammatory bowel disease. Gut 52(1):65–70
Furlan R, Martino G, Galbiati F, Poliani PL, Smiroldo S, Bergami A, Desina G, Comi G, Flavell R, Su MS, Adorini L (1999) Caspase-1 regulates the inflammatory process leading to autoimmune demyelination. J Immunol 163(5):2403–2409
Gaffen SL (2009) Structure and signalling in the IL-17 receptor family. Nat Rev Immunol 9(8):556–567. doi:10.1038/nri2586
Garg AV, Ahmed M, Vallejo AN, Ma A, Gaffen SL (2013) The deubiquitinase a20 mediates feedback inhibition of interleukin-17 receptor signaling. Science Signal 6(278):ra44. doi:10.1126/scisignal.2003699
Gavin MA, Rasmussen JP, Fontenot JD, Vasta V, Manganiello VC, Beavo JA, Rudensky AY (2007) Foxp3-dependent programme of regulatory T-cell differentiation. Nature 445(7129):771–775. doi:10.1038/nature05543
Geboes L, Dumoutier L, Kelchtermans H, Schurgers E, Mitera T, Renauld JC, Matthys P (2009) Proinflammatory role of the Th17 cytokine interleukin-22 in collagen-induced arthritis in C57BL/6 mice. Arthritis Rheum 60(2):390–395. doi:10.1002/art.24220
Genovese MC, Durez P, Richards HB, Supronik J, Dokoupilova E, Mazurov V, Aelion JA, Lee SH, Codding CE, Kellner H, Ikawa T, Hugot S, Mpofu S (2013) Efficacy and safety of secukinumab in patients with rheumatoid arthritis: a phase II, dose-finding, double-blind, randomised, placebo controlled study. Ann Rheum Dis 72(6):863–869. doi:10.1136/annrheumdis-2012-201601
Genovese MC, Van den Bosch F, Roberson SA, Bojin S, Biagini IM, Ryan P, Sloan-Lancaster J (2010) LY2439821, a humanized anti-interleukin-17 monoclonal antibody, in the treatment of patients with rheumatoid arthritis: a phase I randomized, double-blind, placebo-controlled, proof-of-concept study. Arthritis Rheum 62(4):929–939. doi:10.1002/art.27334
Ghoreschi K, Laurence A, Yang XP, Tato CM, McGeachy MJ, Konkel JE, Ramos HL, Wei L, Davidson TS, Bouladoux N, Grainger JR, Chen Q, Kanno Y, Watford WT, Sun HW, Eberl G, Shevach EM, Belkaid Y, Cua DJ, Chen W, O’Shea JJ (2010) Generation of pathogenic T(H)17 cells in the absence of TGF-beta signalling. Nature 467(7318):967–971. doi:10.1038/nature09447
Gopal M, da Cunha AP, Weiner HL (2013) 97: Silencing MicroRNA-21 ameliorates Th17 mediated autoimmune inflammation. Cytokine 63(3):266
Gordon SM, Carty SA, Kim JS, Zou T, Smith-Garvin J, Alonzo ES, Haimm E, Sant’Angelo DB, Koretzky GA, Reiner SL, Jordan MS (2011) Requirements for eomesodermin and promyelocytic leukemia zinc finger in the development of innate-like CD8 + T cells. J Immunol 186(8):4573–4578. doi:10.4049/jimmunol.1100037
Gorelik L, Fields PE, Flavell RA (2000) Cutting edge: TGF-beta inhibits Th type 2 development through inhibition of GATA-3 expression. J Immunol 165(9):4773–4777
Gran B, Zhang GX, Yu S, Li J, Chen XH, Ventura ES, Kamoun M, Rostami A (2002) IL-12p35-deficient mice are susceptible to experimental autoimmune encephalomyelitis: evidence for redundancy in the IL-12 system in the induction of central nervous system autoimmune demyelination. J Immunol 169(12):7104–7110
Gris D, Ye Z, Iocca HA, Wen H, Craven RR, Gris P, Huang M, Schneider M, Miller SD, Ting JP (2010) NLRP3 plays a critical role in the development of experimental autoimmune encephalomyelitis by mediating Th1 and Th17 responses. J Immunol 185(2):974–981. doi:10.4049/jimmunol.0904145
Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, Taniguchi K, Yu GY, Osterreicher CH, Hung KE, Datz C, Feng Y, Fearon ER, Oukka M, Tessarollo L, Coppola V, Yarovinsky F, Cheroutre H, Eckmann L, Trinchieri G, Karin M (2012) Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 491(7423):254–258 nature11465 [pii]10.10348/nature11465
Gulen MF, Bulek K, Xiao H, Yu M, Gao J, Sun L, Beurel E, Kaidanovich-Beilin O, Fox PL, DiCorleto PE, Wang JA, Qin J, Wald DN, Woodgett JR, Jope RS, Carman J, Dongre A, Li X (2012) Inactivation of the enzyme GSK3alpha by the kinase IKKi promotes AKT-mTOR signaling pathway that mediates interleukin-1-induced Th17 cell maintenance. Immunity 37(5):800–812. doi:10.1016/j.immuni.2012.08.019
Gulen MF, Kang Z, Bulek K, Youzhong W, Kim TW, Chen Y, Altuntas CZ, Sass Bak-Jensen K, McGeachy MJ, Do JS, Xiao H, Delgoffe GM, Min B, Powell JD, Tuohy VK, Cua DJ, Li X (2010) The receptor SIGIRR suppresses Th17 cell proliferation via inhibition of the interleukin-1 receptor pathway and mTOR kinase activation. Immunity 32(1):54–66. doi:10.1016/j.immuni.2009.12.003
Guo H, Ingolia NT, Weissman JS, Bartel DP (2010) Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature 466(7308):835–840. doi:10.1038/nature09267
Gutcher I, Donkor MK, Ma Q, Rudensky AY, Flavell RA, Li MO (2011) Autocrine transforming growth factor-beta1 promotes in vivo Th17 cell differentiation. Immunity 34(3):396–408. doi:10.1016/j.immuni.2011.03.005
Hacker H, Redecke V, Blagoev B, Kratchmarova I, Hsu LC, Wang GG, Kamps MP, Raz E, Wagner H, Hacker G, Mann M, Karin M (2006) Specificity in Toll-like receptor signalling through distinct effector functions of TRAF3 and TRAF6. Nature 439(7073):204–207. doi:10.1038/nature04369
Harper EG, Guo C, Rizzo H, Lillis JV, Kurtz SE, Skorcheva I, Purdy D, Fitch E, Iordanov M, Blauvelt A (2009) Th17 cytokines stimulate CCL20 expression in keratinocytes in vitro and in vivo: implications for psoriasis pathogenesis. J Invest Dermatol 129(9):2175–2183. doi:10.1038/jid.2009.65
Harrington LE, Hatton RD, Mangan PR, Turner H, Murphy TL, Murphy KM, Weaver CT (2005) Interleukin 17-producing CD4 + effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat Immunol 6(11):1123–1132. doi:10.1038/ni1254
Harris TJ, Grosso JF, Yen HR, Xin H, Kortylewski M, Albesiano E, Hipkiss EL, Getnet D, Goldberg MV, Maris CH, Housseau F, Yu H, Pardoll DM, Drake CG (2007) Cutting edge: an in vivo requirement for STAT3 signaling in TH17 development and TH17-dependent autoimmunity. J Immunol 179(7):4313–4317
Hartupee J, Liu C, Novotny M, Sun D, Li X, Hamilton TA (2009) IL-17 signaling for mRNA stabilization does not require TNF receptor-associated factor 6. J Immunol 182(3):1660–1666
Hata H, Sakaguchi N, Yoshitomi H, Iwakura Y, Sekikawa K, Azuma Y, Kanai C, Moriizumi E, Nomura T, Nakamura T, Sakaguchi S (2004) Distinct contribution of IL-6, TNF-alpha, IL-1, and IL-10 to T cell-mediated spontaneous autoimmune arthritis in mice. J Clin Invest 114(4):582–588. doi:10.1172/JCI21795
He D, Li H, Yusuf N, Elmets CA, Athar M, Katiyar SK, Xu H (2012) IL-17 mediated inflammation promotes tumor growth and progression in the skin. PLoS ONE 7(2):e32126. doi:10.1371/journal.pone.0032126
He JQ, Zarnegar B, Oganesyan G, Saha SK, Yamazaki S, Doyle SE, Dempsey PW, Cheng G (2006) Rescue of TRAF3-null mice by p100 NF-kappa B deficiency. J Exp Med 203(11):2413–2418 (jem.20061166 [pii]/jem.20061166)
He YW, Deftos ML, Ojala EW, Bevan MJ (1998) RORgamma t, a novel isoform of an orphan receptor, negatively regulates Fas ligand expression and IL-2 production in T cells. Immunity 9(6):797–806
Heinrich PC, Behrmann I, Haan S, Hermanns HM, Muller-Newen G, Schaper F (2003) Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem J 374(Pt 1):1–20. doi:10.1042/BJ20030407
Hemdan NY, Birkenmeier G, Wichmann G (2012) Key molecules in the differentiation and commitment program of T helper 17 (Th17) cells up-to-date. Immunol Lett 148(2):97–109. doi:10.1016/j.imlet.2012.09.007
Herjan T, Yao P, Qian W, Li X, Liu C, Bulek K, Sun D, Yang WP, Zhu J, He A, Carman JA, Erzurum SC, Lipshitz HD, Fox PL, Hamilton TA, Li X (2013) HuR Is Required for IL-17-Induced Act1-Mediated CXCL1 and CXCL5 mRNA Stabilization. J Immunol. doi:10.4049/jimmunol.1203315
Hirano T, Matsuda T, Turner M, Miyasaka N, Buchan G, Tang B, Sato K, Shimizu M, Maini R, Feldmann M et al (1988) Excessive production of interleukin 6/B cell stimulatory factor-2 in rheumatoid arthritis. Eur J Immunol 18(11):1797–1801. doi:10.1002/eji.1830181122
Ho AW, Shen F, Conti HR, Patel N, Childs EE, Peterson AC, Hernandez-Santos N, Kolls JK, Kane LP, Ouyang W, Gaffen SL (2010) IL-17RC is required for immune signaling via an extended SEF/IL-17R signaling domain in the cytoplasmic tail. J Immunol 185(2):1063–1070 jimmunol.0903739 [pii]/jimmunol.0903739
Holland SM, DeLeo FR, Elloumi HZ, Hsu AP, Uzel G, Brodsky N, Freeman AF, Demidowich A, Davis J, Turner ML, Anderson VL, Darnell DN, Welch PA, Kuhns DB, Frucht DM, Malech HL, Gallin JI, Kobayashi SD, Whitney AR, Voyich JM, Musser JM, Woellner C, Schaffer AA, Puck JM, Grimbacher B (2007) STAT3 mutations in the hyper-IgE syndrome. N Engl J Med 357(16):1608–1619. doi:10.1056/NEJMoa073687
Hu Y, Ota N, Peng I, Refino CJ, Danilenko DM, Caplazi P, Ouyang W (2010) IL-17RC is required for IL-17A- and IL-17F-dependent signaling and the pathogenesis of experimental autoimmune encephalomyelitis. J Immunol 184(8):4307–4316. doi:10.4049/jimmunol.0903614
Huang F, Kao CY, Wachi S, Thai P, Ryu J, Wu R (2007) Requirement for both JAK-mediated PI3 K signaling and ACT1/TRAF6/TAK1-dependent NF-kappaB activation by IL-17A in enhancing cytokine expression in human airway epithelial cells. J Immunol 179(10):6504–6513
Huber M, Brustle A, Reinhard K, Guralnik A, Walter G, Mahiny A, von Low E, Lohoff M (2008) IRF4 is essential for IL-21-mediated induction, amplification, and stabilization of the Th17 phenotype. Proc Natl Acad Sci USA 105(52):20846–20851. doi:10.1073/pnas.0809077106
Hueber W, Patel DD, Dryja T, Wright AM, Koroleva I, Bruin G, Antoni C, Draelos Z, Gold MH, Psoriasis Study G, Durez P, Tak PP, Gomez-Reino JJ, Rheumatoid Arthritis Study G, Foster CS, Kim RY, Samson CM, Falk NS, Chu DS, Callanan D, Nguyen QD, Uveitis Study G, Rose K, Haider A, Di Padova F (2010) Effects of AIN457, a fully human antibody to interleukin-17A, on psoriasis, rheumatoid arthritis, and uveitis. Science translational medicine 2 (52):52ra72. doi:10.1126/scitranslmed.3001107
Huffmeier U, Uebe S, Ekici AB, Bowes J, Giardina E, Korendowych E, Juneblad K, Apel M, McManus R, Ho P, Bruce IN, Ryan AW, Behrens F, Lascorz J, Bohm B, Traupe H, Lohmann J, Gieger C, Wichmann HE, Herold C, Steffens M, Klareskog L, Wienker TF, Fitzgerald O, Alenius GM, McHugh NJ, Novelli G, Burkhardt H, Barton A, Reis A (2010) Common variants at TRAF3IP2 are associated with susceptibility to psoriatic arthritis and psoriasis. Nat Genet 42(11):996–999. doi:10.101038/ng.688
Hurst SD, Muchamuel T, Gorman DM, Gilbert JM, Clifford T, Kwan S, Menon S, Seymour B, Jackson C, Kung TT, Brieland JK, Zurawski SM, Chapman RW, Zurawski G, Coffman RL (2002) New IL-17 family members promote Th1 or Th2 responses in the lung: in vivo function of the novel cytokine IL-25. J Immunol 169(1):443–453
Hyun YS, Han DS, Lee AR, Eun CS, Youn J, Kim HY (2012) Role of IL-17A in the development of colitis-associated cancer. Carcinogenesis 33(4):931–936. doi:10.1093/carcin/bgs106
Ichiyama K, Hashimoto M, Sekiya T, Nakagawa R, Wakabayashi Y, Sugiyama Y, Komai K, Saba I, Moroy T, Yoshimura A (2009) Gfi1 negatively regulates T(h)17 differentiation by inhibiting RORgammat activity. Int Immunol 21(7):881–889. doi:10.1093/intimm/dxp054
Ichiyama K, Sekiya T, Inoue N, Tamiya T, Kashiwagi I, Kimura A, Morita R, Muto G, Shichita T, Takahashi R, Yoshimura A (2011) Transcription factor Smad-independent T helper 17 cell induction by transforming-growth factor-beta is mediated by suppression of eomesodermin. Immunity 34(5):741–754. doi:10.1016/j.immuni.2011.02.021
Inoue M, Williams KL, Gunn MD, Shinohara ML (2012) NLRP3 inflammasome induces chemotactic immune cell migration to the CNS in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA 109(26):10480–10485. doi:10.1073/pnas.1201836109
Ishigame H, Kakuta S, Nagai T, Kadoki M, Nambu A, Komiyama Y, Fujikado N, Tanahashi Y, Akitsu A, Kotaki H, Sudo K, Nakae S, Sasakawa C, Iwakura Y (2009) Differential roles of interleukin-17A and -17F in host defense against mucoepithelial bacterial infection and allergic responses. Immunity 30(1):108–119. doi:10.1016/j.immuni.2008.11.009
Ito A, Bebo BF Jr, Matejuk A, Zamora A, Silverman M, Fyfe-Johnson A, Offner H (2001) Estrogen treatment down-regulates TNF-alpha production and reduces the severity of experimental autoimmune encephalomyelitis in cytokine knockout mice. J Immunol 167(1):542–552
Ito R, Kita M, Shin-Ya M, Kishida T, Urano A, Takada R, Sakagami J, Imanishi J, Iwakura Y, Okanoue T, Yoshikawa T, Kataoka K, Mazda O (2008) Involvement of IL-17A in the pathogenesis of DSS-induced colitis in mice. Biochem Biophys Res Commun 377(1):12–16. doi:10.1016/j.bbrc.2008.09.019
Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, Tanoue T, Imaoka A, Itoh K, Takeda K, Umesaki Y, Honda K, Littman DR (2009) Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139(3):485–498. doi:10.1016/j.cell.2009.09.033
Ivanov II, McKenzie BS, Zhou L, Tadokoro CE, Lepelley A, Lafaille JJ, Cua DJ, Littman DR (2006) The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17 + T helper cells. Cell 126(6):1121–1133. doi:10.1016/j.cell.2006.07.035
Iwakura Y, Ishigame H, Saijo S, Nakae S (2011) Functional specialization of interleukin-17 family members. Immunity 34(2):149–162. doi:10.1016/j.immuni.2011.02.012
Jha S, Srivastava SY, Brickey WJ, Iocca H, Toews A, Morrison JP, Chen VS, Gris D, Matsushima GK, Ting JP (2010) The inflammasome sensor, NLRP3, regulates CNS inflammation and demyelination via caspase-1 and interleukin-18. J Neurosci 30(47):15811–15820. doi:10.1523/JNEUROSCI.4088-10.2010
Jiang R, Wang H, Deng L, Hou J, Shi R, Yao M, Gao Y, Yao A, Wang X, Yu L, Sun B (2013) IL-22 is related to development of human colon cancer by activation of STAT3. BMC Cancer 13:59. doi:10.1186/1471-2407-13-59
Johansen C, Usher PA, Kjellerup RB, Lundsgaard D, Iversen L, Kragballe K (2009) Characterization of the interleukin-17 isoforms and receptors in lesional psoriatic skin. Br J Dermatol 160(2):319–324. doi:10.1111/j.1365-2133.2008.08902.x
Johnston A, Fritz Y, Dawes SM, Diaconu D, Al-Attar PM, Guzman AM, Chen CS, Fu W, Gudjonsson JE, McCormick TS, Ward NL (2013) Keratinocyte overexpression of IL-17C promotes psoriasiform skin inflammation. J Immunol 190(5):2252–2262. doi:10.4049/jimmunol.1201505
Joosten LA, Helsen MM, van de Loo FA, van den Berg WB (2008) Anticytokine treatment of established type II collagen-induced arthritis in DBA/1 mice: a comparative study using anti-TNFalpha, anti-IL-1alpha/beta and IL-1Ra. Arthritis Rheum 58(2 Suppl):S110–122. doi:10.1002/art.23363
Kang Z, Swaidani S, Yin W, Wang C, Barlow JL, Gulen MF, Bulek K, Do JS, Aronica M, McKenzie AN, Min B, Li X (2012) Epithelial cell-specific Act1 adaptor mediates interleukin-25-dependent helminth expulsion through expansion of Lin(−)c-Kit(+) innate cell population. Immunity 36(5):821–833. doi:10.1016/j.immuni.2012.03.021
Kawaguchi M, Kokubu F, Matsukura S, Ieki K, Odaka M, Watanabe S, Suzuki S, Adachi M, Huang SK (2003) Induction of C-X-C chemokines, growth-related oncogene alpha expression, and epithelial cell-derived neutrophil-activating protein-78 by ML-1 (interleukin-17F) involves activation of Raf1-mitogen-activated protein kinase kinase-extracellular signal-regulated kinase 1/2 pathway. J Pharmacology and experimental therapeutics 307(3):1213–1220. doi:10.1124/jpet.103.056341
Kim MR, Manoukian R, Yeh R, Silbiger SM, Danilenko DM, Scully S, Sun J, DeRose ML, Stolina M, Chang D, Van GY, Clarkin K, Nguyen HQ, Yu YB, Jing S, Senaldi G, Elliott G, Medlock ES (2002) Transgenic overexpression of human IL-17E results in eosinophilia, B-lymphocyte hyperplasia, and altered antibody production. Blood 100(7):2330–2340. doi:10.1182/blood-2002-01-0012
Kirigin FF, Lindstedt K, Sellars M, Ciofani M, Low SL, Jones L, Bell F, Pauli F, Bonneau R, Myers RM, Littman DR, Chong MM (2012) Dynamic microRNA gene transcription and processing during T cell development. J Immunol 188(7):3257–3267. doi:10.4049/jimmunol.1103175
Kleinewietfeld M, Manzel A, Titze J, Kvakan H, Yosef N, Linker RA, Muller DN, Hafler DA (2013) Sodium chloride drives autoimmune disease by the induction of pathogenic TH17 cells. Nature 496(7446):518–522. doi:10.1038/nature11868
Kleinschek MA, Owyang AM, Joyce-Shaikh B, Langrish CL, Chen Y, Gorman DM, Blumenschein WM, McClanahan T, Brombacher F, Hurst SD, Kastelein RA, Cua DJ (2007) IL-25 regulates Th17 function in autoimmune inflammation. J Exp Med 204(1):161–170. doi:10.1084/jem.20061738
Komiyama Y, Nakae S, Matsuki T, Nambu A, Ishigame H, Kakuta S, Sudo K, Iwakura Y (2006) IL-17 plays an important role in the development of experimental autoimmune encephalomyelitis. J Immunol 177(1):566–573
Korn T, Bettelli E, Gao W, Awasthi A, Jager A, Strom TB, Oukka M, Kuchroo VK (2007) IL-21 initiates an alternative pathway to induce proinflammatory T(H)17 cells. Nature 448(7152):484–487. doi:10.1038/nature05970
Korn T, Bettelli E, Oukka M, Kuchroo VK (2009) IL-17 and Th17 Cells. Annu Rev Immunol 27:485–517. doi:10.1146/annurev.immunol.021908.132710
Kotake S, Udagawa N, Takahashi N, Matsuzaki K, Itoh K, Ishiyama S, Saito S, Inoue K, Kamatani N, Gillespie MT, Martin TJ, Suda T (1999) IL-17 in synovial fluids from patients with rheumatoid arthritis is a potent stimulator of osteoclastogenesis. J Clin Invest 103(9):1345–1352. doi:10.1172/JCI5703
Krajewski S, Zapata JM, Krajewska M, VanArsdale T, Shabaik A, Gascoyne RD, Reed JC (1997) Immunohistochemical analysis of in vivo patterns of TRAF-3 expression, a member of the TNF receptor-associated factor family. J Immunol 159(12):5841–5852
Krakowski M, Owens T (1996) Interferon-gamma confers resistance to experimental allergic encephalomyelitis. Eur J Immunol 26(7):1641–1646. doi:10.1002/eji.1830260735
Krawczyk CM, Shen H, Pearce EJ (2007) Functional plasticity in memory T helper cell responses. J Immunol 178(7):4080–4088
Kreymborg K, Etzensperger R, Dumoutier L, Haak S, Rebollo A, Buch T, Heppner FL, Renauld JC, Becher B (2007) IL-22 is expressed by Th17 cells in an IL-23-dependent fashion, but not required for the development of autoimmune encephalomyelitis. J Immunol 179(12):8098–8104
Kryczek I, Banerjee M, Cheng P, Vatan L, Szeliga W, Wei S, Huang E, Finlayson E, Simeone D, Welling TH, Chang A, Coukos G, Liu R, Zou W (2009) Phenotype, distribution, generation, and functional and clinical relevance of Th17 cells in the human tumor environments. Blood 114(6):1141–1149. doi:10.1182/blood-2009-03-208249
Kryczek I, Wei S, Szeliga W, Vatan L, Zou W (2009) Endogenous IL-17 contributes to reduced tumor growth and metastasis. Blood 114(2):357–359. doi:10.1182/blood-2008-09-177360
Kudo M, Melton AC, Chen C, Engler MB, Huang KE, Ren X, Wang Y, Bernstein X, Li JT, Atabai K, Huang X, Sheppard D (2012) IL-17A produced by alphabeta T cells drives airway hyper-responsiveness in mice and enhances mouse and human airway smooth muscle contraction. Nat Med 18(4):547–554. doi:10.1038/nm.2684
Kuestner RE, Taft DW, Haran A, Brandt CS, Brender T, Lum K, Harder B, Okada S, Ostrander CD, Kreindler JL, Aujla SJ, Reardon B, Moore M, Shea P, Schreckhise R, Bukowski TR, Presnell S, Guerra-Lewis P, Parrish-Novak J, Ellsworth JL, Jaspers S, Lewis KE, Appleby M, Kolls JK, Rixon M, West JW, Gao Z, Levin SD (2007) Identification of the IL-17 receptor related molecule IL-17RC as the receptor for IL-17F. J Immunol 179(8):5462–5473
Lajoie S, Lewkowich IP, Suzuki Y, Clark JR, Sproles AA, Dienger K, Budelsky AL, Wills-Karp M (2010) Complement-mediated regulation of the IL-17A axis is a central genetic determinant of the severity of experimental allergic asthma. Nat Immunol 11(10):928–935. doi:10.1038/ni.1926
Lalor SJ, Dungan LS, Sutton CE, Basdeo SA, Fletcher JM, Mills KH (2011) Caspase-1-processed cytokines IL-1beta and IL-18 promote IL-17 production by gammadelta and CD4 T cells that mediate autoimmunity. J Immunol 186(10):5738–5748. doi:10.4049/jimmunol.1003597
Langowski JL, Zhang X, Wu L, Mattson JD, Chen T, Smith K, Basham B, McClanahan T, Kastelein RA, Oft M (2006) IL-23 promotes tumour incidence and growth. Nature 442(7101):461–465. doi:10.1038/nature04808
Langrish CL, Chen Y, Blumenschein WM, Mattson J, Basham B, Sedgwick JD, McClanahan T, Kastelein RA, Cua DJ (2005) IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J Exp Med 201(2):233–240. doi:10.1084/jem.20041257
Laplante M, Sabatini DM (2012) mTOR signaling in growth control and disease. Cell 149(2):274–293. doi:10.1016/j.cell.2012.03.017
Laurence A, Tato CM, Davidson TS, Kanno Y, Chen Z, Yao Z, Blank RB, Meylan F, Siegel R, Hennighausen L, Shevach EM, O’Shea JJ (2007) Interleukin-2 signaling via STAT5 constrains T helper 17 cell generation. Immunity 26(3):371–381. doi:10.1016/j.immuni.2007.02.009
Lazarevic V, Chen X, Shim JH, Hwang ES, Jang E, Bolm AN, Oukka M, Kuchroo VK, Glimcher LH (2011) T-bet represses T(H)17 differentiation by preventing Runx1-mediated activation of the gene encoding RORgammat. Nat Immunol 12(1):96–104. doi:10.1038/ni.1969
Lee K, Gudapati P, Dragovic S, Spencer C, Joyce S, Killeen N, Magnuson MA, Boothby M (2010) Mammalian target of rapamycin protein complex 2 regulates differentiation of Th1 and Th2 cell subsets via distinct signaling pathways. Immunity 32(6):743–753. doi:10.1016/j.immuni.2010.06.002
Lee YK, Turner H, Maynard CL, Oliver JR, Chen D, Elson CO, Weaver CT (2009) Late developmental plasticity in the T helper 17 lineage. Immunity 30(1):92–107. doi:10.1016/j.immuni.2008.11.005
Leonard JP, Waldburger KE, Goldman SJ (1995) Prevention of experimental autoimmune encephalomyelitis by antibodies against interleukin 12. J Exp Med 181(1):381–386
Lexberg MH, Taubner A, Forster A, Albrecht I, Richter A, Kamradt T, Radbruch A, Chang HD (2008) Th memory for interleukin-17 expression is stable in vivo. Eur J Immunol 38(10):2654–2664. doi:10.1002/eji.200838541
Li H, Chen J, Huang A, Stinson J, Heldens S, Foster J, Dowd P, Gurney AL, Wood WI (2000) Cloning and characterization of IL-17B and IL-17C, two new members of the IL-17 cytokine family. Proc Natl Acad Sci USA 97(2):773–778
Li MO, Flavell RA (2008) TGF-beta: a master of all T cell trades. Cell 134(3):392–404. doi:10.1016/j.cell.2008.07.025
Li MO, Sanjabi S, Flavell RA (2006) Transforming growth factor-beta controls development, homeostasis, and tolerance of T cells by regulatory T cell-dependent and -independent mechanisms. Immunity 25(3):455–471. doi:10.1016/j.immuni.2006.07.011
Li MO, Wan YY, Flavell RA (2007) T cell-produced transforming growth factor-beta1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity 26(5):579–591. doi:10.1016/j.immuni.2007.03.014
Li Q, Han Y, Fei G, Guo Z, Ren T, Liu Z (2012) IL-17 promoted metastasis of non-small-cell lung cancer cells. Immunol Lett 148(2):144–150. doi:10.1016/j.imlet.2012.10.011
Liang SC, Tan XY, Luxenberg DP, Karim R, Dunussi-Joannopoulos K, Collins M, Fouser LA (2006) Interleukin (IL)-22 and IL-17 are coexpressed by Th17 cells and cooperatively enhance expression of antimicrobial peptides. J Exp Med 203(10):2271–2279. doi:10.1084/jem.20061308
Lin Y, Ritchea S, Logar A, Slight S, Messmer M, Rangel-Moreno J, Guglani L, Alcorn JF, Strawbridge H, Park SM, Onishi R, Nyugen N, Walter MJ, Pociask D, Randall TD, Gaffen SL, Iwakura Y, Kolls JK, Khader SA (2009) Interleukin-17 is required for T helper 1 cell immunity and host resistance to the intracellular pathogen Francisella tularensis. Immunity 31(5):799–810. doi:10.1016/j.immuni.2009.08.025
Liu C, Qian W, Qian Y, Giltiay NV, Lu Y, Swaidani S, Misra S, Deng L, Chen ZJ, Li X (2009) Act1, a U-box E3 ubiquitin ligase for IL-17 signaling. Science signaling 2(92):ra63. doi:10.1126/scisignal.2000382
Liu C, Swaidani S, Qian W, Kang Z, Sun P, Han Y, Wang C, Gulen MF, Yin W, Zhang C, Fox PL, Aronica M, Hamilton TA, Misra S, Deng J, Li X (2011) A CC’ loop decoy peptide blocks the interaction between Act1 and IL-17RA to attenuate IL-17- and IL-25-induced inflammation. Science signaling 4(197):ra72. doi:10.1126/scisignal.2001843
Liu SM, King C (2013) IL-21-producing Th cells in immunity and autoimmunity. Journal of immunology 191(7):3501–3506. doi:10.4049/jimmunol.1301454
Liu Y, Helms C, Liao W, Zaba LC, Duan S, Gardner J, Wise C, Miner A, Malloy MJ, Pullinger CR, Kane JP, Saccone S, Worthington J, Bruce I, Kwok PY, Menter A, Krueger J, Barton A, Saccone NL, Bowcock AM (2008) A genome-wide association study of psoriasis and psoriatic arthritis identifies new disease loci. PLoS Genet 4(3):e1000041. doi:10.1371/journal.pgen.1000041
Lock C, Hermans G, Pedotti R, Brendolan A, Schadt E, Garren H, Langer-Gould A, Strober S, Cannella B, Allard J, Klonowski P, Austin A, Lad N, Kaminski N, Galli SJ, Oksenberg JR, Raine CS, Heller R, Steinman L (2002) Gene-microarray analysis of multiple sclerosis lesions yields new targets validated in autoimmune encephalomyelitis. Nat Med 8(5):500–508. doi:10.1038/nm0502-500
Lohoff M, Mittrucker HW, Prechtl S, Bischof S, Sommer F, Kock S, Ferrick DA, Duncan GS, Gessner A, Mak TW (2002) Dysregulated T helper cell differentiation in the absence of interferon regulatory factor 4. Proc Natl Acad Sci USA 99(18):11808–11812. doi:10.1073/pnas.182425099
Lubberts E, Joosten LA, van de Loo FA, Schwarzenberger P, Kolls J, van den Berg WB (2002) Overexpression of IL-17 in the knee joint of collagen type II immunized mice promotes collagen arthritis and aggravates joint destruction. Inflam Res off J Eur Histamine Res Soc 51(2):102–104
Lubberts E, Koenders MI, Oppers-Walgreen B, van den Bersselaar L, Coenen-de Roo CJ, Joosten LA, van den Berg WB (2004) Treatment with a neutralizing anti-murine interleukin-17 antibody after the onset of collagen-induced arthritis reduces joint inflammation, cartilage destruction, and bone erosion. Arthritis Rheum 50(2):650–659. doi:10.1002/art.20001
Luthje K, Kallies A, Shimohakamada Y, Belz GT, Light A, Tarlinton DM, Nutt SL (2012) The development and fate of follicular helper T cells defined by an IL-21 reporter mouse. Nat Immunol 13(5):491–498. doi:10.1038/ni.2261
Ma CS, Chew GY, Simpson N, Priyadarshi A, Wong M, Grimbacher B, Fulcher DA, Tangye SG, Cook MC (2008) Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT3. J Exp Med 205(7):1551–1557. doi:10.1084/jem.20080218
Ma F, Xu S, Liu X, Zhang Q, Xu X, Liu M, Hua M, Li N, Yao H, Cao X (2011) The microRNA miR-29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon-gamma. Nat Immunol 12(9):861–869. doi:10.1038/ni.2073
Ma J, Wang R, Fang X, Ding Y, Sun Z (2011) Critical role of TCF-1 in repression of the IL-17 gene. PLoS ONE 6(9):e24768. doi:10.1371/journal.pone.0024768
Ma Y, Thornton S, Boivin GP, Hirsh D, Hirsch R, Hirsch E (1998) Altered susceptibility to collagen-induced arthritis in transgenic mice with aberrant expression of interleukin-1 receptor antagonist. Arthritis Rheum 41(10):1798–1805. doi:10.1002/1529-0131(199810)41:10<1798::AID-ART11>3.0.CO;2-L
Maitra A, Shen F, Hanel W, Mossman K, Tocker J, Swart D, Gaffen SL (2007) Distinct functional motifs within the IL-17 receptor regulate signal transduction and target gene expression. Proc Natl Acad Sci USA 104(18):7506–7511. doi:10.1073/pnas.0611589104
Male V, Nisoli I, Gascoyne DM, Brady HJ (2012) E4BP4: an unexpected player in the immune response. Trends Immunol 33(2):98–102. doi:10.1016/j.it.2011.10.002
Malfait AM, Butler DM, Presky DH, Maini RN, Brennan FM, Feldmann M (1998) Blockade of IL-12 during the induction of collagen-induced arthritis (CIA) markedly attenuates the severity of the arthritis. Clin Exp Immunol 111(2):377–383
Manel N, Unutmaz D, Littman DR (2008) The differentiation of human T(H)-17 cells requires transforming growth factor-beta and induction of the nuclear receptor RORgammat. Nat Immunol 9(6):641–649. doi:10.1038/ni.1610
Mangan PR, Harrington LE, O’Quinn DB, Helms WS, Bullard DC, Elson CO, Hatton RD, Wahl SM, Schoeb TR, Weaver CT (2006) Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 441(7090):231–234. doi:10.1038/nature04754
Marie JC, Letterio JJ, Gavin M, Rudensky AY (2005) TGF-beta1 maintains suppressor function and Foxp3 expression in CD4+ CD25+ regulatory T cells. J Exp Med 201(7):1061–1067. doi:10.1084/jem.20042276
Marijnissen RJ, Koenders MI, Smeets RL, Stappers MH, Nickerson-Nutter C, Joosten LA, Boots AM, van den Berg WB (2011) Increased expression of interleukin-22 by synovial Th17 cells during late stages of murine experimental arthritis is controlled by interleukin-1 and enhances bone degradation. Arthritis Rheum 63(10):2939–2948. doi:10.1002/art.30469
Martin-Orozco N, Muranski P, Chung Y, Yang XO, Yamazaki T, Lu S, Hwu P, Restifo NP, Overwijk WW, Dong C (2009) T helper 17 cells promote cytotoxic T cell activation in tumor immunity. Immunity 31(5):787–798. doi:10.1016/j.immuni.2009.09.014
Mathur AN, Chang HC, Zisoulis DG, Kapur R, Belladonna ML, Kansas GS, Kaplan MH (2006) T-bet is a critical determinant in the instability of the IL-17-secreting T-helper phenotype. Blood 108(5):1595–1601. doi:10.1182/blood-2006-04-015016
Mathur AN, Chang HC, Zisoulis DG, Stritesky GL, Yu Q, O’Malley JT, Kapur R, Levy DE, Kansas GS, Kaplan MH (2007) Stat3 and Stat4 direct development of IL-17-secreting Th cells. J Immunol 178(8):4901–4907
McGeachy MJ, Bak-Jensen KS, Chen Y, Tato CM, Blumenschein W, McClanahan T, Cua DJ (2007) TGF-beta and IL-6 drive the production of IL-17 and IL-10 by T cells and restrain T(H)-17 cell-mediated pathology. Nat Immunol 8(12):1390–1397. doi:10.1038/ni1539
Medvedev A, Chistokhina A, Hirose T, Jetten AM (1997) Genomic structure and chromosomal mapping of the nuclear orphan receptor ROR gamma (RORC) gene. Genomics 46(1):93–102. doi:10.1006/geno.1997.4980
Mellett M, Atzei P, Horgan A, Hams E, Floss T, Wurst W, Fallon PG, Moynagh PN (2012) Orphan receptor IL-17RD tunes IL-17A signalling and is required for neutrophilia. Nat Commun 3:1119. doi:10.1038/ncomms2127
Meng G, Zhang F, Fuss I, Kitani A, Strober W (2009) A mutation in the Nlrp3 gene causing inflammasome hyperactivation potentiates Th17 cell-dominant immune responses. Immunity 30(6):860–874. doi:10.1016/j.immuni.2009.04.012
Merkenschlager M, Wilson CB (2008) RNAi and chromatin in T cell development and function. Curr Opin Immunol 20(2):131–138. doi:10.1016/j.coi.2008.03.013
Milner JD, Brenchley JM, Laurence A, Freeman AF, Hill BJ, Elias KM, Kanno Y, Spalding C, Elloumi HZ, Paulson ML, Davis J, Hsu A, Asher AI, O’Shea J, Holland SM, Paul WE, Douek DC (2008) Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 452(7188):773–776. doi:10.1038/nature06764
Minegishi Y, Saito M, Tsuchiya S, Tsuge I, Takada H, Hara T, Kawamura N, Ariga T, Pasic S, Stojkovic O, Metin A, Karasuyama H (2007) Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature 448(7157):1058–1062. doi:10.1038/nature06096
Mohawk JA, Green CB, Takahashi JS (2012) Central and peripheral circadian clocks in mammals. Annu Rev Neurosci 35:445–462. doi:10.1146/annurev-neuro-060909-153128
Molet S, Hamid Q, Davoine F, Nutku E, Taha R, Page N, Olivenstein R, Elias J, Chakir J (2001) IL-17 is increased in asthmatic airways and induces human bronchial fibroblasts to produce cytokines. J Allergy Clin Immunol 108(3):430–438. doi:10.1067/mai.2001.117929
Mosmann TR, Coffman RL (1989) TH1 and TH2 cells: different patterns of lymphokine secretion lead to different functional properties. Annu Rev Immunol 7:145–173. doi:10.1146/annurev.iy.07.040189.001045
Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, Cheroutre H (2007) Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science 317(5835):256–260. doi:10.1126/science.1145697
Muljo SA, Ansel KM, Kanellopoulou C, Livingston DM, Rao A, Rajewsky K (2005) Aberrant T cell differentiation in the absence of Dicer. J Exp Med 202(2):261–269. doi:10.1084/jem.20050678
Murphy CA, Langrish CL, Chen Y, Blumenschein W, McClanahan T, Kastelein RA, Sedgwick JD, Cua DJ (2003) Divergent pro- and antiinflammatory roles for IL-23 and IL-12 in joint autoimmune inflammation. J Exp Med 198(12):1951–1957. doi:10.1084/jem.20030896
Murphy E, Shibuya K, Hosken N, Openshaw P, Maino V, Davis K, Murphy K, O’Garra A (1996) Reversibility of T helper 1 and 2 populations is lost after long-term stimulation. J Exp Med 183(3):901–913
Murugaiyan G, Beynon V, Mittal A, Joller N, Weiner HL (2011) Silencing microRNA-155 ameliorates experimental autoimmune encephalomyelitis. J Immunol 187(5):2213–2221. doi:10.4049/jimmunol.1003952
Mycko MP, Cichalewska M, Machlanska A, Cwiklinska H, Mariasiewicz M, Selmaj KW (2012) MicroRNA-301a regulation of a T-helper 17 immune response controls autoimmune demyelination. Proc Natl Acad Sci USA 109(20):E1248–1257. doi:10.1073/pnas.1114325109
Nakae S, Komiyama Y, Nambu A, Sudo K, Iwase M, Homma I, Sekikawa K, Asano M, Iwakura Y (2002) Antigen-specific T cell sensitization is impaired in IL-17-deficient mice, causing suppression of allergic cellular and humoral responses. Immunity 17(3):375–387
Nakayama KI, Nakayama K (2006) Ubiquitin ligases: cell-cycle control and cancer. Nat Rev Cancer 6(5):369–381. doi:10.1038/nrc1881
Naoe Y, Setoguchi R, Akiyama K, Muroi S, Kuroda M, Hatam F, Littman DR, Taniuchi I (2007) Repression of interleukin-4 in T helper type 1 cells by Runx/Cbf beta binding to the Il4 silencer. J Exp Med 204(8):1749–1755. doi:10.1084/jem.20062456
Neurath MF, Weigmann B, Finotto S, Glickman J, Nieuwenhuis E, Iijima H, Mizoguchi A, Mizoguchi E, Mudter J, Galle PR, Bhan A, Autschbach F, Sullivan BM, Szabo SJ, Glimcher LH, Blumberg RS (2002) The transcription factor T-bet regulates mucosal T cell activation in experimental colitis and Crohn’s disease. J Exp Med 195(9):1129–1143
Nishimoto N, Yoshizaki K, Miyasaka N, Yamamoto K, Kawai S, Takeuchi T, Hashimoto J, Azuma J, Kishimoto T (2004) Treatment of rheumatoid arthritis with humanized anti-interleukin-6 receptor antibody: a multicenter, double-blind, placebo-controlled trial. Arthritis Rheum 50(6):1761–1769. doi:10.1002/art.20303
Nograles KE, Zaba LC, Shemer A, Fuentes-Duculan J, Cardinale I, Kikuchi T, Ramon M, Bergman R, Krueger JG, Guttman-Yassky E (2009) IL-22-producing “T22” T cells account for upregulated IL-22 in atopic dermatitis despite reduced IL-17-producing TH17 T cells. J Allergy Clin Immunol 123(6):1244–1252, e1242. doi:10.1016/j.jaci.2009.03.041
Novatchkova M, Leibbrandt A, Werzowa J, Neubuser A, Eisenhaber F (2003) The STIR-domain superfamily in signal transduction, development and immunity. Trends Biochem Sci 28(5):226–229. doi:10.1016/S0968-0004(03)00067-7
Nurieva R, Yang XO, Martinez G, Zhang Y, Panopoulos AD, Ma L, Schluns K, Tian Q, Watowich SS, Jetten AM, Dong C (2007) Essential autocrine regulation by IL-21 in the generation of inflammatory T cells. Nature 448(7152):480–483. doi:10.1038/nature05969
Oganesyan G, Saha SK, Guo B, He JQ, Shahangian A, Zarnegar B, Perry A, Cheng G (2006) Critical role of TRAF3 in the Toll-like receptor-dependent and -independent antiviral response. Nature 439(7073):208–211. doi:10.1038/nature04374
Ogawa A, Andoh A, Araki Y, Bamba T, Fujiyama Y (2004) Neutralization of interleukin-17 aggravates dextran sulfate sodium-induced colitis in mice. Clin Immunol 110(1):55–62. doi:10.1016/j.clim.2003.09.013
Okuda Y, Sakoda S, Bernard CC, Fujimura H, Saeki Y, Kishimoto T, Yanagihara T (1998) IL-6-deficient mice are resistant to the induction of experimental autoimmune encephalomyelitis provoked by myelin oligodendrocyte glycoprotein. Int Immunol 10(5):703–708
Oliver JA, Stolberg VR, Chensue SW, King PD (2012) IL-4 acts as a potent stimulator of IFN-gamma expression in CD8 + T cells through STAT6-dependent and independent induction of Eomesodermin and T-bet. Cytokine 57(1):191–199. doi:10.1016/j.cyto.2011.10.006
Onishi RM, Park SJ, Hanel W, Ho AW, Maitra A, Gaffen SL (2010) SEF/IL-17R (SEFIR) is not enough: an extended SEFIR domain is required for il-17RA-mediated signal transduction. J Biol Chem 285(43):32751–32759. doi:10.1074/jbc.M110.121418
Ono M, Yaguchi H, Ohkura N, Kitabayashi I, Nagamura Y, Nomura T, Miyachi Y, Tsukada T, Sakaguchi S (2007) Foxp3 controls regulatory T-cell function by interacting with AML1/Runx1. Nature 446(7136):685–689. doi:10.1038/nature05673
Oppmann B, Lesley R, Blom B, Timans JC, Xu Y, Hunte B, Vega F, Yu N, Wang J, Singh K, Zonin F, Vaisberg E, Churakova T, Liu M, Gorman D, Wagner J, Zurawski S, Liu Y, Abrams JS, Moore KW, Rennick D, de Waal-Malefyt R, Hannum C, Bazan JF, Kastelein RA (2000) Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 13(5):715–725
Ouyang W (2010) Distinct roles of IL-22 in human psoriasis and inflammatory bowel disease. Cytokine Growth Factor Rev 21(6):435–441. doi:10.1016/j.cytogfr.2010.10.007
Owyang AM, Zaph C, Wilson EH, Guild KJ, McClanahan T, Miller HR, Cua DJ, Goldschmidt M, Hunter CA, Kastelein RA, Artis D (2006) Interleukin 25 regulates type 2 cytokine-dependent immunity and limits chronic inflammation in the gastrointestinal tract. J Exp Med 203(4):843–849. doi:10.1084/jem.20051496
Pan G, French D, Mao W, Maruoka M, Risser P, Lee J, Foster J, Aggarwal S, Nicholes K, Guillet S, Schow P, Gurney AL (2001) Forced expression of murine IL-17E induces growth retardation, jaundice, a Th2-biased response, and multiorgan inflammation in mice. J Immunol 167(11):6559–6567
Papp KA, Leonardi C, Menter A, Ortonne JP, Krueger JG, Kricorian G, Aras G, Li J, Russell CB, Thompson EH, Baumgartner S (2012) Brodalumab, an anti-interleukin-17-receptor antibody for psoriasis. N Engl J Med 366(13):1181–1189. doi:10.1056/NEJMoa1109017
Park H, Li Z, Yang XO, Chang SH, Nurieva R, Wang YH, Wang Y, Hood L, Zhu Z, Tian Q, Dong C (2005) A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin 17. Nat Immunol 6(11):1133–1141. doi:10.1038/ni1261
Parrish-Novak J, Dillon SR, Nelson A, Hammond A, Sprecher C, Gross JA, Johnston J, Madden K, Xu W, West J, Schrader S, Burkhead S, Heipel M, Brandt C, Kuijper JL, Kramer J, Conklin D, Presnell SR, Berry J, Shiota F, Bort S, Hambly K, Mudri S, Clegg C, Moore M, Grant FJ, Lofton-Day C, Gilbert T, Rayond F, Ching A, Yao L, Smith D, Webster P, Whitmore T, Maurer M, Kaushansky K, Holly RD, Foster D (2000) Interleukin 21 and its receptor are involved in NK cell expansion and regulation of lymphocyte function. Nature 408(6808):57–63. doi:10.1038/35040504
Pennino D, Bhavsar PK, Effner R, Avitabile S, Venn P, Quaranta M, Marzaioli V, Cifuentes L, Durham SR, Cavani A, Eyerich K, Chung KF, Schmidt-Weber CB, Eyerich S (2013) IL-22 suppresses IFN-gamma-mediated lung inflammation in asthmatic patients. J Allergy Clin Immunol 131(2):562–570. doi:10.1016/j.jaci.2012.09.036
Pickert G, Neufert C, Leppkes M, Zheng Y, Wittkopf N, Warntjen M, Lehr HA, Hirth S, Weigmann B, Wirtz S, Ouyang W, Neurath MF, Becker C (2009) STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J Exp Med 206(7):1465–1472. doi:10.1084/jem.20082683
Pisitkun P, Ha HL, Wang H, Claudio E, Tivy CC, Zhou H, Mayadas TN, Illei GG, Siebenlist U (2012) Interleukin-17 cytokines are critical in development of fatal lupus glomerulonephritis. Immunity 37(6):1104–1115. doi:10.1016/j.immuni.2012.08.014
Qian Y, Liu C, Hartupee J, Altuntas CZ, Gulen MF, Jane-Wit D, Xiao J, Lu Y, Giltiay N, Liu J, Kordula T, Zhang QW, Vallance B, Swaidani S, Aronica M, Tuohy VK, Hamilton T, Li X (2007) The adaptor Act1 is required for interleukin 17-dependent signaling associated with autoimmune and inflammatory disease. Nat Immunol 8(3):247–256. doi:10.1038/ni1439
Qin H, Wang L, Feng T, Elson CO, Niyongere SA, Lee SJ, Reynolds SL, Weaver CT, Roarty K, Serra R, Benveniste EN, Cong Y (2009) TGF-beta promotes Th17 cell development through inhibition of SOCS3. Journal of immunology 183(1):97–105. doi:10.4049/jimmunol.0801986
Qu F, Gao H, Zhu S, Shi P, Zhang Y, Liu Y, Jallal B, Yao Y, Shi Y, Qian Y (2012) TRAF6 dependent Act1 phosphorylation by the IKK-related kinases suppresses IL-17-induced NF-kappaB activation. Mol Cell Biol. doi: 10.1128/MCB.00268-12
Qui HZ, Hagymasi AT, Bandyopadhyay S, St Rose MC, Ramanarasimhaiah R, Menoret A, Mittler RS, Gordon SM, Reiner SL, Vella AT, Adler AJ (2011) CD134 plus CD137 dual costimulation induces Eomesodermin in CD4 T cells to program cytotoxic Th1 differentiation. J Immunol 187(7):3555–3564. doi:10.4049/jimmunol.1101244
Ramirez-Carrozzi V, Sambandam A, Luis E, Lin Z, Jeet S, Lesch J, Hackney J, Kim J, Zhou M, Lai J, Modrusan Z, Sai T, Lee W, Xu M, Caplazi P, Diehl L, de Voss J, Balazs M, Gonzalez L Jr, Singh H, Ouyang W, Pappu R (2011) IL-17C regulates the innate immune function of epithelial cells in an autocrine manner. Nat Immunol 12(12):1159–1166. doi:10.1038/ni.2156
Rengarajan J, Mowen KA, McBride KD, Smith ED, Singh H, Glimcher LH (2002) Interferon regulatory factor 4 (IRF4) interacts with NFATc2 to modulate interleukin 4 gene expression. J Exp Med 195(8):1003–1012
Renner ED, Rylaarsdam S, Anover-Sombke S, Rack AL, Reichenbach J, Carey JC, Zhu Q, Jansson AF, Barboza J, Schimke LF, Leppert MF, Getz MM, Seger RA, Hill HR, Belohradsky BH, Torgerson TR, Ochs HD (2008) Novel signal transducer and activator of transcription 3 (STAT3) mutations, reduced T(H)17 cell numbers, and variably defective STAT3 phosphorylation in hyper-IgE syndrome. J Allergy Clin Immunol 122(1):181–187. doi:10.1016/j.jaci.2008.04.037
Reynolds JM, Martinez GJ, Nallaparaju KC, Chang SH, Wang YH, Dong C (2012) Cutting edge: regulation of intestinal inflammation and barrier function by IL-17C. J Immunol 189(9):4226–4230. doi:10.4049/jimmunol.1103014
Rickel EA, Siegel LA, Yoon BR, Rottman JB, Kugler DG, Swart DA, Anders PM, Tocker JE, Comeau MR, Budelsky AL (2008) Identification of functional roles for both IL-17RB and IL-17RA in mediating IL-25-induced activities. J Immunol 181(6):4299–4310
Rizzo HL, Kagami S, Phillips KG, Kurtz SE, Jacques SL, Blauvelt A (2011) IL-23-mediated psoriasis-like epidermal hyperplasia is dependent on IL-17A. Journal of immunology 186(3):1495–1502. doi:10.4049/jimmunol.1001001
Rodriguez A, Vigorito E, Clare S, Warren MV, Couttet P, Soond DR, van Dongen S, Grocock RJ, Das PP, Miska EA, Vetrie D, Okkenhaug K, Enright AJ, Dougan G, Turner M, Bradley A (2007) Requirement of bic/microRNA-155 for normal immune function. Science 316(5824):608–611. doi:10.1126/science.1139253
Ron D, Fuchs Y, Chorev DS (2008) Know thy Sef: a novel class of feedback antagonists of receptor tyrosine kinase signaling. Int J Biochem Cell Biol 40(10):2040–2052. doi:10.1016/j.biocel.2008.03.013
Rouvier E, Luciani MF, Mattei MG, Denizot F, Golstein P (1993) CTLA-8, cloned from an activated T cell, bearing AU-rich messenger RNA instability sequences, and homologous to a herpesvirus saimiri gene. J Immunol 150(12):5445–5456
Saijo S, Ikeda S, Yamabe K, Kakuta S, Ishigame H, Akitsu A, Fujikado N, Kusaka T, Kubo S, Chung SH, Komatsu R, Miura N, Adachi Y, Ohno N, Shibuya K, Yamamoto N, Kawakami K, Yamasaki S, Saito T, Akira S, Iwakura Y (2010) Dectin-2 recognition of alpha-mannans and induction of Th17 cell differentiation is essential for host defense against Candida albicans. Immunity 32(5):681–691. doi:10.1016/j.immuni.2010.05.001
Saleh A, Shan L, Halayko AJ, Kung S, Gounni AS (2009) Critical role for STAT3 in IL-17A-mediated CCL11 expression in human airway smooth muscle cells. J Immunol 182(6):3357–3365. doi:10.4049/jimmunol.0801882
Samoilova EB, Horton JL, Hilliard B, Liu TS, Chen Y (1998) IL-6-deficient mice are resistant to experimental autoimmune encephalomyelitis: roles of IL-6 in the activation and differentiation of autoreactive T cells. J Immunol 161(12):6480–6486
Sarkar S, Zhou X, Justa S, Bommireddy SR (2013) Interleukin-22 reduces the severity of collagen-induced arthritis in association with increased levels of interleukin-10. Arthritis Rheum 65(4):960–971. doi:10.1002/art.37849
Schmechel S, Konrad A, Diegelmann J, Glas J, Wetzke M, Paschos E, Lohse P, Goke B, Brand S (2008) Linking genetic susceptibility to Crohn’s disease with Th17 cell function: IL-22 serum levels are increased in Crohn’s disease and correlate with disease activity and IL23R genotype status. Inflamm Bowel Dis 14(2):204–212. doi:10.1002/ibd.20315
Schnyder B, Lima C, Schnyder-Candrian S (2010) Interleukin-22 is a negative regulator of the allergic response. Cytokine 50(2):220–227. doi:10.1016/j.cyto.2010.02.003
Schulz SM, Kohler G, Schutze N, Knauer J, Straubinger RK, Chackerian AA, Witte E, Wolk K, Sabat R, Iwakura Y, Holscher C, Muller U, Kastelein RA, Alber G (2008) Protective immunity to systemic infection with attenuated Salmonella enterica serovar enteritidis in the absence of IL-12 is associated with IL-23-dependent IL-22, but not IL-17. Journal of immunology 181(11):7891–7901
Schwandner R, Yamaguchi K, Cao Z (2000) Requirement of tumor necrosis factor receptor-associated factor (TRAF)6 in interleukin 17 signal transduction. J Exp Med 191(7):1233–1240
Seiderer J, Elben I, Diegelmann J, Glas J, Stallhofer J, Tillack C, Pfennig S, Jurgens M, Schmechel S, Konrad A, Goke B, Ochsenkuhn T, Muller-Myhsok B, Lohse P, Brand S (2008) Role of the novel Th17 cytokine IL-17F in inflammatory bowel disease (IBD): upregulated colonic IL-17F expression in active Crohn’s disease and analysis of the IL17F p.His161Arg polymorphism in IBD. Inflamm Bowel Dis 14(4):437–445. doi:10.1002/ibd.20339
Sekikawa A, Fukui H, Suzuki K, Karibe T, Fujii S, Ichikawa K, Tomita S, Imura J, Shiratori K, Chiba T, Fujimori T (2010) Involvement of the IL-22/REG Ialpha axis in ulcerative colitis. Lab Invest J Tech Methods pathol 90(3):496–505. doi:10.1038/labinvest.2009.147
Serada S, Fujimoto M, Mihara M, Koike N, Ohsugi Y, Nomura S, Yoshida H, Nishikawa T, Terabe F, Ohkawara T, Takahashi T, Ripley B, Kimura A, Kishimoto T, Naka T (2008) IL-6 blockade inhibits the induction of myelin antigen-specific Th17 cells and Th1 cells in experimental autoimmune encephalomyelitis. Proc Natl Acad Sci USA 105(26):9041–9046. doi:10.1073/pnas.0802218105
Shaw PJ, Lukens JR, Burns S, Chi H, McGargill MA, Kanneganti TD (2010) Cutting edge: critical role for PYCARD/ASC in the development of experimental autoimmune encephalomyelitis. J Immunol 184(9):4610–4614. doi:10.4049/jimmunol.1000217
Shen F, Li N, Gade P, Kalvakolanu DV, Weibley T, Doble B, Woodgett JR, Wood TD, Gaffen SL (2009) IL-17 receptor signaling inhibits C/EBPbeta by sequential phosphorylation of the regulatory 2 domain. Science signaling 2(59):ra8. doi:10.1126/scisignal.2000066
Shi P, Zhu S, Lin Y, Liu Y, Liu Y, Chen Z, Shi Y, Qian Y (2011) Persistent stimulation with interleukin-17 desensitizes cells through SCFbeta-TrCP-mediated degradation of Act1. Sci Signal 4(197):ra73. doi:10.1126/scisignal.2001653
Smith KM, Guerau-de-Arellano M, Costinean S, Williams JL, Bottoni A, Mavrikis Cox G, Satoskar AR, Croce CM, Racke MK, Lovett-Racke AE, Whitacre CC (2012) miR-29ab1 deficiency identifies a negative feedback loop controlling Th1 bias that is dysregulated in multiple sclerosis. Journal of immunology 189(4):1567–1576. doi:10.4049/jimmunol.1103171
Sonder SU, Saret S, Tang W, Sturdevant DE, Porcella SF, Siebenlist U (2011) IL-17-induced NF-kappaB activation via CIKS/Act1: physiologic significance and signaling mechanisms. J Biol Chem 286(15):12881–12890. doi:10.1074/jbc.M110.199547
Sonderegger I, Kisielow J, Meier R, King C, Kopf M (2008) IL-21 and IL-21R are not required for development of Th17 cells and autoimmunity in vivo. Eur J Immunol 38(7):1833–1838. doi:10.1002/eji.200838511
Song X, Gao H, Lin Y, Yao Y, Zhu S, Wang J, Liu Y, Yao X, Meng G, Shen N, Shi Y, Iwakura Y, Qian Y (2014) Alterations in the microbiota drive interleukin-17C production from intestinal epithelial cells to promote tumorigenesis. Immunity 40(1):140–152. doi:10.1016/j.immuni.2013.11.018
Song X, Qian Y (2013) The activation and regulation of IL-17 receptor mediated signaling. Cytokine 62(2):175–182. doi:10.1016/j.cyto.2013.03.014
Song X, Qian Y (2013) IL-17 family cytokines mediated signaling in the pathogenesis of inflammatory diseases. Cell Signal 25(12):2335–2347. doi:10.1016/j.cellsig.2013.07.021
Song X, Zhu S, Shi P, Liu Y, Shi Y, Levin SD, Qian Y (2011) IL-17RE is the functional receptor for IL-17C and mediates mucosal immunity to infection with intestinal pathogens. Nat Immunol 12(12):1151–1158. doi:10.1038/ni.2155
Staal FJ, Luis TC, Tiemessen MM (2008) WNT signalling in the immune system: WNT is spreading its wings. Nat Rev Immunol 8(8):581–593. doi:10.1038/nri2360
Steiner DF, Thomas MF, Hu JK, Yang Z, Babiarz JE, Allen CD, Matloubian M, Blelloch R, Ansel KM (2011) MicroRNA-29 regulates T-box transcription factors and interferon-gamma production in helper T cells. Immunity 35(2):169–181. doi:10.1016/j.immuni.2011.07.009
Strange A, Capon F, Spencer CC, Knight J, Weale ME, Allen MH, Barton A, Band G, Bellenguez C, Bergboer JG, Blackwell JM, Bramon E, Bumpstead SJ, Casas JP, Cork MJ, Corvin A, Deloukas P, Dilthey A, Duncanson A, Edkins S, Estivill X, Fitzgerald O, Freeman C, Giardina E, Gray E, Hofer A, Huffmeier U, Hunt SE, Irvine AD, Jankowski J, Kirby B, Langford C, Lascorz J, Leman J, Leslie S, Mallbris L, Markus HS, Mathew CG, McLean WH, McManus R, Mossner R, Moutsianas L, Naluai AT, Nestle FO, Novelli G, Onoufriadis A, Palmer CN, Perricone C, Pirinen M, Plomin R, Potter SC, Pujol RM, Rautanen A, Riveira-Munoz E, Ryan AW, Salmhofer W, Samuelsson L, Sawcer SJ, Schalkwijk J, Smith CH, Stahle M, Su Z, Tazi-Ahnini R, Traupe H, Viswanathan AC, Warren RB, Weger W, Wolk K, Wood N, Worthington J, Young HS, Zeeuwen PL, Hayday A, Burden AD, Griffiths CE, Kere J, Reis A, McVean G, Evans DM, Brown MA, Barker JN, Peltonen L, Donnelly P, Trembath RC (2010) A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat Genet 42(11):985–990. doi:10.1038/ng.694
Stromnes IM, Goverman JM (2006) Active induction of experimental allergic encephalomyelitis. Nat Protoc 1(4):1810–1819. doi:10.1038/nprot.2006.285
Stumhofer JS, Laurence A, Wilson EH, Huang E, Tato CM, Johnson LM, Villarino AV, Huang Q, Yoshimura A, Sehy D, Saris CJ, O’Shea JJ, Hennighausen L, Ernst M, Hunter CA (2006) Interleukin 27 negatively regulates the development of interleukin 17-producing T helper cells during chronic inflammation of the central nervous system. Nat Immunol 7(9):937–945. doi:10.1038/ni1376
Sugihara T, Kobori A, Imaeda H, Tsujikawa T, Amagase K, Takeuchi K, Fujiyama Y, Andoh A (2010) The increased mucosal mRNA expressions of complement C3 and interleukin-17 in inflammatory bowel disease. Clin Exp Immunol 160(3):386–393. doi:10.1111/j.1365-2249.2010.04093.x
Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, Blumberg RS, Xavier RJ, Mizoguchi A (2008) IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest 118(2):534–544. doi:10.1172/JCI33194
Sun CM, Hall JA, Blank RB, Bouladoux N, Oukka M, Mora JR, Belkaid Y (2007) Small intestine lamina propria dendritic cells promote de novo generation of Foxp3 T reg cells via retinoic acid. J Exp Med 204(8):1775–1785. doi:10.1084/jem.20070602
Sun D, Novotny M, Bulek K, Liu C, Li X, Hamilton T (2011) Treatment with IL-17 prolongs the half-life of chemokine CXCL1 mRNA via the adaptor TRAF5 and the splicing-regulatory factor SF2 (ASF). Nat Immunol 12(9):853–860. doi:10.1038/ni.2081
Suto A, Kashiwakuma D, Kagami S, Hirose K, Watanabe N, Yokote K, Saito Y, Nakayama T, Grusby MJ, Iwamoto I, Nakajima H (2008) Development and characterization of IL-21-producing CD4 + T cells. J Exp Med 205(6):1369–1379. doi:10.1084/jem.20072057
Sutton C, Brereton C, Keogh B, Mills KH, Lavelle EC (2006) A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J Exp Med 203(7):1685–1691. doi:10.1084/jem.20060285
Swaidani S, Bulek K, Kang Z, Liu C, Lu Y, Yin W, Aronica M, Li X (2009) The critical role of epithelial-derived Act1 in IL-17- and IL-25-mediated pulmonary inflammation. J Immunol 182(3):1631–1640
Szabo SJ, Dighe AS, Gubler U, Murphy KM (1997) Regulation of the interleukin (IL)-12R beta 2 subunit expression in developing T helper 1 (Th1) and Th2 cells. J Exp Med 185(5):817–824
Takahashi K, Hirose K, Kawashima S, Niwa Y, Wakashin H, Iwata A, Tokoyoda K, Renauld JC, Iwamoto I, Nakayama T, Nakajima H (2011) IL-22 attenuates IL-25 production by lung epithelial cells and inhibits antigen-induced eosinophilic airway inflammation. The Journal of allergy and clinical immunology 128(5):1067–1076, e1061–1066. doi:10.1016/j.jaci.2011.06.018
Tamachi T, Maezawa Y, Ikeda K, Kagami S, Hatano M, Seto Y, Suto A, Suzuki K, Watanabe N, Saito Y, Tokuhisa T, Iwamoto I, Nakajima H (2006) IL-25 enhances allergic airway inflammation by amplifying a TH2 cell-dependent pathway in mice. J Allergy Clin Immunol 118(3):606–614. doi:10.1016/j.jaci.2006.04.051
Teunissen MB, Koomen CW, de Malefyt Waal R, Wierenga EA, Bos JD (1998) Interleukin-17 and interferon-gamma synergize in the enhancement of proinflammatory cytokine production by human keratinocytes. J Invest Dermatol 111(4):645–649. doi:10.1046/j.1523-1747.1998.00347.x
Thai TH, Calado DP, Casola S, Ansel KM, Xiao C, Xue Y, Murphy A, Frendewey D, Valenzuela D, Kutok JL, Schmidt-Supprian M, Rajewsky N, Yancopoulos G, Rao A, Rajewsky K (2007) Regulation of the germinal center response by microRNA-155. Science 316(5824):604–608. doi:10.1126/science.1141229
Tong Z, Yang XO, Yan H, Liu W, Niu X, Shi Y, Fang W, Xiong B, Wan Y, Dong C (2012) A protective role by interleukin-17F in colon tumorigenesis. PLoS ONE 7(4):e34959. doi:10.1371/journal.pone.0034959
Tosolini M, Kirilovsky A, Mlecnik B, Fredriksen T, Mauger S, Bindea G, Berger A, Bruneval P, Fridman WH, Pages F, Galon J (2011) Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res 71(4):1263–1271. doi:10.1158/0008-5472.CAN-10-2907
Toy D, Kugler D, Wolfson M, Vanden Bos T, Gurgel J, Derry J, Tocker J, Peschon J (2006) Cutting edge: interleukin 17 signals through a heteromeric receptor complex. J Immunol 177(1):36–39
Tran EH, Prince EN, Owens T (2000) IFN-gamma shapes immune invasion of the central nervous system via regulation of chemokines. J Immunol 164(5):2759–2768
Tschopp J, Martinon F, Burns K (2003) NALPs: a novel protein family involved in inflammation. Nat Rev Mol Cell Biol 4(2):95–104. doi:10.1038/nrm1019
Tseng PH, Matsuzawa A, Zhang W, Mino T, Vignali DA, Karin M (2009) Different modes of ubiquitination of the adaptor TRAF3 selectively activate the expression of type I interferons and proinflammatory cytokines. Nat Immunol 11(1):70–75. doi:10.1038/ni.1819
Tzartos JS, Friese MA, Craner MJ, Palace J, Newcombe J, Esiri MM, Fugger L (2008) Interleukin-17 production in central nervous system-infiltrating T cells and glial cells is associated with active disease in multiple sclerosis. Am J Pathol 172(1):146–155. doi:10.2353/ajpath.2008.070690
Van Belle AB, de Heusch M, Lemaire MM, Hendrickx E, Warnier G, Dunussi-Joannopoulos K, Fouser LA, Renauld JC, Dumoutier L (2012) IL-22 is required for imiquimod-induced psoriasiform skin inflammation in mice. J Immunol 188(1):462–469. doi:10.4049/jimmunol.1102224
Van Snick J (1990) Interleukin-6: an overview. Annu Rev Immunol 8:253–278. doi:10.1146/annurev.iy.08.040190.001345
Veldhoen M, Hirota K, Westendorf AM, Buer J, Dumoutier L, Renauld JC, Stockinger B (2008) The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 453(7191):106–109. doi:10.1038/nature06881
Veldhoen M, Hocking RJ, Atkins CJ, Locksley RM, Stockinger B (2006) TGFbeta in the context of an inflammatory cytokine milieu supports de novo differentiation of IL-17-producing T cells. Immunity 24(2):179–189. doi:10.1016/j.immuni.2006.01.001
Veldhoen M, Hocking RJ, Flavell RA, Stockinger B (2006) Signals mediated by transforming growth factor-beta initiate autoimmune encephalomyelitis, but chronic inflammation is needed to sustain disease. Nat Immunol 7(11):1151–1156. doi:10.1038/ni1391
Volpe E, Servant N, Zollinger R, Bogiatzi SI, Hupe P, Barillot E, Soumelis V (2008) A critical function for transforming growth factor-beta, interleukin 23 and proinflammatory cytokines in driving and modulating human T(H)-17 responses. Nat Immunol 9(6):650–657. doi:10.1038/ni.1613
Voo KS, Wang YH, Santori FR, Boggiano C, Wang YH, Arima K, Bover L, Hanabuchi S, Khalili J, Marinova E, Zheng B, Littman DR, Liu YJ (2009) Identification of IL-17-producing FOXP3 + regulatory T cells in humans. Proc Natl Acad Sci USA 106(12):4793–4798. doi:10.1073/pnas.0900408106
Wang C, Wu L, Bulek K, Martin BN, Zepp JA, Kang Z, Liu C, Herjan T, Misra S, Carman JA, Gao J, Dongre A, Han S, Bunting KD, Ko JS, Xiao H, Kuchroo VK, Ouyang W, Li X (2013) The psoriasis-associated D10 N variant of the adaptor Act1 with impaired regulation by the molecular chaperone hsp90. Nat Immunol 14(1):72–81. doi:10.1038/ni.2479
Wang K, Zhang H, Kugathasan S, Annese V, Bradfield JP, Russell RK, Sleiman PM, Imielinski M, Glessner J, Hou C, Wilson DC, Walters T, Kim C, Frackelton EC, Lionetti P, Barabino A, Van Limbergen J, Guthery S, Denson L, Piccoli D, Li M, Dubinsky M, Silverberg M, Griffiths A, Grant SF, Satsangi J, Baldassano R, Hakonarson H (2009) Diverse genome-wide association studies associate the IL12/IL23 pathway with Crohn disease. Am J Hum Genet 84(3):399–405. doi:10.1016/j.ajhg.2009.01.026
Wang L, Yi T, Kortylewski M, Pardoll DM, Zeng D, Yu H (2009) IL-17 can promote tumor growth through an IL-6-Stat3 signaling pathway. J Exp Med 206(7):1457–1464. doi:10.1084/jem.20090207
Wang L, Yi T, Zhang W, Pardoll DM, Yu H (2010) IL-17 enhances tumor development in carcinogen-induced skin cancer. Cancer Res 70(24):10112–10120. doi:10.1158/0008-5472.CAN-10-0775
Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, Cui K, Kanno Y, Roh TY, Watford WT, Schones DE, Peng W, Sun HW, Paul WE, O’Shea JJ, Zhao K (2009) Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4 + T cells. Immunity 30(1):155–167. doi:10.1016/j.immuni.2008.12.009
Wilson CB, Rowell E, Sekimata M (2009) Epigenetic control of T-helper-cell differentiation. Nat Rev Immunol 9(2):91–105. doi:10.1038/nri2487
Wilson NJ, Boniface K, Chan JR, McKenzie BS, Blumenschein WM, Mattson JD, Basham B, Smith K, Chen T, Morel F, Lecron JC, Kastelein RA, Cua DJ, McClanahan TK, Bowman EP, de Waal Malefyt R (2007) Development, cytokine profile and function of human interleukin 17-producing helper T cells. Nat Immunol 8(9):950–957. doi:10.1038/ni1497
Wolk K, Haugen HS, Xu W, Witte E, Waggie K, Anderson M, Vom Baur E, Witte K, Warszawska K, Philipp S, Johnson-Leger C, Volk HD, Sterry W, Sabat R (2009) IL-22 and IL-20 are key mediators of the epidermal alterations in psoriasis while IL-17 and IFN-gamma are not. J Mol Med 87(5):523–536. doi:10.1007/s00109-009-0457-0
Wolk K, Kunz S, Witte E, Friedrich M, Asadullah K, Sabat R (2004) IL-22 increases the innate immunity of tissues. Immunity 21(2):241–254. doi:10.1016/j.immuni.2004.07.007
Wolk K, Witte E, Wallace E, Docke WD, Kunz S, Asadullah K, Volk HD, Sterry W, Sabat R (2006) IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: a potential role in psoriasis. Eur J Immunol 36(5):1309–1323. doi:10.1002/eji.200535503
Wright JF, Bennett F, Li B, Brooks J, Luxenberg DP, Whitters MJ, Tomkinson KN, Fitz LJ, Wolfman NM, Collins M, Dunussi-Joannopoulos K, Chatterjee-Kishore M, Carreno BM (2008) The human IL-17F/IL-17A heterodimeric cytokine signals through the IL-17RA/IL-17RC receptor complex. Journal of immunology 181(4):2799–2805
Wu C, Yosef N, Thalhamer T, Zhu C, Xiao S, Kishi Y, Regev A, Kuchroo VK (2013) Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK1. Nature 496(7446):513–517. doi:10.1038/nature11984
Wu S, Rhee KJ, Albesiano E, Rabizadeh S, Wu X, Yen HR, Huso DL, Brancati FL, Wick E, McAllister F, Housseau F, Pardoll DM, Sears CL (2009) A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat Med 15(9):1016–1022. doi:10.1038/nm.2015
Xiao S, Jin H, Korn T, Liu SM, Oukka M, Lim B, Kuchroo VK (2008) Retinoic acid increases Foxp3 + regulatory T cells and inhibits development of Th17 cells by enhancing TGF-beta-driven Smad3 signaling and inhibiting IL-6 and IL-23 receptor expression. J Immunol 181(4):2277–2284
Xie P, Stunz LL, Larison KD, Yang B, Bishop GA (2007) Tumor necrosis factor receptor-associated factor 3 is a critical regulator of B cell homeostasis in secondary lymphoid organs. Immunity 27(2):253–267. doi:10.1016/j.immuni.2007.07.012
Xu L, Kitani A, Fuss I, Strober W (2007) Cutting edge: regulatory T cells induce CD4 + CD25-Foxp3- T cells or are self-induced to become Th17 cells in the absence of exogenous TGF-beta. J immunol 178(11):6725–6729
Xu X, Weiss ID, HZ H, Singh SP, Wynn TA, Wilson MS, Farber JM (2014) Conventional NK Cells Can Produce IL-22 and Promote Host Defense in Klebsiella pneumoniae Pneumonia. J Immunol 192(4):1778–1786. doi:10.4049/jimmunol.1300039
Yamaguchi Y, Fujio K, Shoda H, Okamoto A, Tsuno NH, Takahashi K, Yamamoto K (2007) IL-17B and IL-17C are associated with TNF-alpha production and contribute to the exacerbation of inflammatory arthritis. J Immunol 179(10):7128–7136
Yang L, Anderson DE, Baecher-Allan C, Hastings WD, Bettelli E, Oukka M, Kuchroo VK, Hafler DA (2008) IL-21 and TGF-beta are required for differentiation of human T(H)17 cells. Nature 454(7202):350–352. doi:10.1038/nature07021
Yang XO, Chang SH, Park H, Nurieva R, Shah B, Acero L, Wang YH, Schluns KS, Broaddus RR, Zhu Z, Dong C (2008) Regulation of inflammatory responses by IL-17F. J Exp Med 205(5):1063–1075. doi:10.1084/jem.20071978
Yang XO, Nurieva R, Martinez GJ, Kang HS, Chung Y, Pappu BP, Shah B, Chang SH, Schluns KS, Watowich SS, Feng XH, Jetten AM, Dong C (2008) Molecular antagonism and plasticity of regulatory and inflammatory T cell programs. Immunity 29(1):44–56. doi:10.1016/j.immuni.2008.05.007
Yang XO, Panopoulos AD, Nurieva R, Chang SH, Wang D, Watowich SS, Dong C (2007) STAT3 regulates cytokine-mediated generation of inflammatory helper T cells. J Biol Chem 282(13):9358–9363. doi:10.1074/jbc.C600321200
Yang XO, Pappu BP, Nurieva R, Akimzhanov A, Kang HS, Chung Y, Ma L, Shah B, Panopoulos AD, Schluns KS, Watowich SS, Tian Q, Jetten AM, Dong C (2008) T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity 28(1):29–39. doi:10.1016/j.immuni.2007.11.016
Yang XP, Ghoreschi K, Steward-Tharp SM, Rodriguez-Canales J, Zhu J, Grainger JR, Hirahara K, Sun HW, Wei L, Vahedi G, Kanno Y, O’Shea JJ, Laurence A (2011) Opposing regulation of the locus encoding IL-17 through direct, reciprocal actions of STAT3 and STAT5. Nat Immunol 12(3):247–254. doi:10.1038/ni.1995
Yang Y, Xu J, Niu Y, Bromberg JS, Ding Y (2008) T-bet and eomesodermin play critical roles in directing T cell differentiation to Th1 versus Th17. Journal of immunology 181(12):8700–8710
Yao R, Ma YL, Liang W, Li HH, Ma ZJ, Yu X, Liao YH (2012) MicroRNA-155 modulates Treg and Th17 cells differentiation and Th17 cell function by targeting SOCS1. PLoS ONE 7(10):e46082. doi:10.1371/journal.pone.0046082
Yao Z, Fanslow WC, Seldin MF, Rousseau AM, Painter SL, Comeau MR, Cohen JI, Spriggs MK (1995) Herpesvirus saimiri encodes a new cytokine, IL-17, which binds to a novel cytokine receptor. Immunity 3(6):811–821
Yu Q, Sharma A, Ghosh A, Sen JM (2011) T cell factor-1 negatively regulates expression of IL-17 family of cytokines and protects mice from experimental autoimmune encephalomyelitis. Journal of immunology 186(7):3946–3952. doi:10.4049/jimmunol.1003497
Yu X, Rollins D, Ruhn KA, Stubblefield JJ, Green CB, Kashiwada M, Rothman PB, Takahashi JS, Hooper LV (2013) TH17 cell differentiation is regulated by the circadian clock. Science 342(6159):727–730. doi:10.1126/science.1243884
Zaph C, Du Y, Saenz SA, Nair MG, Perrigoue JG, Taylor BC, Troy AE, Kobuley DE, Kastelein RA, Cua DJ, Yu Y, Artis D (2008) Commensal-dependent expression of IL-25 regulates the IL-23-IL-17 axis in the intestine. J Exp Med 205(10):2191–2198. doi:10.1084/jem.20080720
Zenewicz LA, Yancopoulos GD, Valenzuela DM, Murphy AJ, Stevens S, Flavell RA (2008) Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity 29(6):947–957. doi:10.1016/j.immuni.2008.11.003
Zepp JA, Liu C, Qian W, Wu L, Gulen MF, Kang Z, Li X (2012) Cutting Edge: TNF Receptor-Associated Factor 4 Restricts IL-17-Mediated Pathology and Signaling Processes. J Immunol 189(1):33–37. doi:10.4049/jimmunol.1200470
Zhang F, Meng G, Strober W (2008) Interactions among the transcription factors Runx1, RORgammat and Foxp3 regulate the differentiation of interleukin 17-producing T cells. Nat Immunol 9(11):1297–1306. doi:10.1038/ni.1663
Zhang GX, Gran B, Yu S, Li J, Siglienti I, Chen X, Kamoun M, Rostami A (2003) Induction of experimental autoimmune encephalomyelitis in IL-12 receptor-beta 2-deficient mice: IL-12 responsiveness is not required in the pathogenesis of inflammatory demyelination in the central nervous system. J Immunol 170(4):2153–2160
Zhang JP, Yan J, Xu J, Pang XH, Chen MS, Li L, Wu C, Li SP, Zheng L (2009) Increased intratumoral IL-17-producing cells correlate with poor survival in hepatocellular carcinoma patients. J Hepatol 50(5):980–989. doi:10.1016/j.jhep.2008.12.033
Zhang Y, Wang X, Zhong M, Zhang M, Suo Q, Lv K (2013) MicroRNA let-7a ameliorates con A-induced hepatitis by inhibiting IL-6-dependent Th17 cell differentiation. J Clin Immunol 33(3):630–639. doi:10.1007/s10875-012-9840-7
Zhang Z, Kyttaris VC, Tsokos GC (2009) The role of IL-23/IL-17 axis in lupus nephritis. J Immunol 183(5):3160–3169. doi:10.4049/jimmunol.0900385
Zhao A, Urban JF Jr, Sun R, Stiltz J, Morimoto M, Notari L, Madden KB, Yang Z, Grinchuk V, Ramalingam TR, Wynn TA, Shea-Donohue T (2010) Critical role of IL-25 in nematode infection-induced alterations in intestinal function. Journal of immunology 185(11):6921–6929. doi:10.4049/jimmunol.1000450
Zhao C, Dahlman-Wright K (2010) Liver X receptor in cholesterol metabolism. J Endocrinol 204(3):233–240. doi:10.1677/JOE-09-0271
Zheng Y, Danilenko DM, Valdez P, Kasman I, Eastham-Anderson J, Wu J, Ouyang W (2007) Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature 445(7128):648–651. doi:10.1038/nature05505
Zheng Y, Valdez PA, Danilenko DM, Hu Y, Sa SM, Gong Q, Abbas AR, Modrusan Z, Ghilardi N, de Sauvage FJ, Ouyang W (2008) Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat Med 14(3):282–289. doi:10.1038/nm1720
Zhong B, Liu X, Wang X, Chang SH, Wang A, Reynolds JM, Dong C (2012) Negative regulation of IL-17-mediated signaling and inflammation by the ubiquitin-specific protease USP25. Nat Immunol 13(11):1110–1117. doi:10.1038/ni.2427
Zhou L, Ivanov II, Spolski R, Min R, Shenderov K, Egawa T, Levy DE, Leonard WJ, Littman DR (2007) IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat Immunol 8(9):967–974. doi:10.1038/ni1488
Zhou L, Lopes JE, Chong MM, Ivanov II, Min R, Victora GD, Shen Y, Du J, Rubtsov YP, Rudensky AY, Ziegler SF, Littman DR (2008) TGF-beta-induced Foxp3 inhibits T(H)17 cell differentiation by antagonizing RORgammat function. Nature 453(7192):236–240. doi:10.1038/nature06878
Zhou X, Yu S, Zhao DM, Harty JT, Badovinac VP, Xue HH (2010) Differentiation and persistence of memory CD8(+) T cells depend on T cell factor 1. Immunity 33(2):229–240. doi:10.1016/j.immuni.2010.08.002
Zhou Y, Toh ML, Zrioual S, Miossec P (2007) IL-17A versus IL-17F induced intracellular signal transduction pathways and modulation by IL-17RA and IL-17RC RNA interference in AGS gastric adenocarcinoma cells. Cytokine 38(3):157–164. doi:10.1016/j.cyto.2007.06.002
Zhu J, Davidson TS, Wei G, Jankovic D, Cui K, Schones DE, Guo L, Zhao K, Shevach EM, Paul WE (2009) Down-regulation of Gfi-1 expression by TGF-beta is important for differentiation of Th17 and CD103 + inducible regulatory T cells. J Exp Med 206(2):329–341. doi:10.1084/jem.20081666
Zhu J, Guo L, Min B, Watson CJ, Hu-Li J, Young HA, Tsichlis PN, Paul WE (2002) Growth factor independent-1 induced by IL-4 regulates Th2 cell proliferation. Immunity 16(5):733–744
Zhu S, Pan W, Qian Y (2013) MicroRNA in immunity and autoimmunity. J Mol Med 91(9):1039–1050. doi:10.1007/s00109-013-1043-z
Zhu S, Pan W, Shi P, Gao H, Zhao F, Song X, Liu Y, Zhao L, Li X, Shi Y, Qian Y (2010) Modulation of experimental autoimmune encephalomyelitis through TRAF3-mediated suppression of interleukin 17 receptor signaling. J Exp Med 207(12):2647–2662. doi:10.1084/jem.20100703
Zhu S, Pan W, Song X, Liu Y, Shao X, Tang Y, Liang D, He D, Wang H, Liu W, Shi Y, Harley JB, Shen N, Qian Y (2012) The microRNA miR-23b suppresses IL-17-associated autoimmune inflammation by targeting TAB2, TAB3 and IKK-alpha. Nat Med 18(7):1077–1086. doi:10.1038/nm.2815
Zhu S, Qian Y (2012) IL-17/IL-17 receptor system in autoimmune disease: mechanisms and therapeutic potential. Clin Sci 122(11):487–511. doi:10.1042/CS20110496
Ziolkowska M, Koc A, Luszczykiewicz G, Ksiezopolska-Pietrzak K, Klimczak E, Chwalinska-Sadowska H, Maslinski W (2000) High levels of IL-17 in rheumatoid arthritis patients: IL-15 triggers in vitro IL-17 production via cyclosporin A-sensitive mechanism. J immunol 164(5):2832–2838
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2014 Springer Science+Business Media Dordrecht
About this chapter
Cite this chapter
Song, X., Gao, H., Qian, Y. (2014). Th17 Differentiation and Their Pro-inflammation Function. In: Sun, B. (eds) T Helper Cell Differentiation and Their Function. Advances in Experimental Medicine and Biology, vol 841. Springer, Dordrecht. https://doi.org/10.1007/978-94-017-9487-9_5
Download citation
DOI: https://doi.org/10.1007/978-94-017-9487-9_5
Published:
Publisher Name: Springer, Dordrecht
Print ISBN: 978-94-017-9486-2
Online ISBN: 978-94-017-9487-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)