
Abstract
Scutellaria viscidula Bunge (Labiatae), a perennial herb, is an important medicinal plant that possesses broad pharmacological actions. S. viscidula contains flavonoids with good bioactivities (e.g., baicalin, wogonoside, baicalein, and wogonin) mainly in its dry root, which is used as alternative to Scutellaria baicalensis in the north of China. Furthermore, S. viscidula also has flavones with interesting diverged structures such as panicolin, viscidulin I, viscidulin II, and viscidulin III. Tracing the dynamic process of gene expression will help reveal the mechanism of flavonoid synthesis in S. viscidula, as well as the 4′-deoxyflavone biosynthesis in S. baicalensis. One way is to generate and analyze the expressed sequence tags (ESTs). However, little is known on the transcriptome information of S. viscidula, particularly the key genes involved in flavonoid biosynthesis. In this study, we conducted de novo transcriptome analysis of S. viscidula and obtained 42,310,834 reads and 40,052 unigenes, respectively. We revealed 177 genes relating to flavonoid biosynthesis, where 23 key enzyme-encoding genes including CHS, CHI, F3H, PAL, and 4CL were annotated. Furthermore, we investigated the dynamic expression of SvCHS, SvCHI, SvF3H, SvMYB2, and SvbHLH of stem, root, and leaf of S. viscidula in May, July, and September. Our results showed that these key genes had important regulatory function and exhibited positive correlation with total flavonoid content in different growth stages of S. viscidula. Collectively, this study provides high-quality transcriptome data of S. viscidula, and further gives significant information for understanding the molecular mechanism of gene expression and active ingredients in Scutellaria plants.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
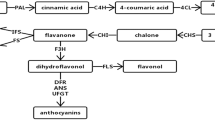
Labiatae is a relatively large family, with an estimated 220 genera and approximately 2200 species (98 genera and 808 species in China), which includes several economically important medicinal plants distributing worldwide, such as Mentha haplocalyx Briq., Salvia miltiorrhiza Bge., Scutellaria baicalensis Georgi., and Scutellaria viscidula Bunge., which is a perennial herb that contains flavonoids (baicalin, baicalein, etc.) that possess effective medicinal functions of inhibiting human immune-deficiency virus (HIV-1), human T-cell leukemia virus (HTLV-1), and cancer cells. S. viscidula has active ingredients including flavonones, flavones, and flavonols, which are also present in S. baicalensis that is commonly used in traditional Chinese medicine (Guo et al. 2016). The dry root of S. viscidula has been used as herbal medicines in north and northeast of China for more than 2000 years. With the decline of wild resource of S. baicalensis and increased demand for baicalin, other Scutellaria plants (e.g., S. viscidula, Scutellaria amoena) are becoming alternatives to extract flavonoids in pharmaceutical industry (Guo et al. 2016; Liu 2016). Flavonoids are the secondary metabolites that have a common basic structure (C6–C3–C6) with one or more hydroxyl substituent. In general, the biosynthesis of flavonoids is regulated by the upstream phenylpropanoid pathway (Vrancken et al. 2013), where the key enzymes synthesizing universal precursors are phenylalanine ammonia lyase (PAL), cinnamate-4-hydroxylase (C4H), and 4-coumarate CoA ligase (4CL) (Fig. 1). To understand the biosynthesis of flavonoids, we characterized the key genes involved in this synthesis pathway in model plants, such as chalcone synthase (CHS), chalcone isomerase (CHI), flavanone-3-hydroxylase (F3H), flavonol synthase (FLS), dihydroflavonal-4-reductase (DFR), and flavone synthase (FNS). However, only 8 DNA and RNA sequences, 6 proteins, 0 EST, and 0 gene could be found in the national center for biotechnology information (NCBI) GenBank database (as of October 1, 2016). The key genes involved in flavonoid biosynthesis of S. viscidula remain unknown, which may further limit gene mining, molecular markers and genetic engineering breeding of S. viscidula.

Flavonoid biosynthesis pathways in plants
The high-throughput next generation RNA sequencing (RNA-seq) technology offers rapid genome-wide transcriptomic studies and is widely used to define gene structure and expression profiles in model organisms. The assembly of de novo transcriptome based on short reads generated from RNA-seq method allows gene discovery in organisms that were not previously studied. High-quality transcriptome data are not only powerful in gene mining and cloning, but also provide useful information for setting molecular breeding strategy in medicinal plants. In addition, RNA-seq can illustrate the identification of novel genes and simple sequence repeat (SSR) molecular markers. For example, Zhao et al. (2017) performed de novo sequencing and identified a number of putative genes involved in flavonoid biosynthesis in Toona sinensis, which is one of the traditional Chinese medicinal plants (Zhao et al. 2017). For Scutellaria plants, Liu et al. (2015) conducted a deep sequencing of S. baicalensis transcriptome and identified four candidate 6-hydroxylase genes for the formation of baicalin (Liu et al. 2015). However, it remains unknown whether interspecific differences exist in the biosynthesis of flavonoids. Moreover, it is intriguing that the key genes involved in flavonoids synthesis have temporal specificity (Ferreyra et al. 2012). Recently, Zhao et al. demonstrated that different synthesis pathways of flavones exist between the aerial parts and the roots of S. baicalensis (Zhao et al. 2016). They found root-specific isoforms of flavone synthase II (FNSII), coenzyme A ligase (SbCLL-7), and chalcone synthase (SbCHS-2) involving in the synthesis of 4′-deoxyflavones in the roots of S. baicalensis. Two cytochromes P450 (CYP450) enzymes which 6- and 8-hydroxylate chrysin to form the 4′-deoxyflavone bioactives in roots of S. baicalensis are also been reported (Zhao et al. 2018). Given the importance of flavonoids in medicine, further investigations are needed to reveal the synthesis pathways in other Scutellaria species. For this purpose, it is necessary to investigate how flavonoid biosynthesis was dynamically regulated in different parts of S. viscidula at different growth stages using transcriptomic approaches.
The previous studies, focusing mainly on collecting germplasms and characterizing active components, have successfully isolated flavonoids and confirmed their structures and medicinal functions (Shang et al. 2010). However, the extraction of flavonoids could not be fully completed because of the inefficient extracting processes, which leads to excessive harvesting and exploitation of medicinal plants in Scutellaria. Therefore, being able to produce stable flavonoids in large quantities is considered to be a promising strategy to meet the high market demand of active ingredients. Here, we characterize the transcriptome of S. viscidula using next generation sequencing (NGS) technology based on Illumina® HiSeq 2000 platform. We would like to answer that: (a) generate high-quality transcripts and unigenes of S. viscidula; (b) identify candidate genes encoding key enzymes involved in flavonoid biosynthesis pathways; (c) produce the primer pairs of SSRs and the transcription factors; and (d) analyze dynamic expression of key genes and their correlation with total flavonoids in different stages. We showed an overview of transcriptome data, where we discovered many candidate genes encoding key enzymes involved in the flavonoid biosynthesis of S. viscidula. Furthermore, we identified several differentially expressed enzyme-encoding genes at spatial and temporal levels and proposed their roles in the regulation process.
Materials and methods
Plant materials
We collected fresh S. viscidula including their roots, stems, and leaves from the Germplasm Resource Garden for Medicinal Plants in Shaanxi Normal University in May, July and September in 2015. The taxonomic identities of voucher specimens were further confirmed by a medicinal botanist to ensure accurate identifications. Part of roots, stems, and leaves collected from five fresh S. viscidula of each period were dried to a constant weight at 60 °C to measure flavonoid content using Lambda 950 spectrophotometer (PerkinElmer, Hopkinton, USA). For analyzing the dynamic expression of genes, the remaining part of the five fresh S. viscidula of each period were used in quantitative real-time PCR (qRT-PCR). For transcriptome analysis, three fresh plants were collected in July, since they contained the highest content of total flavonoids (data shown below) and were cut into small pieces with sufficient mixing, and then immediately frozen in liquid nitrogen and stored at − 80 °C for further experiment.
RNA extraction
Total RNA were isolated from the root of S. viscidula using Total RNA Reagent (Wolact, Hongkong, China) according to the production instructions. Three replicates were sampled and their roots were mixed for RNA preparation. RNA samples were treated with RNase-free DNase (TaKaRa, Dalian, China) to eliminate genomic DNA contamination. Degradation of RNA was monitored on 1% agarose gels. Purity of RNA was checked using NanoPhotometer spectrophotometer (Implen, CA, USA). The concentration of RNA was measured using Qubit RNA Assay Kit in Qubit 2.0 Fluorometer (Life Technologies, CA, USA). The integrity of RNA was assessed using the RNA Nano 6000 Assay Kit of the Agilent Bioanalyzer 2100 system (Agilent Technologies, CA, USA).
cDNA library construction and sequencing
To construct cDNA library, we first used poly-T oligo-linked magnetic beads to isolate mRNAs with poly-A tail. Illumina® proprietary fragmentation buffer (Illumina, San Diego, USA) was then added to cut mRNAs into short fragments (Chen et al. 2014). Using random oligonucleotides, we synthesized the first strand of cDNA. The subsequent cDNA synthesis was performed using Illumina® TruSeq RNA Sample Preparation Kit (Illumina, San Diego, USA) according to manufacturer’s instructions. To select cDNA fragments of preferentially 150–200 bp in length, we purified the library fragments with AMPure XP system (Beckman Coulter, Beverly, USA). The clustering of the index-coded samples was performed on a cBot Cluster Generation System using TruSeq PE Cluster Kit v3-cBot-HS (Illumina, San Diego, USA) according to the manufacturer’s instructions. After cluster generation, the library preparations were sequenced on Illumina® Hiseq 2000 platform (Novogene, Beijing, China) and paired-end reads were generated.
Quality control and transcriptome assembly
Raw reads of fastq format were first processed through inhouse perl scripts. In this step, clean reads were obtained by removing sequencing adapters, poly-N, and low quality reads from raw reads. At the same time, Q20, Q30, GC content, and sequence duplication level of the clean reads were calculated. All the downstream analyses were based on clean reads with high quality. Paired-end reads files from all libraries/samples were pooled into left.fq (read1) and right.fq (read2) files separately. Transcriptome assembly was accomplished based on the left.fq and right.fq using Trinity (Grabherr et al. 2011) with min_kmer_cov set to 2 by default and all other parameters set as default.
Functional annotation
We first carried out homology search against the NCBI non-redundant nucleotide sequences (Nr–Nt) database using BLASTN algorithm (E value < 10−5) to remove ribosomal RNA sequences. For further functional annotation, the remaining sequences were searched against the NCBI non-redundant protein sequences (Nr–P), protein family (Pfam), eukaryotic orthologous groups/clusters of orthologous groups of proteins (KOG/COG), Swiss-Prot, Kyoto encyclopedia of genes and genomes (KEGG) ortholog database (KO) and gene ontology database (GO) by BLASTX (E value < 10−5) (Fig. S1). KOG is a classification system for orthologous gene products, where the proteins were assumed to have the same ancestor proteins. KEGG is a database containing systematic analysis of gene functions, and linking genomic information with functional information of higher order (Kanehisa and Goto 2000). To learn more about the function of genes obtained in S. viscidula, a BLASTX search against KEGG database with an E value of 10−5 was performed. GO is a functional classification system used for annotating and analyzing the functions of genes in any organisms.
SSR detection
SSRs of S. viscidula transcriptome were identified using MISA 1.0 (MIcroSAtellite; http://pgrc.ipk-gatersleben.de/misa/). Default parameters were set for identification of mono-, di-, tri-, tetra-, penta-, and hexa-nucleotide motifs as 10, 6, 5, 5, 5, and 5 repeats, respectively. Primers for each SSR were designed using Primer3 (http://primer3.sourceforge.net/releases.php). SSR motifs were designed with the following parameters: primer sizes ranging from 18 to 25 bp and PCR product sizes ranging from 100 to 500 bp with GC content ranging from 40 to 60%.
Analysis of total flavonoids by UV spectrophotometry
We collected fresh roots, stems, and leaves of S. viscidula at ten o’clock when the plants reached the optimal state of a day, in the middle of May, July, and September. For each month group, five samples of S. viscidula were collected and were dried in dark at 60 °C. The dried roots, stems, and leaves were then separately comminuted with a miller. Each solid sample (40 mesh, 0.40 g) was weighed and extracted with 20 ml 70% ethanol by reflux extraction for 3 times within 60 min. The extracts were cooled to 25 °C and filtered through 0.45 µm Millipore® membrane filters (Merck, Kenilworth, USA), and then, 1 ml of the filtrate was diluted to 100 ml with 70% ethanol. Afterwards, the filtrate was analyzed by Lambda 950 spectrophotometer (PerkinElmer, Hopkinton, USA) at a wavelength of 238 nm. Data were analyzed and evaluated by SPSS Statistics 17.0 (http://www.spss.com.cn/). Duncan’s test was used to distinguish differences between mean values.
qRT-PCR of gene expression during three stages of different parts
The unigenes identified in our study have been deposited in NCBI’s Gene Expression Omnibus (Edgar et al. 2002) and are accessible through GEO Series accession number GSE108555 (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE108555). The sequences for the identified chalcone synthase gene (SvCHS) have been submitted to the NCBI GenBank database with an accession number MG745795. We used Bio-Rad® CFX96 qRT-PCR (Bio-Rad, Hercules, USA) to analyze the transcription levels of putative key enzyme genes (SvCHS, SvCHI, SvF3H, SvMYB2, and SvbHLH) in the roots, stems, and leaves of S. viscidula. The reverse transcription reaction was performed using Prime Script 1st Strand cDNA Synthesis Kit and SYBR Premix Extaq Kits (TaKaRa, Dalian, China). Primer sequences generated by Primer3 for the qRT-PCR assay are listed in Table 1. The Actin gene was chosen as an endogenous control in studying gene expressions in various samples of S. viscidula. Each reaction with a total volume of 25 µl contained 12.5 µl 2 × SYBR Premix Extaq (TaKaRa, Dalian, China), 10 ng of cDNA, and 200 nM gene-specific primers. The cycling conditions were as follows: 95 °C for 30 s followed by 40 cycles of 95 °C for 10 s and then 61 °C for 30 s. A melting curve was performed from 55 to 95 °C to check the specificity of the amplified product. The mean value of three replicates was normalized using Actin as the internal control.
Results and discussion
Transcriptome sequence assembling and functional classification
In total, 42,310,834 clean reads used for subsequent analysis were produced. These reads had an average GC content of 45.39%. After assembling, we got 40,052 unigenes ranging from 201 to 13,361 bp with an N50 length of 1577 bp. After searching against seven databases, we got 24,892 assembled unigenes (62.14%) out of 40,052 sequences (Table 2) that were annotated.
Total of 9732 unigenes (24.29% of total unigenes) were clustered into 26 groups using KOG (Fig. 2). The largest part was assigned to the cluster of general function prediction only (1609, 14.75%) followed by posttranslational modification, protein turnover, and chaperones (1322, 12.12%) and the third category was signal transduction mechanisms (871, 7.98%).

Histogram presentation of KOG classification. (1) RNA processing and modification; (2) chromatin structure and dynamics; (3) energy production and conversion; (4) cell cycle control, cell division, chromosome partitioning; (5) amino acid transport and metabolism; (6) nucleotide transport and metabolism; (7) carbohydrate transport and metabolism; (8) coenzyme transport and metabolism; (9) lipid transport and metabolism; (10) translation, ribosomal structure and biogenesis; (11) transcription; (12) replication, recombination and repair; (13) cell wall/membrane/envelope biogenesis; (14) cell motility; (15) posttranslational modification, protein turnover, chaperones; (16) inorganic ion transport and metabolism; (17) secondary metabolites biosynthesis, transport and catabolism; (18) general function prediction only; (19) FUNCTION unknown; (20) signal transduction mechanisms; (21) Intracellular trafficking, secretion, and vesicular transport; (22) defense mechanisms; (23) extracellular structures; (24) unnamed protein; (25) nuclear structure; (26) Cytoskeleton
With using GO, 18,796 unigenes (46.92% of total unigenes) were classified into three functional categories: biological process (50,627 genes), cellular component (37,187 genes), and molecular function (24,482 genes) (Fig. 3). Based on GO annotation, cellular process (11,568 genes), cell (7502 genes), and binding (11,167 genes) were the largest GO terms in biological process of cellular component and molecular function, respectively. We also found that metabolic process (10,986 genes), catalytic activity (9488 genes), and cell part (7486 genes) consist of considerable parts in the three classifications. In addition, 291 genes were assigned to the secondary metabolic process and provide abundance data to reveal plant secondary metabolism (Fig. 4). These results indicated that most of the sequences were categorized into fundamental pathways of biological regulation and metabolism.

Histogram of GO classification of annotated unigenes from S. viscidula transcriptome. Biological process: (1) biological adhesion; (2) biological regulation; (3) cellular component organization or biogenesis; (4) cellular process; (5) developmental process; (6) establishment of localization; (7) growth; (8) immune system process; (9) localization; (10) locomotion; (11) metabolic process; (12) multicellular organismal process; (13) multi-organism process; (14) negative regulation of biological process; (15) positive regulation of biological process; (16) regulation of biological process; (17) reproduction; (18) reproductive process; (19) response to stimulus; (20) signaling; (21) single-organism process. Cellular component: (22) cell; (23) cell junction; (24) cell part; (25) extracellular matrix; (26) extracellular matrix part; (27) extracellular region; (28) extracellular region part; (29) macromolecular complex; (30) membrane; (31) membrane-enclosed lumen; (32) membrane part; (33) organelle; (34) organelle part; (35) virion; (36) virion part. Molecular function: (37) antioxidant activity; (38) binding; (39) catalytic activity; (40) channel regulator activity; (41) enzyme regulator activity; (42) molecular transducer activity; (43) nucleic acid binding transcription factor activity; (44) protein binding transcription factor activity; (45) receptor activity; (46) structural molecule activity; (47) transporter activity

Unigenes from S. viscidula related to secondary metabolism according to KEGG. (1) aflatoxin biosynthesis; (2) anthocyanin biosynthesis; (3) betalain biosynthesis; (4) butirosin and neomycin biosynthesis; (5) caffeine metabolism; (6) flavone and flavonol biosynthesis; (7) flavonoid biosynthesis; (8) glucosinolate biosynthesis; (9) isoflavonoid biosynthesis; (10) isoquinoline alkaloid biosynthesis; (11) novobiocin biosynthesis; (12) phenylpropanoid biosynthesis; (13) stilbenoid, diarylheptanoid and gingerol biosynthesis; (14) streptomycin biosynthesis; (15) tropane, piperidine and pyridine alkaloid biosynthesis
We mapped 9173 unigenes to 262 KEGG pathways. Of these, 4014 were involved in metabolism processes (Fig. 5). These genes are related to pathways involved in the biosynthesis of other secondary metabolites, including phenylpropanoid biosynthesis (136 genes, ko00940), flavonoid biosynthesis (35 genes, ko00941), stilbenoid, diarylheptanoid and gingerol biosynthesis (40 genes, ko00945) and flavone and flavonol biosynthesis (7 genes, ko00944). Given the number of mapped unigenes, it is obvious that flavonoid biosynthesis lies just in the downstream of phenylpropanoid biosynthesis, which also plays a key role in the secondary metabolic activity. This result will contribute to deciphering the metabolomics pathways and the application of drug manufacture in S. viscidula.

KEGG classification of unigenes in S. viscidula. (A) Cell process. (B) Environment information processing. (C) Genetic information processing. (D) Metabolism. (E) Organismal
In addition, we compared the 11 enzymes of flavonoid biosynthesis with the results of Liu et al. (2015) obtained in S. baicalensis (Liu et al. 2015). We found that an enzyme, anthocyanidin reductase (ANR), is absent in S. viscidula (Fig. 6). It is noteworthy that ANR involved in the process of catalyzing pelargonidin, cyanidin, and delphinidin, which will further generate epiafzelechin, epicatechin, and epigallocatechin. The absence of ANR will lead to metabolic, physiological, and phenotypic diversity of flavonoid biosynthesis between S. viscidula and S. baicalensis. For example, there is high flavone/flavonol biosynthesis in flowers of S. viscidula caused by high expression of MYB12, a transcription factor that regulates proanthocyanidin and flavonol synthesis (Yamagishi 2011; Wang et al. 2017). This could reduce anthocyanin biosynthesis due to high levels of flavonol/flavone biosynthesis which compete for flavanone precursors. Thus, the flower color of S. viscidula is usually light yellow, while that of S. baicalensis is typically purple.

Metabolic pathway of flavonoid biosynthesis in S. viscidula according to KEGG annotation. 11 enzymes from the transcriptome sequences data set are marked in red boxes. The box filled in red (EC. 1.3.1.77) represents anthocyanidin reductase (ANR), which is absent in S. viscidula but present in S. baicalensis
Frequency and distribution of SSRs in the transcriptome
As a resource for random candidate markers in population genetics studies, SSRs are polymorphic stretches of 1–6 nucleotide units repeated in tandem and randomly spread in eukaryotic genomes (Krumholz et al. 2009). SSRs are generally associated with functional and phenotypic variations. In plants, SSRs have been used as a powerful genetic resource for genetic mapping (Cipriani et al. 1999) and phylogenetic study (Guilford et al. 1997). In this study, 8925 SSR loci, distributing in 7298 unigenes (18.22%), were obtained from all the 40,052 unigenes. On average, SSRs occurs every 3960 bp in length. Dinucleotide repeats (4614, 51.69%) were the most common SSRs in our data sets (Table 3), and followed by mononucleotide (2591, 29.03%) and trinucleotide (1633, 8.32%). Among the dinucleotide repeat motif, AG/CT (3640, 40.78%) appears as the largest class, followed by AC/GT (523, 5.86%) and AT/AT (450, 5.04%). These results have important practical value for analyzing the genetic diversity and molecular breeding of S. viscidula using SSR molecular markers.
Accumulation of total flavonoids in root, stem, and leaf of three periods
Our results indicated that total flavonoids’ content of roots in July was significantly higher than those in May and September (Fig. 7). This could be related to the growth characteristics of S. viscidula. In general, S. viscidula reaches the vigorous stage in July and August when it begins to accumulate secondary metabolites. For leaves and stems, the content of total flavonoids increases gradually from May to September, indicating a different pattern of flavonoid synthesis in them. For different parts of S. viscidula, root has the highest flavonoid content, followed by stem, and leaf has the lowest content of total flavonoids (Fig. S5). This result supported with the fact that root is usually used as the main medicinal part of S. viscidula (Yamamoto 1991).

Changes of total flavone content in each periods of the same part of S. viscidula. Total flavone content showed significant difference among May, July and September in roots, stems and leaves. Samples were collected from five replicates. Data were analyzed by Duncan’s test. *P < 0.05
Dynamic gene expression in flavonoid biosynthesis
Using transcriptomic analysis, we identified a series of transcription factor families (TFs) involved in the flavonoid biosynthesis of S. viscidula. Several well-known families of Myb transcription factors (MYB), basic helix–loop-transcription factors (bHLH), and WD40 repeat proteins (WD40) have 88, 34, and 10 unigenes in S. viscidula transcriptome, respectively (Table 4) (Hichri et al. 2010). It has been confirmed that MYB and bHLH regulate the gene expression of flavonoid synthesis (Zhao et al. 2013; Xu et al. 2015). In addition, MYB, bHLH, and WD40 can form a protein complex structure named MBW (MYB–bHLH–WD40). This structure (TT2/TT8/TTG1) controls the accumulation of flavonoid biosynthesis in Arabidopsis thaliana (Baudry et al. 2004, 2006; Xu et al. 2015).
To investigate how the flavonoid biosynthesis was spatially and temporally regulated at transcription level, we identified several key genes including SvCHS, SvCHI, SvF3H, SvMYB2, and SvbHLH and analyzed the expression levels of these genes by qRT-PCR. SvCHS and SvCHI are the first two genes involved in the early, upstream of the flavonoid biosynthesis pathways. SvCHS encodes the first key enzyme that catalyzed Malonyl-CoA and 4-coumarate CoA to chalcone pathway (Koes et al. 1994). SvCHI encodes another key enzyme that catalyzed chalcone to flavanone, which further synthesized many other flavonoid compounds (Fig. 1). Muir et al. (2001) found that overexpression of Petunia CHI leads flavonoid compounds to increase markedly (Muir et al. 2001). In root, as presented, the relative expression of SvCHS all exhibited higher level in July than in May and the expression level decreased in September (Fig. 8). On the contrary, SvCHI exhibited lower level of gene expression in root in each month. SvMYB2 was down expressed in each period, indicating possible regulations to SvCHS and SvCHI, which was positive to SvCHS while negative to SvCHI. These results were similar to other findings in S. baicalensis that MYB2 negatively regulates the expression of CHI and positively regulates the expression of CHS (Yuan et al. 2013). In addition, SvbHLH and SvF3H had no remarkable temporal regularity, indicating that there is no regulation relationship between the two genes, which is consistent with the results of Grotewold et al. (Grotewold et al. 1998). Given the expression level of SvCHS, SvF3H, and SvCHI, we concluded that these genes were highly expressed in July when the content of total flavonoids reached high level, indicating a possible positive correlation between gene expression level and flavonoid content.

Changes of the expression of several genes in S. viscidula. Quantitative real-time PCR analysis of key enzyme genes involving in flavonoid synthesis in roots, stems and leaves of different periods. a SvCHS, b SvF3H, c SvCHI, d SvbHLH, e SvMYB2. Actin gene was amplified as an internal control. Data are expressed as the mean ± SD of five replicates
Furthermore, we observed obvious organ-specific expression of these genes. In May, July, and September, the expression level of SvCHS and SvMYB2 was higher in root than in stem and leaf, while SvCHI was highly expressed in leaf. In July and September, SvF3H has the highest expression level in stem. These results were similar to the work of S. baicalensis by Liu et al. (2015), suggesting that flavonoid synthesis was regulated in different manners in plants (Liu et al. 2015). There is also strong evidence that roots responded more rapidly and accumulated higher levels of defense-related hormones than leaves (Balmer et al. 2013; Larbat et al. 2012). Recently, Zhao et al. described a root-specific 4′-deoxyflavone synthesis pathway and identified specific isoforms of SbCHS-2 and CYP82D in S. baicalensis (Zhao et al. 2016, 2018). In addition, Lei et al. cloned a new root-specific gene CHS in S. viscidula and confirmed that it is structurally close to CHS in S. baicalensis (Lei et al. 2010). Furthermore, we conducted nucleotide alignment between SvCHS, SbCHS-1, and SbCHS-2 using NCBI BLAST tool. We found that the SvCHS characterized in our study was closer to the root-specific SbCHS-2, comparing with SbCHS-1 in S. baicalensis (Table 5; Fig. S2-S4). Given the structure as well as expression level of SvCHS and SvMYB2 in our study, it is, therefore, reasonable to conjecture that the two genes involve in flavonoid synthesis in the same manner, i.e., root-specific 4′-deoxyflavone pathway, in S. viscidula. In addition, light, which is one of the most important environmental factors, affects flavonoid biosynthesis in plants. Numerous studies have revealed that R2R3 MYB transcription factors can regulate differential expression in the biosynthesis of distinct flavonoids in response to specific light (Zoratti et al. 2014). As the major light-sensing organ, leaves absorb light in different wavelengths, and further regulate the metabolic pathways in plants. In our study, we found that SvCHI was highly expressed in leaves in each period, indicating a possible role of this gene in light-dependent flavonoid synthesis. Recently, Sheehan et al. (2016) demonstrated that another R2R3-type MYB transcription factor, MYB–FL, is the major determinant of differences in flavonol levels and can affect pollinator preference in Petunia (Sheehan et al. 2016). For S. viscidula, the different expressions of flavonoid synthesis genes are considered to be correlated with positive evolutionary significance (e.g., pollination and stress resistance). In conclusion, these results help to gain more insight into genetic manipulation of organ-specific gene expression (e.g., genome editing) and thereby enhance flavonoid contents in plants.
Conclusions
In this study, we used transcriptome sequencing to provide functional information of genes that are related to flavonoid synthesis in several secondary metabolism pathways of S. viscidula. Total 24,892 unigenes were functionally annotated using KO, Swiss-Prot, Pfam, GO, and KOG. Furthermore, we identified 178 unigenes as candidate genes involved in the biosynthesis of flavonoids. The results will deepen our understanding of candidate genes involved in flavonoid biosynthesis, which will ultimately contribute to the improvement of medicinal quality of S. viscidula. Meanwhile, we demonstrated the identification of flavonoid genes and compared these genes among species in Scutellaria to reveal the molecular mechanism and gene evolution in flavonoid synthesis pathways. These high-quality unigenes and candidate genes will be useful in trait related gene mining. In addition, results from SSRs will help analyze genetic diversity and improve medicinal active ingredients. These data sets can serve as reference transcriptome for further analyses, e.g., quantitative gene expression profiling, to broaden our understanding of S. viscidula and to improve the content of active ingredients in this medicinal plant.
Author contribution statement
JX and CKB carried out the experiments, data analysis, and preparation of figures. CKB and BC participated in the experiments and data analysis. BC and XL participated in the data analysis and preparation of figures, and contributed with consultation. GSL contributed to sample collection and data analysis. CKB managed and designed the research and experiments.
References
Balmer D, Papajewski DV, Planchamp C, Glauser G, Mauch-Mani B (2013) Induced resistance in maize is based on organ-specific defence responses. Plant J 74:213–225
Baudry A, Heim MA, Dubreucq B, Caboche M, Weisshaar B, Lepiniec L (2004) TT2, TT8, and TTG1 synergistically specify the expression of BANYULS and proanthocyanidin biosynthesis in Arabidopsis thaliana. Plant J 39:366–380
Baudry A, Caboche M, Lepiniec L (2006) TT8 controls its own expression in a feedback regulation involving TTG1 and homologous MYB and bHLH factors, allowing a strong and cell-specific accumulation of flavonoids in Arabidopsis thaliana. Plant J 46:768–779
Chen J, Hou K, Qin P, Liu H, Yi B, Yang W, Wu W (2014) RNA-seq for gene identification and transcript profiling of three Stevia rebaudiana genotypes. BMC Genom 15:571
Cipriani G, Lot G, Huang WG, Marrazzo MT, Peterlunger E, Testolin R (1999) AC/GT and AG/CT microsatellite repeats in peach [Prunus persica (L) Batsch]: isolation, characterization and cross-species amplification in Prunus. Theor Appl Genet 99:65–72
Edgar R, Domrachev M, Lash AE (2002) Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30:207–210
Ferreyra MLF, Rius S, Casati P (2012) Flavonoids: biosynthesis, biological functions, and biotechnological applications. Front Plant Sci 3:222
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, Chen Z, Mauceli E, Hacohen N, Gnirke A, Rhind N, Federica P, Birren BW, Nusbaum C, Lindblad-Toh K, Friedman N, Regev A (2011) Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat Biotechnol 29:644–652
Grotewold E, Chamberlin M, Snook M, Siame B, Butler L, Swenson J, Maddock S, Clairb GS, Bowen B (1998) Engineering secondary metabolism in maize cells by ectopic expression of transcription factors. Plant Cell 10:721–740
Guilford P, Prakash S, Zhu JM, Rikkerink E, Gardiner S, Bassett H, Forster R (1997) Microsatellites in Malus X domestica (apple): abundance, polymorphism and cultivar identification. Theor Appl Genet 94:249–254
Guo L, Lei CK, Yang FL, Duan CY, Bai CK (2016) Similarity and diversity evaluation of bioactive ingredients in S. baicalensis and S. viscidula by HPLC. Northwest Pharm J 31:115–118
Hichri I, Heppel SC, Pillet J, Léon C, Czemmel S, Delrot S, Lauvergeat V, Bogs J (2010) The basic helix-loop-helix transcription factor MYC1 is involved in the regulation of the flavonoid biosynthesis pathway in grapevine. Mol Plant 3:509–523
Kanehisa M, Goto S (2000) KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28:27–30
Koes RE, Quattrocchio F, Mol JNM (1994) The flavonoid biosynthetic pathway in plants: function and evolution. Bioessays 16:123–132
Krumholz MR, Klein RI, McKee CF, Offner SSR, Cunningham AJ (2009) The formation of massive star systems by accretion. Science 323:754–757
Larbat R, Bot JL, Bourgaud F, Robin C, Adamowicz S (2012) Organ-specific responses of tomato growth and phenolic metabolism to nitrate limitation. Plant Biol 14:760–769
Lei W, Tang SH, Luo KM, Sun M (2010) Molecular cloning and expression profiling of a chalcone synthase gene from hairy root cultures of Scutellaria viscidula Bunge. Genet Mol Biol 33:285–291
Liu HO (2016) Studies on the chemical components and biological activity of Scutellaria amoena Wright CH. Dissertation, Yunnan University of Traditional Chinese Medicine, China
Liu J, Hou J, Jiang C, Li G, Lu H, Meng FY, Shi LC (2015) Deep sequencing of the Scutellaria baicalensis Georgi transcriptome reveals flavonoid biosynthetic profiling and organ-specific gene expression. PloS One 10:e0136397
Muir SR, Collins GJ, Robinson S, Hughes S, Bovy A, Ric De Vos CH, van Tunen AJ, Verhoeyen ME (2001) Overexpression of petunia chalcone isomerase in tomato results in fruit containing increased levels of flavonols. Nat Biotechnol 19:470–474
Shang X, He X, He X, Li M, Zhang R, Fan P, Zhang Q, Jia Z (2010) The genus Scutellaria an ethnopharmacological and phytochemical review. J Ethnopharmacol 128:279–313
Sheehan H, Moser M, Klahre U, Korinna E, Alexandre DO, Therese M, Sabine M, Michiel V, Loreta F, Cris K (2016) MYB-FL controls gain and loss of floral UV absorbance, a key trait affecting pollinator preference and reproductive isolation. Nat Genet 48:159–166
Vrancken K, Holtappels M, Schoofs H, Deckers T, Treutter D, Valcke R (2013) Erwinia amylovora affects the phenylpropanoid-flavonoid pathway in mature leaves of Pyruscommunis cv. Conférence. Plant Physiol Biochem 72:134–144
Wang N, Xu H, Jiang S, Zhang Z, Lu N, Qiu H, Qu C, Wang Y, Wu S, Chen X (2017) MYB12 and MYB22 play essential roles in proanthocyanidin and flavonol synthesis in red-fleshed apple (Malus sieversii f. niedzwetzkyana). Plant J 90:276–292
Xu W, Dubos C, Lepiniec L (2015) Transcriptional control of flavonoid biosynthesis by MYB-bHLH-WDR complexes. Trends Plant Sci 20:176–185
Yamagishi M (2011) Oriental hybrid lily Sorbonne homologue of LhMYB12 regulates anthocyanin biosyntheses in flower tepals and tepal spots. Mol Breed 28:381–389
Yamamoto H (1991) Biotechnology in agriculture and forestry. In: Bajaj YPS (ed) Medicinal and aromatic plants III. Springer, Berlin, pp 398–418
Yuan Y, Wu C, Liu Y, Yang J, Huang L (2013) The Scutellaria baicalensis R2R3-MYB transcription factors modulates flavonoid biosynthesis by regulating GA metabolism in transgenic tobacco plants. PloS One 8:e77275
Zhao L, Gao L, Wang H, Chen X, Wang Y, Yang H, Wei C, Wan X, Xia T (2013) The R2R3-MYB, bHLH, WD40, and related transcription factors in flavonoid biosynthesis. Funct Integr Genom 13:75–98
Zhao Q, Zhang Y, Wang G, Hill L, Weng JK, Chen XY, Xue H, Martin C (2016) A specialized flavone biosynthetic pathway has evolved in the medicinal plant, Scutellaria baicalensis. Sci Adv 2:e1501780
Zhao H, Ren L, Fan X, Tang K, Li B (2017) Identification of putative flavonoid-biosynthetic genes through transcriptome analysis of Taihe Toona sinensis bud. Acta Physiol Plant 39:122
Zhao Q, Cui MY, Levsh O, Yang D, Liu J, Li J, Hill L, Yang L, Hu Y, Weng JK, Chen XY, Martin C (2018) Two CYP82D enzymes function as flavone hydroxylases in the biosynthesis of root-specific 4′-deoxyflavones in Scutellaria baicalensis. Mol Plant 11:135–148
Zoratti L, Karppinen K, Escobar AL, Häggman H, Jaakola L (2014) Light-controlled flavonoid biosynthesis in fruits. Front Plant Sci 5:534
Acknowledgements
We would like to thank the reviewers, whose work greatly improve the manuscript. We thank Dr. Yinghua Zha for her helpful revision on the manuscript. This work was supported by the Innovation Team Project of Breeding and Standardized Production of New Varieties of Traditional Chinese Medicine in Fundamental Research Funds of the Central Universities [GK201801008 to CKB]; the National Natural Science Foundation of China [31100241 to CKB]; and the Fundamental Research Funds for the Central Universities [GK201503046 to GSL].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declared that no competing interests exist.
Additional information
Communicated by M H. Walter.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Bai, C., Xu, J., Cao, B. et al. Transcriptomic analysis and dynamic expression of genes reveal flavonoid synthesis in Scutellaria viscidula. Acta Physiol Plant 40, 161 (2018). https://doi.org/10.1007/s11738-018-2733-5
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11738-018-2733-5