
Abstract
Toona sinensis, a member of Meliaceae family, is a traditional Chinese woody vegetable widely used as food and in health since ancient times. T. sinensis bud has extensive clinical uses because of its high flavonoid content. However, the literature lacks information on flavonoid metabolism and characterization of the corresponding genes in T. sinensis. In this study, we constructed two cDNA libraries of green (GYC-2) and purple toons (BYC-2) distributed in Taihe County of Anhui Province in China. A total of 9.48 Gb of raw sequencing reads were generated using Illumina technology. Obtained raw reads were assembled into 66,331 non-redundant unigenes with mean length 1076 bp. A total of 50,582 unigenes (accounting for 76. 26% of all-unigenes) could be matched to public database using BLASTx. Through alignment against KEGG database, a total of 34,183 were annotated into 135 KEGG pathways. Among such pathways, many candidate genes that are associated with flavonoid biosynthesis were discovered in our transcriptome data. In total, 9541 unigenes identified were differentially expressed genes (DEGs), including 5408 up-regulated unigenes and 4133 down-regulated unigenes in green toon vs purple toon. Moreover, numerous transcription factors (MYB, bHLH, and WD40) were found in our data. Many genes related to flavonoid biosynthesis showed preferential expression in BYC-2 cultivar. Therefore, de novo transcriptome analysis of unique transcripts provides an invaluable resource for exploring vital gene related to flavonoid biosynthesis of T. sinensis bud.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Taihe Toona sinensis Roem, which belongs to Toona Roem of Meliaceae, is a potentially important medicinal and woody vegetable that is native to Taihe County in Anhui, China. Commonly known as Chinese toon bud, buds and leaves of the plant are crispy and juicy, aromatic, taste uniquely, and have high consumption value (Zhou et al. 2010). Various natural products, such as triterpenes, phenolics, and flavonoids, were isolated and identified from T. sinensis buds and leaves (Mu et al. 2007; Kakumu et al. 2014). Studies showed that T. sinensis leaf extracts have many functions, such as suppression of ovarian cancer cell proliferation, anti-inflammation, analgesic functions, boil growth inhibition, antioxidant activity, and apoptotic induction of cancer cells (Huang et al. 2012; Yang et al. 2013). Bud color evolved via breeding with natural pollination. Moreover, bud color variation ranges from green to deep purple rachis and leaves (Fig. 1). Per bud or leaf color, Taihe T. sinensis are sorted into two clusters: purple (BYC-2) and green toons (GYC-2). Leaves or young shoots of the former are flavorful and edible vegetables, whereas the latter is used for timbering and forestation (Wang et al. 2008). Flavonoids are important in T. sinensis bud quality, and their contents or components have high variability among plant cultivars (Yang et al. 2010). Anthocyanin is the primary pigment responsible for T. sinensis bud color (Jin and Dong 1994). The compound is also critical for its nutrient contribution and health benefits (Zhang et al. 2014). Flavonoids, which increase antioxidant capacity of cells and tissues, are responsible for the antioxidant properties of T. sinensis bud. In vitro studies revealed the powerful antioxidant properties of polyphenols from T. sinensis buds (Wang et al. 2007; Vinodhini and Lokeswari 2014). Toon buds are used to prepare Chinese toon tea, which reduces risks of cardiovascular diseases and cancer. Different toon varieties have different flavonoid contents, and causes of such differences remain unknown. Multi-species transcriptome sequencing offers a shortcut for research on complex transcriptional regulation and metabolic pathways of different flavonoid contents (Shi et al. 2014; Wang et al. 2015; Zhang et al. 2015). Given rapid developments in bioinformatics tools, transcriptome studies are now possible for species, such as T. sinensis, even in the case of unknown reference genomes.

GYC-2 and BYC-2 buds of Taihe T. sinensis. a GYC-2 buds; b BYC-2 buds. GYC-2: green toon buds; BYC-2: purple toon buds)
Previous research on T. sinensis included mining and validation of molecular markers (Liu et al. 2012), isolation and characterization of important enzyme genes responsible for secondary metabolites (Hsu et al. 2012), and transcriptome studies involving in lysine biosynthesis in T. sinensis leaflets (Zhang et al. 2016). However, the literature still lacks information on flavonoid biosynthesis of T. sinensis bud. De novo sequencing offers a powerful tool for effectively obtaining entire plant transcriptome information. Among the new-generation, high-throughput RNA-Seq methods, Illumina HiSeq 4000 System is prevailing because of its low price and high output (Reuter et al. 2015). HiSeq 4000 sequencing technology is valuable for studying species, such as Anoectochilus roxburghii and Arachis hypogaea (Liu et al. 2015; Zhou et al. 2016).
In this report, we have demonstrated de novo transcriptome data in two cultivars of Taihe T. sinensis bud using Illumina HiSeq 4000 sequencing platform. Moreover, transcriptomic changes between GYC-2 and BYC-2 were investigated to characterize molecular regulation of flavonoid biosynthesis. This study provides insight into molecular mechanisms of flavonoid biosynthesis on this species, and sequencing outcomes could provide important basic data for further gene function studies or cultivation of T. sinensis for yielding high levels of flavonoid for medicinal purposes or for human consumption.
Materials and methods
Plant sample collection
Toon seedlings were grown for 5 years in the T. sinensis industry demonstration zone in Taihe County, Anhui, China. Two cultivars (GYC-2 and BYC-2) were planted using maternal plant stem with axillary bud as propagule, guaranteeing genetic background consistency of different individuals of the same cultivar. Ten buds of each cultivar were selected as RNA-seq sample. Another three buds of each cultivar were selected for quantitative real-time (RT-qPCR) confirmation. We removed the weak buds, and robust samples were collected based on bud color. For the two cultivars, all buds of the same variety were mixed for RNA-seq and six buds of the two cultivars were separated from one another. All samples were rapidly immersed in liquid nitrogen and frozen immediately for subsequent RT-qPCR confirmation.
cDNA library construction, sequencing, and de novo assembly
Total RNA was extracted from toon bud samples with TRIzol Reagent (Invitrogen, USA) according to manufacturer’s protocol. RNA concentration and quality were determined by Nanodrop. mRNA was isolated from the total RNA with oligo (dT) magnetic beads and then was fragmented using the fragmentation buffer. mRNA fragments were reverse-transcribed into cDNA. Purified short fragments were used for end repair, and then were ligated with adaptors, and the resulting cDNA were enriched by PCR amplification. Quality of the cDNA library was confirmed by Bioanalyzer and real-time PCR was performed to quantify the library. Finally, cDNA library was sequenced on the Illumina sequencing platform (Illumina HiSeq™ 4000, BGI, Shenzhen, China). The raw reads were preprocessed by discarding the adapter and low-quality fragments (including redundant sequences) using the filter-fq software. Clean reads were assembled to obtain unigenes in the Trinity software (Grabherr et al. 2011). Total unigenes were sorted into two classes. One class was gene family clustering named after “CL” with a cluster id behind it. In this cluster, similarity between unigenes was more than 70%. The other one was identified as singleton, with the prefix, “Unigene.”
Gene annotation and analysis
All unigene sequences were identified and functionally annotated in seven functional databases, including Nr, Nt, GO, COG, KEGG, Swiss-Prot, and Interpro database for unigenes. We used BLASTx (E value <10−5) to align unigenes to protein databases. BlastN (E value <10−5) was used to align unigenes to NT. Sequences were aligned with above databases to predict and classify functions. We used the ESTScan software to determine sequence direction. Blast2GO, which is a popular GO annotation software, was used to obtain GO annotation against GO database for unigenes annotated by Nr (Conesa et al. 2005), whereas InterProScan5 was used to acquire Interpro annotation. GO functional classification of all-unigenes was performed using the WEGO software and to characterize gene function distribution within different pathways from overall level (Ye et al. 2006).
Differential expression analysis
To compare transcript abundance between the two cultivars’ toon bud, the protocol by Audic and Claverie (1997) was applied to analyze transcript count information for each unigene. Unigene expression level was calculated following Fragments Per Kilobase Million (FPKM) formula. FPKM is derived using the following:
where C and N represent the counts of mappable reads uniquely aligned to a unigene and sum of reads sequenced uniquely aligned to total unigenes, respectively; L represents the sum of a unigene in base pairs. False discovery rate (FDR) is used to evaluate significantly differential expressive unigenes of the two cultivars’ toon bud (Kim and van de Wiel 2008). FDR ≤0.001 was threshold P value, and log2 fold change ≥2, indicating that differentially expressed genes (DEGs) have significant differences. After that, DEGs were further incorporated into GO functional categories and KEGG pathway analysis.
RT-qPCR
Relative expression levels of putative flavonoid-biosynthetic genes were evaluated by RT-qPCR. First-strand cDNA synthesis was performed using TransScript All-in-One cDNA supermix for qPCR (TransGene Biotech, Beijing) per the manufacturer’s instruction. Primers (Supplementary file 1) for RT-qPCR were designed by Generay Biotechnology Company (Shanghai). Actin gene of T. sinensis, an internal housekeeping gene, was used as control. PCR was performed in an Applied Biosystems 7300 Real-time PCR System (ABI, USA) using GoTaq® qPCR Master Mix (Promega) with the following parameters: 95 °C for 20 min followed by 40 cycles of 95 °C for 15 s, 60 °C for 30 s, and 72 °C for 30 s. After the reactions, dissociation curve analysis was conducted to evaluate primer specificity. All reactions were performed in triplicate per experiment on 96-well PCR plates. Relative mRNA levels were calculated using 2−ΔΔCt method against the actin gene.
Measurement of anthocyanin content
The quantification of anthocyanin was determined following the protocol of Mehrtens et al. (2005). Anthocyanin was extracted from 0.3 g fresh Taihe T. sinensis bud using 1 mL extraction reagent (1% HCl in methanol), incubated at 21 °C for 18 h with slight shaking. Extracts were then centrifuged at 21,500g for 3 min at room temperature. To determine anthocyanin content, 0.4 mL supernatant was drawn and mixed with 0.6 mL extraction reagent. Extract absorbance was assayed at 530 and 657 nm using UV-2800 double beam UV/Vis spectrophotometer (Zhejiang Scientific Instruments, China). The anthocyanin concentration was calculated following the protocol of Mano et al. (2007).
Results
Sequencing and de novo assembly of Taihe T. sinensis bud transcriptome
To generate complete profile of Taihe T. sinensis bud transcriptome, two cDNA libraries from GYC-2 and BYC-2 were, respectively, constructed and sequenced using Illumina, generating 9.48 Gb of raw RNA-seq data. Table 1 presents an overview of sequencing and assembly. After the deletion of poor quality sequences (including adaptor-polluted, redundant sequences), 36.77 and 36.16 Mb clean reads for GYC-2 and BYC-2, respectively, were retained and assembled. The Q20 scores were 97.72 and 97.85%, and GC contents were 41.35 and 41.16%, generated from GYC-2 and BYC-2, respectively. Trinity tool was used to assemble independently clean sequences from each library, which generated 87,527 and 86,870 contigs for GYC-2 and BYC-2. Their mean sizes were 885 and 926 bp with N50s of 1607 and 1649 bp for GYC-2 and BYC-2, respectively. Contigs were then assembled into 51,723 and 55,850 transcripts for GYC-2 and BYC-2. Their mean lengths were 1003 bp with N50 of 1677 bp and 1013 bp with N50 of 1680 bp, respectively. Finally, transcript sets from two libraries were further merged with 66,331 unigenes, comprising 71,420,182 bp with an average size of 1076 bp and N50 of 1756 bp. Unigene size distribution showed the following: 58.9% (39,049) of the unigenes were between 300 and 1000 bp in length; 36.3% (24,064) were between 1000 and 3000 bp; and unigenes with length more than 3000 bp accounted for only 4.9% (3218) (Fig. 2).

Sequence sizes distribution of All-unigenes in Taihe T. sinensis bud transcriptome
Functional annotation and species distribution
Based on the public databases available and sequence homologies, all generated unigenes were aligned using a serial blast with cut-off E value 10−5. This process provided functional annotations for the obtained unigenes. 50,582 unigenes (accounted for 76.26% of the total unigenes) were annotated in this way. Of these, 47,288 and 44,861 unigenes were annotated to Nr and Nt database, respectively, 31,555 hits via Swiss-Prot, 34,183 for KEGG; 18,044 for COG; 34,284 for Interpro; and 24,011 for GO (Table 2).
COG analysis showed that 35,897 unigenes were aligned to COG database for functional classification (Fig. 3). The most frequently identified classes were the following: category function accounted for 17.93% (6435); transcription for 9.73% (3493); replication related for 8.48% (3043); signal transduction for 7.19% (2580); posttranslational modification related for 7.05% (2530); and translation related for 6.33% (2274). Moreover, carbohydrate transport and metabolism was occupied an important position with 2013 unigenes (5.61% of COG-annotated unigenes).

COG functional classification of All-unigenes
GO annotation assigned 132,719 unigenes to molecular function, cellular process, and biological process categories (Fig. 4). The two most abundant unigene sequences were metabolic process (13,432, 10.12%) and cellular process (12,355, 9.31%) within biological process. At the location level of cellular process, 21,396 unigenes (47.48%) were distributed in cell and cell parts, 7897 (17.54%) in organelle, and 5728 (12.73%) in membrane. Those unigenes involving in molecular function modules were more widely distributed in GO categories. For example, catalytic activity (12,855, 45.59%) and binding (11,582, 41.08%) proteins comprised the majority, whereas 7.89% is attributed to activity proteins, such as transporter, structural molecule, molecular transducer, enzyme regulator, receptor, antioxidant, electron carrier, and transcription factor.

GO functional classification of All-unigenes
To identify the metabolic pathways genes and their biological functions in T. sinensis bud, assembled unigenes were assigned to various metabolic pathways in the KEGG database based on sequence similarity. Overall, 34,183 (51.53%) T. sinensis unique sequences were mapped to 135 predicted metabolic pathways using KEGG, and among such sequences, 6805 (19.91%) unique ones were assigned to metabolic pathways. A total of 3799 (11.11%) unique sequences were classified under biosynthesis of secondary metabolites, 1332 (3.9%) in plant–pathogen interaction, 1255 (3.67%) in RNA transport, 1185 (3.47%) in spliceosome, and 1140 (3.33%) in plant hormone signal transduction. A total of 181 (0.53%) unigenes were assigned to flavonoid biosynthesis (Supplementary file 2).
Species distribution results based on BLASTx searches showed that Taihe T. sinensis bud unigenes have the greatest homology with sequences from Citrus sinensis (56.46%) followed by other species, including Citrus clementina (24.03%), Theobroma cacao (4.16%), and Vitis vinifera (1.93%). The other 13.42% of unigenes was blasted to other species first (Fig. 5), indicating that Taihe T. sinensis is closer to C. sinensis than other species in the transcriptome databases.

Distribution of annotated species
Transcripts differentially expressed between GYC-2 and BYC-2 buds
To identify genes with different expression levels between GYC-2 and BYC-2 buds, we used FPKM method to calculate unigene expression levels (Fig. 6). A total of 9541 unigenes showed differential expression (with fold changes larger than two, and FDR ≤ 0.001) between the two cultivars. Among the unigenes, 5408 were identified to be up-regulated genes, whereas 4133 were down-regulated genes (Supplementary file 3). Then, all DEGs were mapped to each GO database term and counted within the corresponding GO term categories. A total of 52 functional groups, including molecular function, cellular component, and biological process, showed remarkable enrichment in DEGs compared with the genomic background using hypergeometric test (Supplementary file 4). Signal transduction and metabolic pathways remarkably enriched were identified using KEGG enrichment analysis of DEGs. 133 pathways by DEGs were shown in Supplementary file 5, and 30 metabolic pathways were significantly overrepresented. The top ten enriched pathways were mainly related to secondary metabolisms, including plant hormone signal transduction (ko04075), phenylpropanoid biosynthesis (ko00940), monoterpenoid biosynthesis (ko00902), and carotenoid biosynthesis (ko00906).

Gene transcription profile between GYC-2 and BYC-2 libraries. Red points represent up-regulated DEGs. Blue points represent down-regulated DEGs. Black points represent non-DEGs
Candidate genes related to flavonoid biosynthesis and their regulation in toon buds
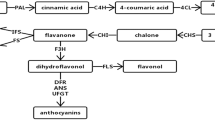
As shown in Fig. 7, a large number of unigenes related to flavonoid biosynthesis were identified in our transcriptome. Transcriptome analysis revealed 181 enzymes genes related to the flavonoid biosynthesis (Supplementary file 6), including genes related to general phenylpropanoid pathway [C3H (6), C4H (4), HCT (59), CCoAOMT (5)], genes related to the “early” step of flavonoid biosynthesis [CHS (8), CHI (4), F3′H (9)], and genes related to the “late” step [F3H (3), FLS (34), DFR (8), LAR (16), LDO (5), ANR (6)]. Almost major enzyme genes involved in flavonoid biosynthesis were annotated in this pathway. In addition to the main enzyme genes involved in the flavonoid biosynthesis, many regulatory genes that encoding transcription factors were found in the transcriptome data, they were thought as more widely regulatory function than enzyme genes in flavonoid biosynthesis. In particular, the enzyme gene transcription involved in flavonoid synthesis is regulated by bHLH, MYB, and WD40 (Jaakola 2013; Xu et al. 2015). A number of unigenes encoding for MYB, bHLH, and WD40 were found in T. sinensis bud transcriptome. The expression level of candidate genes related to flavonoid biosynthesis and regulation was altered in GYC-2 and BYC-2, including FLS, DFR, C4H, LAR, LDO, HCT, MYB, and MYC (Table 3). Molecular mechanism complexity of flavonoid biosynthesis remained uncertain in T. sinensis bud. Further investigation is needed to ascertain vital candidate regulators involved in flavonoid biosynthesis of T. sinensis bud.

Simplified pathway of flavonoid biosynthesis. Numbers of putative unigenes encoding enzymes were given in parentheses
RT-qPCR validation of DEGs
To confirm the differences in expression of unigenes in FPKM analysis, nine putative genes related to flavonoid formation of toon buds were selected for RT-qPCR and validation. Figure 8 displays the RT-qPCR expression patterns of these transcripts. The expression profiles of the genes assayed showed higher gene expression in BYC-2 than that in GYC-2, and confirming the RNA-seq data. The selected unigenes related to flavonoid biosynthesis were also validated as the most highly expressed in BYC-2 by RT-qPCR analysis.

RT-qPCR for validating RNA-Seq data between GYC-2 and BYC-2. Relative mRNA levels were normalized with respect to inner control gene (actin), and the corresponding values were expressed relative to GYC-2 bud (control), which was given a value of 1. Values represent mean ± SD of independent biological triplicates. Primer sequences used for RT-qPCR were shown in Table S1. CL9131.Contig1_All: flavonol synthase; CL4232.Contig2_All: dihydroflavonol reductase; Unigene2826_All: cinnamate-4-hydroxylase; CL11381.Contig1_All: leucoanthocyanidin reductase; Unigene3323_All: leucoanthocyanidin dioxygenase; Unigene26619_All: Shikimate-O-hydroxycinnamoyltransferase; CL7255.Contig1_All: MYB; CL4723.Contig2_All: MYB; CL4474.Contig1_All: MYC
Accumulation of anthocyanin in toon buds
Toon bud color changes are closely related to anthocyanin accumulation, which is an important phenotypic characteristic for identification of Taihe T. sinensis cultivars. To examine anthocyanin accumulation in GYC-2 and BYC-2 buds, bud extracts were subjected to spectrophotometer analysis. Our results indicate that BYC-2 buds accumulate total anthocyanin that is 6.25-fold higher than that of GYC-2 buds (Fig. 9).

Anthocyanin accumulation in GYC-2 and BYC-2 buds. GYC-2: anthocyanin concentrations in GYC-2 buds; BYC-2: anthocyanin concentrations in BYC-2 buds. Concentrations in GYC-2 and BYC-2 buds were determined by measuring absorbances at A530 and A657 nm using a spectrophotometer. Data are mean ± SD of three replicates
Discussion
To characterize the key genes involved in flavonoid biosynthesis of T. sinensis bud, two commonly cultivated Taihe T. sinensis cultivars (GYC-2 and BYC-2) were subjected to cDNA sample sequencing to construct T. sinensis bud transcriptome. In total, 36.77 and 36.16 Mb non-redundant reads were obtained. By de novo assembly, 51,723 and 55,850 unigenes were generated for GYC-2 and BYC-2, respectively. We obtained 66,331 all-unigenes with mean length of 1076 bp from T. sinensis bud transcriptomes of the two cultivars. This number of all-unigenes is more than that of another T. sinensis variety from Zhang’s report (54,628 unigenes) (Zhang et al. 2016). Illumina sequencing data and Trinity assembler yielded the same number of contigs as those from other studies (Galli et al. 2014; Huo et al. 2016), suggesting that these data fully matched transcriptome analysis requirements.
Among the 66,331 unigenes, 50,582 (76.26%) were annotated by comparison with public databases. The present study obtained more complete annotation information compared with a previous study, which only included 25,570 (46.76%) annotated unigenes from a total of 54,682 (Zhang et al. 2016). The higher matching ratio of unigenes to public databases in our study might be partially caused by higher ratio of long sequences to our all-unigenes (1076 bp mean length in our study to 764 bp mean length in Zhang’s study) (Wang et al. 2010). The majority of unigenes exhibited significant similarity with sweet orange sequences, indicating that molecular functions are relatively conserved between the two species. Sweet orange is an important crop of flavonoid-rich fruit, implying that it may have similar molecular mechanisms for flavonoid biosynthesis with T. sinensis (Pan et al. 2014; Assefa et al. 2016; Wang et al. 2016). Another possible reason for the high matching rate of species is the fact that the draft of the orange genome has been completed and more sequences information has been annotated (Xu et al. 2013). Similar to T. sinensis bud, tea plant is another economically important crop that is a rich source of flavonoid. Shi et al. (2011) obtained approximately 34.5 million reads of a tea plant variety and assembled them into 127,094 unigenes. Wu et al. (2014) constructed a database of 146,342 unigenes from four varieties of tea plant and aligned approximately 68,890 unigenes to public database sequences. Improvement in our T. sinensis transcriptome data set is needed based on comparisons with volume of transcriptome data from tea plants. More varieties, different seasons, and different tissues from T. sinensis should be selected for constructing a unified T. sinensis unigene database. Matching percentage reflects similar degrees of molecular genetic background between the two species (Toon sinensis and Citrus sinensis) and represents the scope of available sequence information in the existing T. sinensis database.
Based on public databases and GO and COG classification system, numerous unigenes were annotated to known proteins in the current study, indicating that our transcriptome data represent diverse transcripts. 18,930 (58.38%) unigenes were mapped to KEGG pathway and mainly distributed to ten subcategories. Results indicate that metabolic processes in Taihe T. sinensis bud are active and similar with sweet orange (Yu et al. 2012).
Given that T. sinensis bud is a rich source of flavonoids, putative genes related to flavonoid biosynthesis were focused on mining. Flavonoids are composed of a series of polyphenolic products with over 6000 members in plants, many of which play diverse physiological functions. Many reports on flavonoid biosynthesis pathway concentrated mainly on model plant, crops, and fruit trees, such as Arabidopsis, maize, and grape (Boss et al. 1996; McMullen et al. 2001; Routaboul et al. 2006). However, the current information lacks data on molecular mechanism of flavonoid biosynthesis and regulation in T. sinensis. We obtained from our annotated T. sinensis bud transcriptome numerous unigenes that encode for almost all known enzymes in flavonoid biosynthesis (Fig. 7). Biosyntheses of anthocyanins, proanthocyanins (PAs), and flavonols share a common phenylpropanoid pathway, which is activated by phenylalanine ammonia lyase (PAL), C4H, and 4CL. Phenylalanines are converted to p-Coumaroyl-CoA or continually converted to caffeoyl-CoA by the enzymes HCT and C3H. Subsequently, p-Coumaroyl-CoA/caffeoyl-CoA was converted to naringenin/eriodictyol under CHS and CHI via “early step” of the flavonoid pathway. In “late step,” F3H catalyzes hydroxylation of naringenin/eriodictyol to form dihydrokaempferol/dihydroquercetin. Eriodictyol/dihydroquercetin is synthesized from naringenin/dihydrokaempferol under catalysis by F3′H. Dihydrokaempferol is continually hydroxylated into dihydromyricetin by F3′5′H. Subsequently, the dehydrogenation reaction of dihydroquercetin and dihydromyricetin to flavonol was catalyzed by FLS. In the anthocyanin pathway, DFR catalyzes the conversion of dihydroquercetin or dihydromyricetin to leucocyanidin or leucodelphinidin, which are further converted to cyanidin or delphinidin by LDO or anthocyanidin synthase. Cyanidin or delphinidin is further reacted by various tailoring processes, including glycosylation and methylation modification. A branch pathway of PAs synthesis comes from the intermediate products of anthocyanin formation via reduction of leucocyanidin or leucodelphinidin by ANR or of cyanidin or delphinidin by LAR.
Comparative analysis of GYC-2 and BYC-2 bud transcriptome profiles showed that 9541 unigenes are differentially expressed, and DEGs are significantly enriched in secondary metabolism and plant hormone signal transduction processes, such as flavonoid biosynthesis. In “early step” of the flavonoid pathway, expression levels of enzyme genes were slightly higher in BYC-2 than the GYC-2 (Supplementary file 7), and level of transcript that encode C4H was significantly higher in BYC-2 than the GYC-2 (Fig. 8). In “late step” of the flavonoid pathway, expression abundance of four genes that encode enzymes, including FLS, DFR, LDO, and LAR, was markedly higher in BYC-2 than GYC-2. Real-time PCR analysis confirmed that expression levels of the four genes in BYC-2 bud are significantly higher than those in GYC-2. Moreover, HCT catalyzes the conversion of p-Coumaroyl-CoA to caffeoyl-CoA in the phenylpropanoid pathway. Transcript level of the enzyme gene was higher in BYC-2 than in GYC-2, indicating that more dihydroquercetin can be synthesized via this pathway. Result was consistent with those of previous report, indicating that dihydroquercetin content was higher in BYC-2 than that in GYC-2 (Yang et al. 2010). Regulatory genes encoding TFs associated with flavonoid biosynthesis were identified in our study; these TFs include MYBs (150), bHLHs (113), and WD40s (159) (Dixon et al. 2013; Xu et al. 2015). Of these factors, through RT-qPCR detection, we observed greater changes in expressions of two MYB genes and one MYC gene in BYC-2 buds compared with GYC-2 buds. This result was consistent with the significantly different anthocyanin accumulations in two T. sinensis cultivar buds. Specifically, BYC-2 buds accumulate more total anthocyanin than GYC-2 buds. Studies on regulation of flavonol biosynthesis showed that in Arabidopsis, AtMYB11, AtMYB12, and AtMYB111 regulate AtFLS1 and other steps (Mehrtens et al. 2005). In Prunus persica, MYB10 positively regulates promoters of DFR, and MYBPA1 trans-activates the promoters of DFR and LAR (Ravaglia et al. 2013). Co-expression of GbMYB1 and GbMYC1 (or bHLH transcription factor) activated GbDFR and GbANS gene promoters, which resulted in anthocyanin accumulation of Gynura bicolor roots (Shimizu et al. 2011). In the current study, we observed that expression-level relevance between four key structural genes (FLS, DFR, LDO, and LAR) and regulatory genes (MYBs and MYCs) in two T. sinensis cultivars indicates that candidate MYBs and MYCs possibly activate key structural gene promoters and positively regulate their gene expressions, thereby leading to flavonoid/anthocyanin biosynthesis in T. sinensis bud.
However, further studies should investigate the specific molecular events regarding candidate gene interactions and their products to regulate flavonoid biosynthesis in T. sinensis bud. Such information would clarify the interrelation between molecular mechanism of flavonoid biosynthesis and color formation of T. sinensis bud.
Conclusions
Here, we obtained a high-quality transcriptome data of Taihe T. sinensis bud using Illumina sequencing. This is the first reports on generation and de novo assembly of Taihe T. sinensis bud transcriptome. The results provide a significant number of important unigenes associated with flavonoid biosynthesis of T. sinensis buds. In the long term, our data would facilitate key gene functional study of flavonoid biosynthesis and elucidate the mechanism of T. sinensis bud color formation. Moreover, our data will improve the production of medicinal components obtained from T. sinensis through future genetic engineering.
Author contribution statement
HZ, LR: conceived and designed the experiments. XF, KT: performed the experiments and analyzed data, BL: contributed reagents and materials, HZ: wrote the paper.
References
Assefa AD, Ko EY, Moon SH, Keum YS (2016) Antioxidant and antiplatelet activities of flavonoid-rich fractions of three citrus fruits from Korea. 3 Biotech 6:109
Audic S, Claverie JM (1997) The significance of digital gene expression profiles. Genome Res 7:986–995
Boss PK, Davies C, Robinson SP (1996) Analysis of the expression of anthocyanin pathway genes in developing Vitis vinifera L. cv shiraz grape berries and the implications for pathway regulation. Plant Physiol 111:1059–1066
Conesa A, Cötz S, García-Gómez JM, Talón M, Robles M (2005) Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21:3674–3676
Dixon RA, Liu CG, Jun JH (2013) Metabolic engineering of anthocyanins and condensed tannins in plants. Curr Opin Biotechnol 24(2):329–335
Galli V, Guzman F, Messias RS, Körbes AP, Silva SDA, Margis-Pinheiro M, Margis R (2014) Transcriptome of tung tree mature seeds with an emphasis on lipid metabolism genes. Tree Genet Genomes 10:1353–1367
Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q (2011) Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol 29:761–780
Hsu CY, Huang PL, Chen CM, Mao CT, Chaw SM (2012) Tangy Scent in Toona sinensis (Meliaceae) leaflets: isolation, functional characterization, and regulation of TsTPS1 and TsTPS2, two key terpene synthase genes in the biosynthesis of the scent compound. Curr Pharm Biotechnol 13:1–12
Huang PJ, Hseu YC, Lee MS, Kumar KJS, Wu CR, Hsu LS, Liao JW, Cheng IS, Kuo YT, Huang SY, Yang HL (2012) In vitro and in vivo activity of gallic acid and Toona sinensis leaf extracts against HL-60 human premyelocytic leukemia. Food Chem Toxicol 50(10):3489–3497
Huo JW, Liu P, Wang Y, Qin D, Zhao LJ (2016) De novo transcriptome sequencing of blue honeysuckle fruit (Lonicera caerulea L.) and analysis of major genes involved in anthocyanin biosynthesis. Acta Physiol Plant 38:180
Jaakola L (2013) New insights into the regulation of anthocyanin biosynthesis in fruits. Trends Plant Sci 18(9):477–483
Jin B, Dong HR (1994) Nutritious substances and pigments in Chinese toon sprouts and physiological changes during storage. Acta Agric Shanghai 10(4):84–88
Kakumu A, Ninomiya M, Efdi M, Adfa M, Hayashi M, Tanaka K, Koketsu M (2014) Phytochemical analysis and antileukemic activity of polyphenolic constituents of Toona sinensis. Bioorg Med Chem Lett 24:4286–4290
Kim KI, van de Wiel MA (2008) Effects of dependence in high-dimensional multiple testing problems. BMC Bioinform 9:114
Liu J, Sun ZX, Chen YT, Jiang JM (2012) Isolation and characterization of microsatellite loci from an endangered tree species. Toona ciliata var. pubescens. Genet Mol Res 11(4):4411–4417
Liu SS, Chen J, Li SC, Zeng X, Meng ZX, Guo SX (2015) Comparative transcriptome analysis of genes involved in GA-GID1-DELLA regulatory module in symbiotic and asymbiotic seed germination of Anoectochilus roxburghii (Wall.) Lindl. (Orchidaceae). Int J Mol Sci 16(12):30190–30203
Mano H, Ogasawara F, Sato K, Higo H, Minobe Y (2007) Isolation of a regulatory gene of anthocyanin biosynthesis in tuberous roots of purple-fleshed sweet potato. Plant Physiol 143:1252–1268
McMullen MD, Snook M, Lee EA, Byrne PF, Kross H, Musket TA, Houchins K, Coe EH Jr (2001) The biological basis of epistasis between quantitative trait loci for flavone and 3-deoxyanthocyanin synthesis in maize (Zea mays L.). Genome 44:667–676
Mehrtens F, Kranz H, Bednarek P, Weisshaar B (2005) The Arabidopsis transcription factor MYB12 is a flavonol-specific regulator of phenylpropanoid biosynthesis. Plant Physiol 138:1083–1096
Mu RM, Wang XR, Liu SX, Yuan XL, Wang SB, Fan ZQ (2007) Rapid determination of volatile compounds in Toona sinensis (A. Juss.) Roem. by MAE-HS-SPME followed by GC–MS. Chromatographia 65:463–467
Pan ZY, Li Y, Deng XX, Xiao SY (2014) Non-targeted metabolomic analysis of orange (Citrus sinensis [L.] Osbeck) wild type and bud mutant fruits by direct analysis in real-time and HPLC-electrospray mass spectrometry. Metabolomics 10(3):508–523
Ravaglia D, Espley RV, Henry-Kirk RA, Andreotti C, Ziosi V, Hellens RP, Costa G, Allan AC (2013) Transcriptional regulation of flavonoid biosynthesis in nectarine (Prunus persica) by a set of R2R3 MYB transcription factors. BMC Plant Biol 13:68
Reuter JA, Spacek DV, Snyder MP (2015) High-throughput sequencing technologies. Mol Cell 58(4):586–597
Routaboul JM, Kerhoas L, Debeaujon I, Pource L, Caboche M, Einhorn J, Lepiniec L (2006) Flavonoid diversity and biosynthesis in seed of Arabidopsis thaliana. Planta 224(1):96–107
Shi CY, Yang H, Wei CL, Yu O, Zhang ZZ, Jiang CJ, Sun J, Li YY, Chen Q, Xia T, Wan XC (2011) Deep sequencing of the Camellia sinensis transcriptome revealed candidate genes for major metabolic pathways of tea-specific compounds. BMC Genom 12:131
Shi SG, Yang M, Zhang M, Wang P, Kang YX, Liu JJ (2014) Genome-wide transcriptome analysis of genes involved in flavonoid biosynthesis between red and white strains of Magnolia sprengeri pamp. BMC Genom 15:706
Shimizu Y, Maeda K, Kato M, Shimomura K (2011) Co-expression of GbMYB1 and GbMYC1 induces anthocyanin accumulation in roots of cultured Gynura bicolor DC. plantlet on methyl jasmonate treatment. Plant Physiol Biochem 49:159–167
Vinodhini V, Lokeswari TS (2014) Antioxidant activity of the isolated compounds, methanolic and hexane extracts of Toona ciliata leaves. Int J Eng Technol 4(3):135–138
Wang KJ, Yang CR, Zhang YJ (2007) Phenolic antioxidants from Chinese toon (fresh young leaves and shoots of Toona sinensis). Food Chem 101(1):365–371
Wang CL, Cao JW, Tian SR, Wang YR, Chen ZQ, Chen MH, Gong GL (2008) Germplasm resources research of Toona sinensis with RAPD and isoenzyme analysis. Biologia 63(3):320–326
Wang XW, Luan JB, Li JM, Bao YY, Zhang CX, Liu SS (2010) De novo characterization of a whitefly transcriptome and analysis of its gene expression during development. BMC Genom 11:400
Wang X, Li ST, Li J, Li CF, Zhang YS (2015) De novo transcriptome sequencing in Pueraria lobata to identify putative genes involved in isoflavones biosynthesis. Plant Cell Rep 34(5):733–743
Wang JH, Liu JJ, Chen KL, Li HW, He J, Guan B, He L (2016) Anthocyanin biosynthesis regulation in the fruit of Citrus sinensis cv. Tarocco. Plant Mol Biol Rep 34(6):1043–1055
Wu ZJ, Li XH, Liu ZW, Xu ZS, Zhuang J (2014) De novo assembly and transcriptome characterization: novel insights into catechins biosynthesis in Camellia sinensis. BMC Plant Biol 14:277
Xu Q, Chen LL, Ruan XA, Chen DJ, Zhu AD, Chen CL, Bertrand D, Jiao WB, Hao BH, Lyon MP, Chen JJ, Gao S, Xing F, Lan H, Chang JW, Ge XH, Lei Y, Hu Q, Miao Y, Wang L, Xiao SX, Biswas MK, Zeng WF, Guo F, Cao HB, Yang XM, Xu XW, Cheng YJ, Xu J, Liu JH, Luo OJH, Tang ZH, Guo WW, Kuang HH, Zhang HY, Roose ML, Nagarajan N, Deng XX, Ruan YJ (2013) The draft genome of sweet orange (Citrus sinensis). Nat Genet 45(1):59–66
Xu W, Dubos C, Lepiniec L (2015) Transcriptional control of flavonoid biosynthesis by MYB-bHLH-WDR complexes. Trends Plant Sci 20:176–185
Yang JX, Yang Y, Li WY, Qu CQ (2010) Determination of quercetin in Taihe Toona Sinensis by high performance liquid chromatography. Pharm Biotechnol 17(6):513–515
Yang SJ, Zhao Q, Xiang HM, Liu MJ, Zhang QY, Xue W, Song BA, Yang S (2013) Antiproliferative activity and apoptosis-inducing mechanism of constituents from Toona sinensis on human cancer cells. Cancer Cell Int 13:12
Ye J, Fang L, Zheng H, Zhang Y, Chen J, Zhang Z, Wang J, Li S, Li R, Bolund L (2006) WEGO: a web tool for plotting GO annotations. Nucleic Acids Res 34:293–297
Yu KQ, Xu Q, Da XL, Guo F, Ding YD, Deng XX (2012) Transcriptome changes during fruit development and ripening of sweet orange (Citrus sinensis). BMC Genom 13:10
Zhang W, Li C, You LJ, Fu X, Chen YS, Luo YQ (2014) Structural identification of compounds from Toona sinensis leaves with antioxidant and anticancer activities. J Funct Foods 10:427–435
Zhang MF, Jiang LM, Zhang DM, Jia GX (2015) De novo transcriptome characterization of Lilium ‘Sorbonne’ and key enzymes related to the flavonoid biosynthesis. Mol Genet Genom 290:399–412
Zhang X, Song ZQ, Liu T, Guo LL, Li XF (2016) De Novo assembly and comparative transcriptome analysis provide insight into lysine biosynthesis in Toona sinensis Roem. Int J Genom 3:1–9
Zhou GX, Zhang BG, Lin L, Zhu Q, Guo L, Pu YY, Cao X (2010) Study on the relationship between Toona sinensis Roem stand productivity and site conditions in Sichuan Basin. Ecol Econ 6:387–394
Zhou XJ, Dong Y, Zhao JJ, Huang L, Ren XP, Chen YN, Huang SM, Liao BS, Lei Y, Yan LY, Jiang HF (2016) Genomic survey sequencing for development and validation of single-locus SSR markers in peanut (Arachis hypogaea L.). BMC Genom 17:420
Acknowledgements
This research was supported by a grant from the Natural Science Key Foundations of the Anhui Bureau of Education (Nos. KJ2016A552, KJ2015KJ006, and 2014KJ019) and the Innovation Program for College Students (No. 201510371061).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by M. H. Walter.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Zhao, H., Ren, L., Fan, X. et al. Identification of putative flavonoid-biosynthetic genes through transcriptome analysis of Taihe Toona sinensis bud. Acta Physiol Plant 39, 122 (2017). https://doi.org/10.1007/s11738-017-2422-9
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1007/s11738-017-2422-9