
Abstract
DNA methylation plays an important role in gene expression regulation during biological development in plants. To explore the mechanism of chemically induced male sterility (CIMS) in wheat, using cDNA-amplified fragment length polymorphism (cDNA-AFLP) and methylation-sensitive amplification polymorphism (MSAP) approaches, 6.66 and 3.42 % of the sequences showed changes in gene expression and DNA methylation in 1376-CIMS as compared to its fertility line 1376. We sequenced 54 fragments that differed in cDNA-AFLP and 26 DNA fragments which differentially displayed in MSAP comparisons of CIMS and 1376. Our results provided evidences for genome-wide changes in gene expression and DNA methylation occurring in the development of the 1376-CIMS system induced by chemical hybridizing agent SQ-1, and its counterpart fertility line 1376. Moreover, this study contributed to the elucidation of CIMS effects on responses of transcriptome and methylome in the development of the anther.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
The intricate and precise regulation of gene expression in space and time is fundamental for normal development in all organisms. The spatial and temporal orchestration of gene expression trajectories is primarily controlled genetically by specific DNA sequences including cis- and trans-acting elements. However, increasing evidence suggests that many aspects of development also involve epigenetic regulations. Epigenetics refers to a variety of processes which have long-term effects on gene expression programs without changes in DNA sequence. Key players in epigenetic control are DNA methylation and histone modifications which, in concert with chromatin remodeling complexes, nuclear architecture and microRNAs, define the chromatin structure of a gene and its transcriptional activity. It has been proposed that cytosine DNA methylation represents an important epigenetic modification of eukaryotic chromatin, which plays an essential role in orchestrating gene expression across plant development (Ingelbrecht et al. 1994; Zhang et al. 2011). In addition, DNA methylation is important in silencing transposons and other repetitive (Cantu et al. 2010; Yan et al. 2010). Moreover, it has been showed that the inheritance of epigenetic state in plants relies largely on the maintenance of cytosine methylation (particularly CG methylation) through sporophytic mitosis, meiosis and postmeiotic mitosis, giving rise to gametophytes (Takeda and Paszkowski 2006). Significant changes of cytosine methylation have been observed in the processes of reproductive development in species such as Arabidopsis (Finnegan et al. 1996; Saze et al. 2003), poplar (Song et al. 2012), Malus domestica Borkh (Forino et al. 2003), and maize (Garcia-Aguilar et al. 2010). Furthermore, abnormal DNA methylation will lead to male sterility in species such as Arabidopsis (Ronemus et al. 1996), maize (Lu et al. 2010), and rice (Ding et al. 2012a, b; Zhou et al. 2012), which implies that DNA methylation plays a vital role in plant fertility.
Hybrid wheat has attracted considerable research effort spanning several decades, and wheat hybrids have shown significantly higher yields and better adaptation to adverse environments than the best homozygous genotypes (Pickett and Galwey 1997; Singh et al. 2010; Zhang et al. 2001). Since the autogamous nature of wheat makes hybrid seed production a challenging task, it continues to be a major constraint in a wider application of hybrids. Although several seed production systems (both genetic and chemical based) have been developed in wheat, chemical hybridizing agents (CHAs) could provide an alternate, workable system for inducing male sterility, which is rapid, flexible and would not require fertility restoration, and the major advantage of using CHAs is that almost any inbred line may be used as a female parent. So, exploitation of chemically induced male sterility (CIMS) is the best available strategy to break the existing yield barriers in wheat. However, how do CHAs cause male sterility in wheat? Despite the some progress in cytoplasmic male sterility (CMS) (Klindworth et al. 2002; Liu et al. 2002; Sasakuma et al. 2001; Yu et al. 2011; Zhang et al. 2009, 2010b), little information on regulatory mechanism of CIMS (Adugna et al. 2004; Chakraborty and Devakumar 2006; Parodi and Gaju 2009; Rao et al. 2000), in particular, the epigenetic switch system for their concerted regulation available is currently not known. Furthermore, we should get more information on how CHAs (SQ-1) alter gene expression and DNA methylation in the development of the anther.
The first objective of the present work was to compare changes in gene expression and DNA methylation between a typical CIMS line, Xi’nong 1376-CIMS induced by SQ-1 (“1376-CIMS” for short) and its fertile line, Xi’nong 1376 (“1376” for short) in the development of the anther. An additional objective was to shed light on how SQ-1 affected gene expression and DNA methylation changes between CIMS and its fertile line 1376. Unbiased and global cDNA-amplified fragment length polymorphism (cDNA-AFLP) and methylation-sensitive amplification polymorphism (MSAP) approaches were performed on anthers during anther development in 1376-CIMS and corresponding fertile line 1376. Thus, the fragments displaying changes in gene expression and/or DNA methylation as a result of CIMS were cloned and characterized.
Materials and methods
Plant materials and microscopic observation
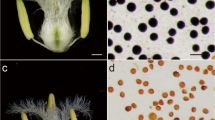
A typical CIMS line, Xi’nong CIMS-1376 induced by chemical hybridization agents SQ-1 (“1376-CIMS” for short) and its fertile line, Xi’nong 1376 (“1376” for short), both of the two lines with common genetic background, were grown in experimental fields of Northwest A&F University in the summer from 2010 to 2012. SQ-1, at the rate of 5.0 kg ha−1, was sprayed when wheat development on average was at the 8.5 stage (about 175 mm spikes) of the Feekes' scale. Anthers at three developmental stages were picked out from the corresponding young spikes and collected for experiments. To establish cytological controls, the purity and developmental stage of pollen were checked using acetic acid magenta staining of nuclei and microscopy. The whole anther development in this study was divided into three stages according to the microsporogenesis progress by checking under the microscope, i.e., (stage 1) uninucleate anther stage, (stage 2) binucleate anther stage, and (stage 3) trinucleate anther stage (anther obtained from spikelets immediately on ice) (Fig. 1a–c). Pollen fertility was tested with iodine-potassium iodide (KI-I2) assays (Fig. 1d, e). In addition, the morphology of anther and pollen was observed using a JSM-6360LV (Japan) scanning electron microscope (SEM). And around 800 anthers were needed to obtain 100 mg anther material for each sample.

Microspore development and pollen fertility of 1376-CIMS as compared to 1376. a–c Stained with acetocarmine. d, e Stained with potassium iodide–starch. a Uninucleate microspore; b binucleate microspore; c trinucleate pollen; d pollens from 1376. e Pollens from 1376-CIMS. Scanning electron microscopy analysis of the anther surface (f, g) and of the pollen exine (h, i); f, h from 1376; g, i from 1376-CIMS. 1376-CIMS, chemically induced male sterility line; 1376, fertile line
DNA and RNA extraction and cDNA synthesis
Total genomic DNA was isolated from fresh anthers using the method as described by Murray and Thompson (1980). Total RNA was extracted using TRIzol reagent (Invitrogen). Before reverse transcription, total RNA was treated with RNase-free DNase I (Takara) at 37 °C for 30 min to avoid genomic DNA contamination. First-strand cDNA was synthesized from total RNA using oligo-(dT)18 primer and reverse transcriptase (RT) SuperScript (Takara) according to the manufacturer’s specifications. Second-strand cDNA was synthesized using 10 U DNA polymerase I (Takara) and 3 U RNase H (Takara) according to standard procedures. The resulted double-stranded cDNA was purified by phenol–chloroform extraction and ethanol precipitation, and then resuspended in a final volume of 40 μl ddH2O. Approximately a quarter of this volume was checked on an agarose gel. If the expected smear between 100 and 4,000 bp was observed, the rest of the cDNA was stored at −20 °C for future use.
cDNA-AFLP analysis
The cDNA-AFLP technique was modified from published methods (Bachem et al. 1996). Equal amounts of cDNA from the same organ but different individuals of the same plant material were mixed, respectively. Approximately 500 ng of mixed cDNA was double-digested with 5 U Mse I and 5 U EcoR I (Takara) at 37 °C for 5 h. The digestion was inactivated by heating at 65 °C for 45 min. The digested fragments were ligated to 10 pmol of EcoR I adaptor, 100 pmol of Mse I adaptor, and 1 U T4 DNA ligase (Takara) at 22 °C for 6 h. The pre-amplification step was carried out using 50 ng of the ligation products as templates, with 0.2 μM EcoR I and Mse I preselection primers, in a final volume of 25 μl containing 1× polymerase chain reaction (PCR) buffer, 1.5 mM MgCl2, 0.2 mM dNTP, and 2 U Taq polymerase (Takara). The PCR reactions were performed with the following program: 3 min at 94 °C, 26 cycles consisting of 30 s at 94 °C, 45 s at 56 °C, 1 min at 72 °C, and a final extension step of 10 min at 72 °C. The pre-amplification PCR product was diluted 1:50 with ddH2O, and 2 μl was used as template for selective amplification with 0.2 μM of the primers “EcoR I + 2” (containing two selective bases) and “Mse I + 2” (two selective bases). The other components were the same as in the pre-amplification reactions. Selective PCR reactions were performed with the following procedure: 94 °C for 4 min, 30 cycles of 30 s denaturing at 94 °C, 30 s annealing and 60 s extensions at 72 °C, ending with 10 min at 72 °C to complete extension. Annealing was initiated at a temperature of 65 °C, which was then reduced by 1 °C for the next 10 cycles, and maintained at 55 °C for the remaining 26 cycles. The sequences of adaptors and primers are listed in Table 1.
5 μl selective PCR products were mixed with 5 μl of formamide dye (98 % formamide, 10 mM EDTA, 0.05 % w/v bromophenol blue and xylene cyanol), denatured at 94 °C for 5 min and separated by electrophoresis on 6 % denaturing polyacrylamide (20:1 acrylamide: bisacrylamide, 7.5 M urea, and 1× Tris–borate-EDTA buffer, pH 7.8). The gels were pre-run at 100 W for about 30 min before 10 μl of the mix was loaded, then run at 250 V for about 2.5 h, and silver stained according to the DNA silver-staining system procedure.
Methylation-sensitive amplification polymorphism assay
The MSAP technique was modified from published methods (Xiong et al. 1999). The protocol involved use of the isoschizomers Hpa II and Msp I instead of Mse I as ‘frequent cutter’ enzymes. The adapter and primer for the ‘rare-cutter’ enzyme EcoR I were the same as that used in standard AFLP analysis, while the Hpa II/Msp I adapter was designed as in Table 2.
To detect MSAP, restriction and ligation were done concurrently and two sets of digestion/ligation reactions were carried out simultaneously. In the first reaction, 2 μl of the extracted DNA (120 ng DNA) was added to 18 μl buffer (10 mM Tris–HCl pH 7.5, 10 mM MgAc, 50 mM KAc) containing 6 units EcoR I, 5 units Hpa II (Takara), 4 units T4 DNA ligase (Takara), 5 pmol EcoR I adapter, 50 pmol Hpa II/Msp I adapter and 10 mM ATP. The mixture was then incubated at 37 °C for 6 h. The reaction was stopped by incubating at 65 °C for 10 min and diluting 10 times in 0.1 × TE (1 mM Tris–HCl, 0.1 mM EDTA, pH 8.0) for PCR amplification. The second digestion/ligation reaction was carried out in the same way, except that Msp I was used in place of Hpa II.
We used two consecutive PCRs to selectively amplify the EcoR I–Hpa II and EcoR I–Msp I DNA fragments. The pre-selective amplification (first PCR) was performed using 5 μl of the above mentioned diluted mixture added to a 15 μl mixture giving a final concentration of 10 mM Tris–HCl pH 8.3, 50 mM KCl, 1.5 mM MgCl2, 0.2 mM of each dNTP, 50 ng of EcoR I and Hpa II/Msp I adapter-directed primer, each possessing a single selective base (E + 1; HM + 1) and 1 unit of Taq polymerase (Takara). PCR reactions were performed with the following profile: 94 °C for 60 s, 25 cycles of 30 s denaturing at 94 °C, 30 s annealing at 55 °C and 60 s extension at 72 °C, ending with 10 min at 72 °C to complete extension. After checking for the presence of a smear of fragments (100–1,000 bp in length) by agarose electrophoresis, the amplification product was diluted 10 times in 0.1 × TE.
Selective amplification (second PCR) of the diluted pre-amplification products was carried out using a total of 30 primer combinations obtained with four EcoR I primers in combination with three Hpa II/Msp I primers with three selective bases each (E + 3, HM + 3). The EcoR I and Hpa II/MspI adapters and primers were synthesized by Invitrogen Life Technologies. Selective PCR reactions were performed with the following procedure: 94 °C for 60 s, 36 cycles of 30 s denaturing at 94 °C, 30 s annealing and 60 s extension at 72 °C, ending with 10 min at 72 °C to complete extension. Annealing was initiated at a temperature of 65 °C, which was then reduced by 0.7 °C for the next 13 cycles and maintained at 56 °C for the subsequent 23 cycles.
The second PCR products were mixed with 15 μl of formamide dye (98 % formamide, 10 mM EDTA, 0.01 % w/v bromophenol blue and 0.01 % w/v xylene cyanol), denatured at 95 °C for 5 min and separated by electrophoresis on 6 % denaturing polyacrylamide sequencing gels (6 % acrylamide 19:1, 7M Urea) in 1× TBE buffer. The gels were pre-run at 150 W for about 30 min before 10 μl of the mix was loaded.
Gels were run at 250 V for about 2.5 h, and silver stained. The gel was fixed in 10 % acetic acid for 10 min, washed twice with a large quantity of ultrapure water for 1 min, transferred to a silver impregnation solution (1 g/l AgNO3, 0.75 % formaldehyde) for 8 min and then rinsed with ultrapure water for 20 s. All the above steps were performed with slow agitation on a shaker. Image development was carried out with manual agitation in developer (30 g/l Na2CO3, 0.02 % formaldehyde). To stop development and fix the gel, 10 % acetic acid was added directly to the developing solution and incubated with shaking for 2–3 min.
Scoring of cDNA-AFLP and MSAP bands
Each cDNA-AFLP and MSAP gel was run twice. For the cDNA-AFLP and MSAP analyses, the replicate gel was run from the same cDNA and DNA sample but from a different digestion–ligation-amplification reaction, only the reactions showing full reproducible results between replicas were used for data analysis. Moreover, with both cDNA-AFLP and MSAP gels, the upper part and the lower part of the gel, where the resolution is not satisfactory, were not used for band scoring. The scored cDNA-AFLP and MSAP bands were transformed into a binary character matrix, using “1” and “0” to indicate the presence and absence, respectively, of a band at a particular position.
Cloning and sequence analysis of differentially displayed fragment
Bands that displayed DNA methylation differences during the anther development were excised from the gel and re-amplified using the selective amplification PCR conditions. The amplified fragments were ligated into pGEM-T easy vector (Takara) and sequenced. Homology search were performed on the National Center Biotechnology Information Server (http://www.ncbi.nlm.nih.gov/BLAST/).
Results
Changes in gene expression during the development of anthers induced by SQ-1
In order to collect synchronous anther samples for analysis of each discrete developmental stage and to establish cytological controls, it was essential to confirm the precise cytological stage of microspore development. Cytological results of wheat microspore development using acetocarmine staining techniques are shown (Fig. 1a–c). We observed the pollen fertility of 1376-CIMS and its fertile line 1376 (Fig. 1d, e), suggesting that SQ-1 can induce complete male sterility. The anther exine pattern in 1376-CIMS was further analyzed using scanning electron microscopy (SEM). We observed an orderly linear-shaped surface of the anther in 1376, and yet a confused and disordered surface of the CIMS anther (Fig. 1f, g). In the meantime, pollens in 1376 exhibited characteristic round and smooth surface, pollens from the 1376-CIMS showed a complete misshapen and shrunken extine pattern (Fig. 1h, i). These results implied SQ-1 had a major impact on the development of the anthers.
To determine how SQ-1 causes the transcriptome of 1376-CIMS to diverge from that of its fertile line 1376, cDNA-AFLP display was performed on the anther in the development of 1376-CIMS and its fertile line 1376. The band patterns were expected to be comparisons of the cDNA-AFLP profiles of between 1376-CIMS and 1376, and its fertile line 1376 was served as control, and all cases of deviation from its fertile line 1376 were scored as gene expression changes in this study. An example of this analysis was presented in Fig. 2. Thirty-two pairs of selective primers were used in the cDNA-AFLP assay.

cDNA-AFLP patterns of 1376-CIMS as compared to their corresponding fertility line 1376. B1, B2 and B3 are uninuclear pollen stage, binuclear pollen stage and trinucleater pollen stage in 1376, respectively. While A1, A2 and A3 are the corresponding stage in 1376-CIMS. 1, 2 and 3 bands detected in 1376-CIMS but disappeared in 1376; 4, 5 and 6 bands detected in 1376 but disappeared in 1376-CIMS. M, DM10-100 bp ladder DNA marker; a 500 bp; b 300 bp. 1376-CIMS, chemically induced male sterility line; 1376, fertile line
In anther at uninucleate stage, 896 transcripts were obtained with 72 (8.04 %) transcripts showing expression alterations in 1376-CIMS. Among these 72 transcripts, 23 transcripts (31.94 %) disappeared in 1376-CIMS, while 49 new transcripts (68.06 %) appeared. At binucleate stage, 53 (5.52 %) of 960 cDNA fragments were altered in their expression patterns in 1376-CIMS with 14 (26.42 %) transcripts were suppressed, and 39 (73.58 %) were activated. At trinucleate stage, 55 upon 848 (6.49 %) cDNA fragments were altered in their expression patterns in 1376-CIMS as compared to its fertile line 1376 with 19 (34.55 %) transcripts silenced and 36 (65.55 %) transcripts activated (Table 3).
Combining data from the three stages, 180 of 2,704 transcripts 6.66 %) were altered in their expression in 1376-CIMS. About 2.07 % (56 of 2,704) of the transcripts were silenced in 1376-CIMS, while 4.59 % (124 of 2,704) of the transcripts were activated (Table 3). The proportion of gene activating was significantly higher than that of gene silencing.
Alterations of DNA methylation during the anther development in 1376-CIMS
Isoschizomers Hpa II and Msp I recognize and digest 5′-CCGG-3′ sites, but display differential sensitivity to DNA methylation. Hpa II is inactive if either cytosine is fully methylated (Methylation of both strands), even as it cleaves the hemi-methylated sequence (only one strand methylation). Msp I is sensitive only to methylation at the external cytosine. It cuts in the case of inner cytosine methylation (CmCGG), but not in the case of outer cytosine methylation (mCCGG). Therefore, different band patterns from PCR amplification can reflect the methylation status and level at the site.
To explore changes in DNA methylation induced by SQ-1, we employed MSAP analysis to assess cytosine methylation at specific restriction sites throughout the genome (Fig. 3). Examples of some MSAP profiles were shown in Fig. 4a–g. In the case of absence of methylation changes, 1376-CIMS were expected to have the same methylation patterns as 1376 which were generated due to the DNA methylation at the CCGG sites and did not switched during the anther development (Fig. 4a–c). Polymorphic bands between 1376-CIMS and 1376 were scored as methylation changes (Fig. 4d–g). A total of 24 pairs of EcoR I + Hpa II/Msp I selective primer combinations were used in MSAP analysis. Table 4 summarized the methylation status in the 1376-CIMS relative to its fertility line 1376.

Methylation polymorphism between the maintainer line and its male sterility line shown in methylation-sensitive amplified polymorphism (MSAP) analysis. Uni, Bi and Tri stand for uninucleate phase, binucleate phase and trinucleate phase, respectively. And, H and M refer to digestion with EcoR I + Hpa II and EcoR I + Msp I, respectively. The polymorphism bands amplified by the primer combination HM + TGC/E + ACT are shown. 1376-CIMS, chemically induced male sterility line; 1376, fertile line

Examples of MSAP profiles in 1376-CIMS and its fertility line 1376. EcoR I-HpaII (H) and EcoR I–MspI (M) were used for digestion in MSAP analysis. Example of additive pattern (a–c). Examples of DNA methylation changes derived from 1376-CIMS (d–g). 1376-CIMS, chemically induced male sterility line; 1376, fertile line
In anther at uninucleate stage, comparisons of the MSAP profiles revealed a total of 466 fragments which were detected in 1376-CIMS and its fertility line 1376. Among them, 19 (4.08 %) showed methylation changes between 1376-CIMS and its fertility line 1376. 11 bands were from pattern of hypermethylation, 6 bands from pattern of demethylation. At binucleate stage, 15 of 423 fragments (3.54 %) showed methylation alterations between 1376-CIMS and their fertility line 1376. Of the altered bands, 11 cases were from pattern of hypermethylation MSAP markers, and 4 cases from pattern of demethylation. At trinucleate stage, 12 (2.63 %) out of 456 bands showed methylation changes between 1376-CIMS and its fertility line 1376. 7 bands were hypermethylated, while 5 bands were demethylated.
Combining the data for these three stages, 46 of 1,342 bands (3.42 %) showed methylation alterations in 1376-CIMS as compared to its fertile line 1376. Among the 46 changed bands, 31 bands were from pattern of hypermethylation and 15 bands from pattern of demethylation. Thus, the proportion of 1376-CIMS methylation alterations was significantly different from that of 1376 methylation alterations.
Characterization of fragments showing changes in gene expression or methylation
Some of the fragments showing changes in gene expression or methylation associated with anther development from 1376-CIMS and its fertile line 1376 were cloned and sequenced. The homologies of these sequences to known protein and gene sequences were detected using the BLASTX suite of programs.
Fifty-four upon 168 cDNA-AFLP fragments showing changes in gene expression from 1376-CIMS and its fertile line 1376 were cloned and sequenced. Among the 31 candidate genes, 7 had no similarity to GenBank entries. The other candidate genes had a variety of functions, including metabolic enzymes, kinases, F-box proteins, proteins of the proton-dependent oligopeptide transport family, proteins involved in plant hormonal regulation, proteins participating in signal transduction pathways, transcription factors, anti-apoptotic proteins, and proteins of unknown function (Supplementary Table 1).
Twenty-six upon 46 fragments showing changes in methylation status in 1376-CIMS and its fertility line 1376 were randomly selected, cloned, and sequenced. About 9 sequences had no gene annotation in GenBank with BLASTX. The candidate fragments encode a variety of proteins, including metabolic enzymes, F-box proteins, leucine-rich-like protein, hormonal regulation, and bHLH proteins, retroelement, and proteins of unknown functions. Notably, 15.4 % (4 of 26) of the sequenced fragments that underwent methylation changes in 1376-CIMS showed similarity to retroelement sequences (Supplementary Table 2).
Discussion
Effects by SQ-1 in 1376-CIMS on gene expression and DNA methylation
As more plant transcriptomes and methylomes become available, it shall be a major priority for future research to elucidate the molecular and cellular control of cytosine methylation dynamics and inheritance, and to relate the contributions of this epigenetic modification to gene expression as well as its role in controlling plant development.
The cDNA-AFLP provides the expression profile of whole genomes, allowing the monitoring of transcriptional changes related to outside stimuli (Wang et al. 2011). And the advantage of not requiring previous genome knowledge makes this technique a powerful tool for identifying novel genes in non-model organisms. In addition, cDNA-AFLP has high sensitivity and specificity, comparable to microarrays (Levterova et al. 2010). Several studies have demonstrated the effectiveness of cDNA-AFLP to analyze gene expression (Aubry et al. 2003; Baisakh et al. 2006; Durrant et al. 2000; Hsu et al. 2008; Mondego et al. 2003; Paris et al. 2012). And many previous studies also have confirmed that the MSAP technique is highly efficient for large-scale detection of cytosine methylation in plant genomes (Baurens et al. 2008; Chen et al. 2009; Hanai et al. 2010; Portis et al. 2004; Shan et al. 2012; Wu et al. 2000; Xiong et al. 1999). Thus, these technological improvements have the potential of unlocking the complexity of epigenetic modifications just as profoundly as whole genome sequencing has impacted genetics.
In flowering plants, the male gametophyte (or pollen grain) plays a pivotal role in plant fertility and crop production through the generation and delivery of the male gametes to the embryo sac for double fertilization. Male gametophyte development in higher plants is a complex process that requires the coordinated participation of various cell and tissue types and their associated specific gene expression patterns. Our data indicated that the expression and DNA methylation of the anthers in 1376-CIMS differed in their ability to be modified in gene expression and methylome. Although the underlying mechanisms for these phenomena are yet to be not determined, we supposed that genome-biased alterations in gene expression and DNA methylation might be a way to overcome incompatibilities after CHA treated, thus might contribute to rapidly adapt to adversity. Our result showed that in individual stages the number of transcripts varied from 896 (uninucleate microspores, UNM) through 960 (bicellular pollen, BCP) to 848 (tricellular pollen, TCP) (Table 3), whose trend was consistent with that in Arabidopsis (Honys and Twell 2004), but there’s a great gulf between the number of transcripts of both, and that may be due in part to different analysis technique. And our data also revealed that in individual stages a net new number of transcripts (new transcripts in 1376-CIMS minus transcripts absent in 1376-CIMS) varied from 26 (UNM) through 25 (BCP) to 17 (TCP), which suggested that SQ-1 should have increased the net number of the transcripts in the development of the anther.
Moreover, MSAP analysis also revealed that 1376-CIMS and 1376 have distinct methylation alteration patterns (Table 4). About 4.92 % of the methylation changes occurred in Hpa II lanes (hemi-methylated at mCHG), and 21.68 % occurred in Msp I lanes (methylated at mCG), while 73.47 % occurred in both Msp I and Hpa II lanes in 1376-CIMS. In 1376, 5.59 % of the DNA methylation changes occurred in Hpa II lanes, and 22.21 % occurred in Msp I lanes, while 72.28 % occurred in both Msp I and Hpa II lanes. The results suggested that there were different effects on DNA methylation pattern between 1376-CIMS and 1376. The most striking observation revealed that the relative methylation level of mCG and total (mCG + mCHG) in 1376-CIMS was lower than that of 1376, which, implied more extensive demethylation (mCG + mCHG) in 1376-CIMS was than that in 1376.
However, the tissues assayed in maize included seedlings and tassels of C-type cytoplasmic male sterility line were more methylated than their corresponding maintainers not only on MSAP ratios but also on the full methylation levels (Lu et al. 2010), which are inconsistent with our data. The possible cause of this gap is that the pattern of DNA cytosine methylation has tissue-, organ-, developmental stage- and species-specific gene expression across plant development. In especial, it is not the same that the regulation mechanism between the CIMS and CMS (cytoplasmic male sterility).
Combining the data between cDNA-AFLP and MSAP analyses demonstrated up to 6.66 % of the transcripts in the 1376-CIMS showed changes in expression, and 3.42 % of the sequences in the 1376-CIMS showed changes in DNA methylation as compared to its fertile line 1376. Moreover, SQ-1 decreased the number of the methylated fragments and increased the net number of the transcripts in the development of anther in 1376-CIMS, which were consistent with previous work that genome demethylation causes increased the rates of transcription (Zilberman et al. 2007). Since methylation-demethylation switches can control regulated gene expression, our results also indicated that extent and pattern of gene expression and DNA methylation alteration took place during anther development induced by SQ-1. Therefore, we infer that the variation in the extent of methylation and characterization of molecular epigenetic events such as demethylation and hypermethylation in male sterility lines relative to its fertile line may help explain the phenotypic variation/deviation observed in 1376-CIMS as compared to 1376, and thereby may help in understanding the relationship between DNA methylation and CIMS.
Sequences affected by SQ-1 in 1376-CIMS
Despite just part of the sequences showing changes in gene expression or methylation were sequenced, the results provided valuable information about types of sequences affected in 1376-CIMS, In 1376-CIMS, the spectrum of genes whose expression affected by CHA was broad. And the DNA methylation changes were found in various types of sequences including retrotransposons, functional genes, and non-coding DNA. In any case, the sequencing results indicated CHA might affect changes in gene expression and DNA methylation in genome-wide scale.
Approximately 8.70 % of the sequenced fragments that underwent methylation changes in 1376-CIMS showed similarity to retrotransposons. This result indicated that male sterility is accompanied by frequent epigenetically modification of retrotransposons. Retrotransposons can generate mutations by inserting within or near functional genes, and these elements may provide regulatory sequences for gene expression and then alter the expression of the adjacent genes (Fedoroff 2000). All these data suggested that retrotransposons might have played an important role in anther development. Besides, our results also indicated some differentially methylated DNA fragments including ethylene receptor and lipid transfer protein could play an important role in anther development (Supplementary Table 2). It was also confirmed that the abnormal expression of ethylene receptor genes could induce stable male sterility in Nicotiana tabacum (Ishimaru et al. 2006). As is well-known that synthesis of lipidic components in anthers, including the pollen exine, is essential for plant male reproductive development and that plant lipid transfer proteins (LTPs) are small, abundant lipid-binding proteins have the ability to exchange lipids between membranes. OsC6, encoding a LTPs and positively regulated by a basic helix-loop-helix transcription factor, plays a crucial role in the development of lipidic orbicules and pollen exine during anther development, and silencing of OsC6 could reduce pollen fertility in rice (Zhang et al. 2010a).
As is well-known, in the development of pollen, meiosis in the anthers yields haploid unicellular microspores, subsequently, pollen mitosis I (PM I) yields a larger vegetative cell and a smaller generative cell (GC), and the GC undergoes PM II, a symmetric division that yields two sperm cells. In this process, our data showed that the patterns of gene expression and DNA methylation alterations induced by SQ-1 can occur. And, the results also demonstrated that the rapid and numerous changes in gene expression and DNA methylation of the anthers observed here may be programmed responses to SQ-1, and may be disadvantageous for formation and development of the pollen. To promote a better understanding of the male sterility induced by CHA, an important question needed to be clarified is whether the observed epigenetic and gene expression changes in CIMS are stochastic or non-random (or part of the changes are stochastic and part of the changes are non-random). Answer to this question should come from analyzing a large amount of mutational analysis, which is a powerful approach to identify structural and regulatory components that participate in a particular developmental pathway.
Author contribution
Conceived and designed the experiments: QS Ba, GS Zhang. Performed the experiments: QS Ba JS Wang. Analyzed the data: QS Ba. Wrote the paper: QS Ba GS Zhang. Provided experimental material: N Niu, SC Ma, JW Wang.
References
Adugna A, Nanda GS, Singh K, Bains NS (2004) A comparison of cytoplasmic and chemically-induced male sterility systems for hybrid seed production in wheat (Triticum aestivum L.). Euphytica 135:297–304
Aubry C, Paven MCML, Chateigner-Boutin AL, Teulat-Merah B, Ricoult C, Peltier D, Jalouzot R, Limami AM (2003) A gene encoding a germin-like protein, identified by a cDNA-AFLP approach, is specifically expressed during germination of Phaseolus vulgaris. Planta 217:466–475
Bachem CWB, van der Hoeven RS, de Bruijn SM, Vreugdenhil D, Zabeau M, Visser RGF (1996) Visualization of differential gene expression using a novel method of RNA fingerprinting based on AFLP: Analysis of gene expression during potato tuber development. Plant J 9:745–753
Baisakh N, Subudhi PK, Parami NP (2006) cDNA-AFLP analysis reveals differential gene expression in response to salt stress in a halophyte Spartina alterniflora Loisel. Plant Sci 170:1141–1149
Baurens FC, Causse S, Legavre T (2008) Methylation-sensitive amplification polymorphism (MSAP) protocol to assess CpG and CpNpG methylation in the banana genome. Fruits 63:117–123
Cantu D, Vanzetti LS, Sumner A, Dubcovsky M, Matvienko M, Distelfeld A, Michelmore RW, Dubcovsky J (2010) Small RNAs, DNA methylation and transposable elements in wheat. BMC Genomics 11:408
Chakraborty K, Devakumar C (2006) Evaluation of chemical compounds for induction of male sterility in wheat (Triticum aestivum L.). Euphytica 147:329–335
Chen XQ, Ma Y, Chen F, Song WQ, Zhang L (2009) Analysis of DNA methylation patterns of PLBs derived from Cymbidium hybridium based on MSAP. Plant Cell Tiss Org 98:67–77
Ding J, Shen J, Mao H, Xie W, Li X, Zhang Q (2012a) RNA-directed DNA methylation is involved in regulating photoperiod-sensitive male sterility in rice. Mol Plant 6:1210–1216
Ding JH, Lu Q, Ouyang YD, Mao HL, Zhang PB, Yao JL, Xu CG, Li XH, Xiao JH, Zhang QF (2012b) A long noncoding RNA regulates photoperiod-sensitive male sterility, an essential component of hybrid rice. Proc Natl Acad Sci USA 109:2654–2659
Durrant WE, Rowland O, Piedras P, Hammond-Kosack KE, Jones JDG (2000) cDNA-AFLP reveals a striking overlap in race-specific resistance and wound response gene expression profiles. Plant Cell 12:963–977
Fedoroff N (2000) Transposons and genome evolution in plants. Proc Natl Acad Sci USA 97:7002–7007
Finnegan EJ, Peacock WJ, Dennis ES (1996) Reduced DNA methylation in Arabidopsis thaliana results in abnormal plant development. Proc Natl Acad Sci USA 93:8449–8454
Forino LMC, Andreucci AC, Del Tredici I, Felici C, Giraldi E, Tagliasacchi AM (2003) DNA methylation of tapetum cells during microsporogenesis in Malus domestica Borkh. Israel J Plant Sci 51:91–100
Garcia-Aguilar M, Michaud C, Leblanc O, Grimanelli D (2010) Inactivation of a DNA methylation pathway in maize reproductive organs results in apomixis-like phenotypes. Plant Cell 22:3249–3267
Hanai LR, Floh EIS, Fungaro MHP, Santa-Catarina C, de Paula FM, Viana AM, Vieira MLC (2010) Methylation patterns revealed by MSAP profiling in genetically stable somatic embryogenic cultures of Ocotea catharinensis (Lauraceae). In Vitro Cell Dev Pl 46:368–377
Honys D, Twell D (2004) Transcriptome analysis of haploid male gametophyte development in Arabidopsis. Genome Biol 5(11):R85
Hsu TW, Tsai WC, Wang DP, Lin S, Hsiao YY, Chen WH, Chen HH (2008) Differential gene expression analysis by cDNA-AFLP between flower buds of Phalaenopsis Hsiang Fei cv. H. F. and its somaclonal variant. Plant Sci 175:415–422
Ingelbrecht I, Vanhoudt H, Vanmontagu M, Depicker A (1994) Posttranscriptional silencing of reporter transgenes in tobacco correlates with DNA methylation. Proc Natl Acad Sci USA 91:10502–10506
Ishimaru K, Takada K, Watanabe S, Kamada H, Ezura H (2006) Stable male sterility induced by the expression of mutated melon ethylene receptor genes in Nicotiana tabacum. Plant Sci 171:355–359
Klindworth DL, Williams ND, Maan SS (2002) Chromosomal location of genetic male sterility genes in four mutants of hexaploid wheat. Crop Sci 42:1447–1450
Levterova V, Panaiotov S, Brankova N, Tankova K (2010) Typing of genetic markers involved in stress response by fluorescent cDNA-amplified fragment length polymorphism technique. Mol Biotechnol 45:34–38
Liu HL, Cui P, Zhan KH, Lin Q, Zhuo GY, Guo XL, Ding F, Yang WL, Liu DC, Hu SN, Yu J, Zhang AM (2011) Comparative analysis of mitochondrial genomes between a wheat K-type cytoplasmic male sterility (CMS) line and its maintainer line. BMC Genomics 12:163–177
Liu BS, Sun QX, Sun LZ, Gao QR, Xie CJ, Dou BD, Ni ZF, Wei YL, Zhang YC (2002) RAPD and ISSR markers of fertility restoring gene for Aegilops kotschyi cytoplasmic male sterility in wheat. Acta Bot Sin 44:446–450
Lu YL, Liu YX, Wang J, Cao MJ, Rong TZ (2010) Variation and patterns of DNA methylation in maize C-type CMS lines and their maintainers. J Plant Biochem Biotechnol 19:43–50
Mondego JMC, Simoes-Araujo JL, de Oliveira DE, Alves-Ferreira M (2003) A gene similar to bacterial translocase I identified by cDNA-AFLP is expressed during flower bud development of Arabidopsis thaliana. Plant Sci 164:323–331
Murray MG, Thompson WF (1980) Rapid isolation of high molecular weight plant DNA. Nucleic Acids Res 8:4321–4325
Paris R, Dondini L, Zannini G, Bastia D, Marasco E, Gualdi V, Rizzi V, Piffanelli P, Mantovani V, Tartarini S (2012) dHPLC efficiency for semi-automated cDNA-AFLP analyses and fragment collection in the apple scab-resistance gene model. Planta 235:1065–1080
Parodi PC, Gaju MD (2009) Male sterility induced by the chemical hybridizing agent clofencet on wheat, Triticum aestivum and T. turgidum var. durum. Cienc Investig Agrar 36:267–276
Pickett AA, Galwey NW (1997) A further evaluation of hybrid wheat. Plant Var Seeds 10:15–32
Portis E, Acquadro A, Comino C, Lanteri S (2004) Analysis of DNA methylation during germination of pepper (Capsicum annuum L.) seeds using methylation-sensitive amplification polymorphism (MSAP). Plant Sci 166:169–178
Rao NVPRG, Mahajan V, Nagarajan S (2000) Effect of chemical hybridizing agent on floret opening and seed set in wheat (Triticum aestivum). Indian J Agric Sci 70:689–690
Ronemus MJ, Galbiati M, Ticknor C, Chen JC, Dellaporta SL (1996) Demethylation-induced developmental pleiotropy in Arabidopsis. Science 273:654–657
Sasakuma T, Ahmed TA, Tsujimoto H (2001) QTL analysis of fertility-restoration against cytoplasmic male sterility in wheat. Genes Genet Syst 76:33–38
Saze H, Scheid OM, Paszkowski J (2003) Maintenance of CpG methylation is essential for epigenetic inheritance during plant gametogenesis. Nat Genet 34:65–69
Shan XH, Li YD, Liu XM, Wu Y, Zhang MZ, Guo WL, Liu B, Yuan YP (2012) Comparative analyses of genetic/epigenetic diversities and structures in a wild barley species (Hordeum brevisubulatum) using MSAP, SSAP and AFLP. Genet Mol Res 11:2749–2759
Singh SK, Chatrath R, Mishra B (2010) Perspective of hybrid wheat research: a review. Indian J Agric Sci 80:1013–1027
Song YP, Ma KF, Bo WH, Zhang ZY, Zhang DQ (2012) Sex-specific DNA methylation and gene expression in andromonoecious poplar. Plant Cell Rep 31:1393–1405
Takeda S, Paszkowski J (2006) DNA methylation and epigenetic inheritance during plant gametogenesis. Chromosoma 115:27–35
Wang L, Zhou BR, Wu LL, Guo BZ, Jiang TB (2011) Differentially expressed genes in Populus simonii × Populus nigra in response to NaCl stress using cDNA-AFLP. Plant Sci 180:796–801
Wu C, Sayos J, Nguyen K, Wang N, Coyle A, Biron C, Terhorst C (2000) Genomic organization and characterization of msap: the murine homologue of human XLP gene. FASEB J 14:926
Xiong LZ, Xu CG, Maroof MAS, Zhang QF (1999) Patterns of cytosine methylation in an elite rice hybrid and its parental lines, detected by a methylation-sensitive amplification polymorphism technique. Mol Gen Genet 261:439–446
Yan HH, Kikuchi S, Neumann P, Zhang WL, Wu YF, Chen F, Jiang JM (2010) Genome-wide mapping of cytosine methylation revealed dynamic DNA methylation patterns associated with genes and centromeres in rice. Plant J 63:353–365
Zhang AM, Nie XL, Liu DC, Guo XL (2001) Advances of hybrid wheat breeding in China. Cereal Res Commun 29:343–350
Zhang GS, Chen RH, Ye JX, Wang JS, Niu N, Ma SC, Zhao JX, Zhu JC (2009) Differential proteomic analysis of anther proteins between cytoplasmic-nuclear male sterility line and its maintainer in wheat (Triticum aestivum L.). Progress Biochem Biophys 36:431–440
Zhang DS, Liang WQ, Yin CS, Zong J, Gu FW, Zhang DB (2010a) OsC6, encoding a lipid transfer protein, is required for postmeiotic anther development in rice. Plant Physiol 154:149–162
Zhang GS, Chen RH, Liu W, Ye JX (2010b) Mitochondrial proteomic analysis of cytoplasmic male sterility line and its maintainer in wheat (Triticum aestivum L.). Agric Sci China 9:771–782
Zhang ZH, Chen H, Huang XH, Xia R, Zhao QZ, Lai JB, Teng KL, Li Y, Liang LM, Du QS, Zhou XP, Guo HS, Xie Q (2011) BSCTVC2 attenuates the degradation of SAMDC1 to suppress DNA methylation-mediated gene silencing in Arabidopsis. Plant Cell 23:273–288
Zhou H, Liu QJ, Li J, Jiang DG, Zhou LY, Wu P, Lu S, Li F, Zhu LY, Liu ZL, Chen LT, Liu YG, Zhuang CX (2012) Photoperiod- and thermo-sensitive genic male sterility in rice are caused by a point mutation in a novel noncoding RNA that produces a small RNA. Cell Res 22:649–660
Zilberman D, Gehring M, Tran RK, Ballinger T, Henikoff S (2007) Genome-wide analysis of Arabidopsis thaliana DNA methylation uncovers an interdependence between methylation and transcription. Nat Genet 39:61–69
Acknowledgments
The research was supported by National High Technology Research and Development Program of China (Nos. 2011AA10A106), National Natural Science Foundation of China (Nos. 31071477, 31171611, 31371697), The Key Scientific and Technological Innovation Special Projects of Shaanxi “13115” (No. 2010ZDKG-68, 2011KTZB02-01-01).
Author information
Authors and Affiliations
Corresponding author
Additional information
Communicated by T. Moriguchi.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article
Cite this article
Ba, Q., Zhang, G., Wang, J. et al. Gene expression and DNA methylation alterations in chemically induced male sterility anthers in wheat (Triticum aestivum L.). Acta Physiol Plant 36, 503–512 (2014). https://doi.org/10.1007/s11738-013-1431-6
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11738-013-1431-6