
Abstract
Phylogeographic perspectives incorporating multiple classes of characters, especially those relating to sexual signals, are promising for the elucidation of recent evolutionary mechanisms driving speciation. Here, forest frogs were used as a model system to access distinct stages in the process of evolutionary differentiation. We studied 280 individuals assigned to three species: Allobates paleovarzensis, A. nidicola and A. masniger. Samples were collected at 20 localities arranged in two study systems, along the middle Amazon and the lower Madeira Rivers, in Central Amazonia. Mantel tests, analyses of molecular variance, and the spatial distribution of haplogroups indicated that the distribution of genetic variability, as inferred from a mitochondrial DNA marker, was determined by a combination of isolation-by-distance effects and the transposition of large Amazonian rivers. These two factors had contrasting relative influences in each of the study systems, which also differed regarding the estimated time of the major cladogenetic events. Pronounced population genetic structure was observed. However, multivariate discriminant function analyses revealed that the phenotypic (morphological and acoustic) divergence was loosely related with genetic differentiation and did not successfully predict assignment of individuals to genetic groups. The observed distribution of genetic variability showed the important role of genetic drift in the diversification of the mitochondrial marker studied. The phenotypic conservatism among populations was surprising in view of the high genetic structuring observed, and indicates a prevailing role of stabilizing selective forces in the process of sexual signal and morphological differentiation.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Historically isolated populations are premier candidates for the study of the origin of differences in genetic and phenotypic characters because they would have been potentially exposed to long term genetic drift or divergent selection pressures, without the homogenizing influences of gene flow (Avise 2000; Nosil et al. 2009). Therefore, it is expected that spatially structured populations, which experience restricted demographic connectivity, might show concordant divergences among different sets of characters. Nevertheless, decoupled patterns of population differentiation can be observed. This indicates that either stabilizing or directional selection pressures acting differentially on specific characteristics may lead to distinct patterns of the distribution of variation among populations and have important implications for the speciation process (Santos et al. 2006; Adams et al. 2009; Lemmon 2009).
Recent theoretical and analytical advances enabled by the integration of distinct disciplines have led to a better understanding of patterns and mechanisms of differentiation among populations of diverse organisms (Crisci et al. 2003; Santini et al. 2012). Nonetheless, the relationship between genotypic and phenotypic differentiation in the early stages of speciation remains poorly understood, directing research to questions regarding ecological and historical mechanisms that promote or maintain variation at this level of the evolutionary process (Diniz-Filho et al. 2008). Important advances may be obtained by investigating the geographic pattern in which genetic variability is distributed among populations, as well as the order according to which ecological and phenotypic differentiation appears (West-Eberhard 1989; Dall 1997; Panhuis et al. 2001).
The extent and shape of the spatial structure of intraspecific genealogical relationships are typically unknown for natural populations, especially in megadiverse tropical regions, where not even the species boundaries are clearly defined (e.g., Groeneveld et al. 2009; Vieites et al. 2009; Hubert et al. 2012). Beyond the challenge of delimiting species, a better comprehension of the speciation process requires the study of geographical factors which may underpin the origin and diversification of lineages within species, such as vicariance and isolation by distance (Hutchison and Templeton 1999; Zink et al. 2000). In Amazonia, species distribution patterns (e.g., Wallace 1852; Haffer 1969; Ayres and Clutton-Brock 1992) and fine-scale genealogical structure assessments (e.g., Cohn-Haft 2000; Funk et al. 2007; Solomon et al. 2008; Amézquita et al. 2009) have inferred the effect of both vicariance and isolation by distance in terrestrial organisms. Nevertheless, the relative contribution of each of them varied greatly, perhaps due to idiosyncratic particularities of the study systems. To date, no clear geographical pattern or palaeoenvironmental force has emerged as a general explanation for the diversification of most of the animal groups (Antonelli et al. 2010; Ribas et al. 2011).
Phylogeographic perspectives involving more than one class of character are particularly promising for the elucidation of recent evolutionary mechanisms driving differentiation, given that the history inferred from DNA sequences may provide a historical demographic context to evaluate divergence in various phenotypic attributes (Wiens 2008; Campbell et al. 2010; Guillot et al. 2012). These appraisals might be conducted with any kind of genetically based phenotypic marker, not necessarily of molecular origin (Avise 2004). This is because many phenotypic attributes carry phylogenetic and even phylogeographic signals (Wycherley et al. 2002; Erdtmann and Amézquita 2009; Goicochea et al. 2009) that might, together with gene trees, reciprocally illuminate the history of populations and the mechanisms responsible for evolutionary divergence. Integrative, multicharacter approaches are particularly important for our knowledge of tropical organisms, which urgently need detection of significant evolutionary units for conservation, assessment of cryptic diversity, and determination of species limits (Moritz et al. 2000; Dayrat 2005; Wiens 2007; Crawford et al. 2010).
Phylogeographic structure is commonly observed among species of frogs, and is often credited to their low vagility when compared to other vertebrates (Avise 2000; Vences and Wake 2007). Frogs are suitable for population-level multi-character evolutionary approaches because of their high abundance, which allows dense sampling along their distributional ranges, and their conspicuous advertisement calls, which can be used as population markers (Wycherley et al. 2002; Lougheed et al. 2006). Given that anurans use advertisement calls for sexual recognition and mate choice, differences in these signals dramatically increase the evolutionary potential for speciation due to the possibility of pre-zygotic isolation (Panhuis et al. 2001; Pröhl et al. 2006; Boul et al. 2007).
Allobates (Anura, Dendrobatoidea, Aromobatidae) frogs were used here as a model system to access distinct stages of the evolutionary differentiation process through the examination of morphological, acoustic and genetic characters. We studied individuals assigned to three nominal species: Allobates paleovarzensis Lima et al. (2010), A. nidicola (Caldwell and Lima 2003) and A. masniger (Morales 2002). Sampling was performed at 20 localities arranged in two distinct geographic study systems. The choice of these systems was based on studies of phenotypic variability that suggested that these populations may have experienced initial, but distinct stages in the process of allopatric speciation (Kaefer and Lima 2012; Kaefer et al. 2012; Tsuji-Nishikido et al. 2012). This is the first population genetic assessment of the three aforementioned species. This study focuses on the use of gene trees and phenotypic data in order to disentangle the most recent events that led to the spatial organization of the multi-trait variation currently observed.
We expected that a neutral, stochastic process plays a central role in the emergence of evolutionary differentiation which may lead to the formation of new species. Thus, we tested the premise that the distribution of genetic variability is determined by a combination of the effects of isolation by distance (i.e., geographic distance among individual samples) and the transposition of vicariant barriers (Amazonian rivers). Our null hypothesis was that the phenotypic differentiation is related to the genetic pattern observed. Therefore, we predicted that phenotypic divergence would be correlated with genetic differentiation and not incongruent with the groupings indicated by molecular analyses. In order to test this hypothesis, we accessed the genealogical relationship among individual samples through phylogeographic analyses, including parameters related to historical demography inferred from the non-coding 16S gene, which is presumed to meet the assumption of selective neutrality (Hafner and Upham 2011). We also evaluated the extent of population differentiation in advertisement calls and morphology by their correlation with geographic and genetic distances, as well as the roles of both acoustic and morphological traits in predicting the assignment of individuals to genetic groups.
Methods
Study Area
This study was conducted in two adjacent areas of the Central Amazon lowlands, in the Brazilian states of Amazonas and Pará, on a region characterized by wide tracts of tropical rainforests. These areas are crossed by the two major rivers of the biome: the Amazon, at its middle course (Fig. 1a), and the Madeira, at its lower portion (Fig. 2a). The Madeira River is the main southern tributary of the Amazon, which represents the largest river system on Earth. The white, sediment-rich water channels of these rivers are characterized by intense sedimentary dynamics in the Central Amazon, permitting the formation of marginal lakes, islands and channels and by extensive floodplains covered with seasonally flooded forests (várzeas) on its riverbanks (Sioli 1984; Irion and Kalliola 2010). The region also has extensive plateaus harboring non-flooded rainforest (terra firme), as well as transitional forests, called palaeo-várzeas, that are ancient floodplains of the Amazon River and its tributaries (Irion and Kalliola 2010).

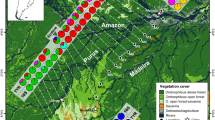
Geographic location of sampling sites (a) and haplotype network for Allobates paleovarzensis (b). The network was built from 142 16S rDNA sequences. The size and color of each ellipse indicate the frequency and geographic origin of the individuals bearing that haplotype. White dots and transverse bars represent not sampled (missing) intermediate haplotypes. Sample localities are numbered according to Table 1

Geographic location of sampling sites (a) and haplotype network for Allobates nidicola and A. masniger (b). The network was built from 134 16S rDNA sequences. The size and color of each ellipse indicate the frequency and geographic origin of the individuals bearing that haplotype. White dots and transverse bars represent not sampled (missing) intermediate haplotypes. Sample localities are numbered according to Table 1
Studied Species
The three sampled species pertain to the Allobates trilineatus group (sensu Morales 2002). Published phylogenetic reconstructions include only A. paleovarzensis (named as “SãoFrancisco”) and A. nidicola (Grant et al. 2006; Santos et al. 2009). According to these studies, A. paleovarzensis is not closely related to A. nidicola within the group. Nevertheless, A. nidicola and A. masniger constitute sister, reciprocally monophyletic mitochondrial clades in relation to other species within the genus (Kaefer et al. in press). Here we study these two species jointly, given that (1) there are no diagnostic phenotypic characters that can distinguish A. nidicola from A. masniger, raising questions regarding the validity of the two taxa (Tsuji-Nishikido et al. 2012); (2) even if both are valid species, they are more closely related to one another than with any other within the genus, which makes them an evolutionary unit that enables the conduction of phylogeographic analyses. This integrated sample design permits addressing the objectives presented in this paper, including the appraisal of the Madeira River as a barrier to gene flow. We call the set of populations of A. paleovarzensis as paleovarzensis system, while the populations of A. nidicola and A. masniger are referred to as nidicola–masniger system.
The Paleovarzensis System
The Amazonian palaeo-várzea forests constitute the habitat for Allobates paleovarzensis, a diurnal aromobatid frog which deposits its eggs in the forest floor and whose tadpole development must be completed in water bodies. This is a recently described species with a distribution that encompasses its type and adjacent localities south of the Amazon River, and also localities to the north, reaching the municipality of Barcelos, at the western margin of the Negro River (Kaefer and Lima 2012). Our sampling design includes all known areas of occurrence of this species, including its type locality (Fig. 1a).
The Nidicola-Masniger System
Allobates nidicola and Allobates masniger are diurnal leaf-litter frogs that occur in terra firme forests of Central Amazonia, having been described without reference to each other. Recently, it was found that these species share all the available diagnostic characters, including the direct, terrestrial development of endotrophic larvae into froglets (Tsuji-Nishikido et al. 2012). Nevertheless, this study detected significant differences in morphology and calls between populations from both type localities and from opposite sides of the lower Madeira River, the main biogeographic barrier in the studied area. This report also suggests that they constitute allopatrically distributed sister species. In order to better understand the potential speciation process occurring in this study system, we analyze these two species jointly, according to the sample design presented in Tsuji-Nishikido et al. (2012) (Fig. 2a).
Data Collection
Data were collected at 20 sites (10 per study system), which will be referred to as populations hereafter. The fieldwork procedures employed for the acquisition of the phenotypic data analyzed here are fully described in recent studies which evaluated the taxonomic identity and the variation in phenotypic characters of individuals belonging to both study systems (Kaefer and Lima 2012; Kaefer et al. 2012; Tsuji-Nishikido et al. 2012). The collection of specimens constituted the first assessment of the taxonomic identity of individuals through the distributional ranges of the three species involved. Therefore, the present study did not involve new collections, in addition to those described in these previous publications. The number of individuals analyzed for each data set, and the geographic coordinates of the sample sites, are provided in Table 1. Muscular tissue samples from each individual were dissected out and preserved in 95 percent ethanol prior to fixation of voucher specimens in formol. Tissue samples were housed at the Coleção de Tecidos de Genética Animal of the Universidade Federal do Amazonas (CTGA–UFAM), Manaus, Brazil. Studied individuals were deposited in the Herpetological Collection of the Instituto Nacional de Pesquisas da Amazônia, in Manaus (INPA-H).
Acoustic Data
Call parameters from individuals of the paleovarzensis (10 calls/individual) and nidicola–masniger (20 calls/individual) systems were obtained from recordings with Raven Pro 1.3 (Charif et al. 2008) and Raven 1.2 (Charif et al. 2004), respectively. Temporal and spectral call traits analysed for each species are described on Online Resource 1 and Online Resource 2. These measurements were averaged in order to represent each of the populations in the statistical analyses. The call structure of each of the species, including graphic representations, and the detailed description of call traits analyzed are provided elsewhere (Kaefer and Lima 2012; Kaefer et al. 2012; Tsuji-Nishikido et al. 2012). Summary statistics of the call measurements are given on Online Resource 1 and Online Resource 2.
Morphological Data
External morphometric measurements (in mm) were taken from the left side of the specimens, with a stereoscopic microscope with a graduated ocular lens (precision 0.10 mm) and a digital caliper (to the nearest 0.01 mm). All individuals were analyzed according to the twenty-three measurements presented and described in detail in Online Resource 3. Among these measurements, snout-vent length (SVL) was used exclusively as a covariate in analyses of phenotypic differentiation. Descriptive statistics for the morphometric measurements in each sampling locality are given in Online Resource 3 and Online Resource 4.
Molecular Data
Genomic DNA was extracted using a standard cetyl trimethyl ammonium bromide (CTAB) protocol. For the amplification of 16S ribosomal RNA gene, the primers 16Sar and 16Sbr were used according to Palumbi (1996). This gene is the most commonly used marker for amphibian systematics (Fouquet et al. 2007), including studies on the superfamily Dendrobatoidea (Vences et al. 2000; Grant et al. 2006; Santos et al. 2009). It shows high rates of amplification success (Vences et al. 2005), having satisfactory performance at the intraspecific level, as well as among closely related species (Lougheed et al. 2006; Vences et al. 2005; Simões 2010).
Polymerase Chain Reaction (PCR) amplifications were performed in 15 μL reaction volumes containing 5.8 μL ddH2O, 1.5 μL of 25 mM MgCl2, 1.5 μL of 10× amplification buffer (75 mM Tris HCl, 50 mM KCl, 20 mM (NH4)2SO4), 1.5 μL of a 2 μM solution of each primer, 1.5 μL of 10 mM dNTPs (2.5 mM each dNTP), 0.7 μL of Taq DNA polymerase 2.5 U/μL (Biotools, Spain) and 1 μL of template DNA (about 40 ng/μL). The temperature profile consisted of (1) preheating at 90 °C for 60 s, (2) denaturation at 92 °C for 60 s, (3) primer annealing at 50 °C for 50 s, (4) extension at 72 °C for 90 s, and (5) a final extension at 72 °C for 5 min. Steps 2–4 were repeated 35 times. Amplicons were purified with exonuclease and alkaline phosphatase (Fermentas Life Sciences, Canada), and sequenced using forward primers and ABI BigDye Terminator Cycle Sequencing Kit protocols, as indicated by the manufacturer. The resulting single-stranded products were read in an ABI 3130xl automatic capillary sequencer. The DNA sequences had their homologous regions aligned in BioEdit (Hall 1999), where a preliminary alignment was generated by the ClustalW algorithm (Thompson et al. 1994), and were finally checked and corrected by eye in comparison with the original chromatograms. The final alignments were 527 bp long for Allobates paleovarzensis, and 539 bp long for A. nidicola and A. masniger. Newly determined sequences were deposited in GenBank (Online Resource 5).
Data Analysis
Population Analyses
The genealogical relationships among individual samples and populations were estimated through haplotype networks using statistical parsimony (Templeton et al. 1992) in TCS 1.21 (Clement et al. 2000). Gaps were considered as 5th character state, and the connection limit was fixed at 23 steps to allow association among all sampled haplotypes. We estimated the relative partitioning of genetic variation (within populations, among populations, and between riversides) through standard molecular analysis of variance (AMOVA; Excoffier et al. 1992), in Arlequin version 3.5 (Excoffier and Lischer 2010). The relative genetic structuring among sampling localities was estimated through the calculation of distance-based fixation indices (F ST; Wright 1951), equivalent to N ST of Lynch and Crease (1990). The statistical significance of F ST values was obtained after 1,023 haplotype permutations in Arlequin. Genetic differentiation among sampling localities was estimated via Kimura 2-parameter corrected genetic distances (Kimura 1980) in MEGA version 5 (Tamura et al. 2011).
The most likely number of genetic clusters formed by the sampled mitochondrial DNA sequences was inferred through Bayesian analysis of population structure, in BAPS version 5 (Corander et al. 2008). Based on nucleotide frequencies, the model attempts to create k groups of individuals, such that those allocated in the same group resemble each other genetically as much as possible. The upper limit to the number of clusters was set from one to the number of sample localities on each study system. The log-likelihood values of the best models were used to select the most probable cluster arrangement.
Within each of the genetic clusters inferred by the Bayesian approach, we calculated haplotype and nucleotide diversities, as well as the number of segregating sites, and performed the neutrality tests Tajima’s D, Fu’s Fs, and Ramos-Onsis & Rozas’ R2 (Tajima 1989; Fu 1997; Ramos–Onsins and Rozas 2002). The statistical significance of these tests was estimated through coalescent simulations with 10,000 replicates in DnaSP 5.10 (Librado and Rozas 2009). In addition to the neutrality tests, past demographic expansion events were inferred from the distribution of the observed pairwise nucleotide site differences (mismatch distributions) in each cluster, as calculated in DnaSP 5.10. We used coalescent simulation procedures in order to test the validity of the estimated expansion models. The parameters of these models were re-estimated in order to obtain their empirical confidence intervals, and the empirical distribution of the sum of squared deviations between the observed and the expected mismatch values (SSD; Schneider and Excoffier 1999) and the raggedness index (Hri; Harpending 1994). These calculations were obtained through 10,000 parametric bootstrap replicates in Arlequin.
Divergence Time Estimation
Divergence times were estimated using BEAST 1.6.2 (Bayesian Evolutionary Analysis Sampling Trees; Drummond and Rambaut 2007). We analyzed only unique haplotypes recovered from the population analyses conducted in Arlequin. An available 16S sequence of Allobates talamancae (GenBank AY843577) was considered as an external group for both systems. In addition, 16S sequences of A. trilineatus (GenBank DQ502118) and A. brunneus (GenBank EU342522) were used as outgroups for the paleovarzensis and nidicola–masniger systems, respectively. As priors (calibration points), we used average means and confidence intervals of the most recent common ancestors of both the external groups and the studied systems obtained from Santos et al. (2009). Models of nucleotide substitution were estimated for the reduced mitochondrial DNA datasets in MEGA version 5 (Tamura et al. 2011). For the two study systems, we applied an uncorrelated lognormal relaxed clock model, a lognormal prior distribution, a randomly generated starting tree, and length of chain of 10 million generations with samples taken every 10 thousand generations, and discarding 10 % of the trees as burn-in, resulting in 900 sampled trees in the Markov Chain Monte Carlo (MCMC). The stationarity of the posterior distributions for all model parameters, including medians and 95 % Highest Posterior Density intervals (HPD) of the nodes was checked on Tracer 1.5 (Rambaut and Drummond 2007). From the MCMC output, we generated the final consensus tree using Tree Annotator 1.6.2 (Drummond and Rambaut 2007).
Phenotypic Differentiation
In order to test whether the genetic clusters assigned via Bayesian analysis of population structure were phenotypically distinguishable, we used discriminant function analyses (DFA) with acoustic and morphometric traits as predictor variables. The discriminant function was used to calculate the probability of classification of each individual collected in its genetic cluster (grouping variable) by a Jackknifed classification matrix. This approach included all DNA-sequenced individuals with available measurements regarding acoustic and/or morphometric traits. Acoustic data were both temperature and temperature-SVL adjusted through linear regression. In order to account for the effect of body size on morphometric measurements, we used 12 morphological ratios as predictor variables (Verdade and Rodrigues 2007): LL/SVL, HAND3/SVL, FL/SVL, HW/HL, EN/HL, EL/HL, TYM/HL, TYM/EL, IN/HW, HAND1/HAND2, HAND2/HAND3, HAND4/HAND1 (see Online Resource 3 for acronyms).
Geographic Correlates of Phenotypic and Genetic Variation
We tested correlations between the linear geographic distances (measured in km) between sampling sites and genetic and phenotypic distances between the populations of each of the study systems by applying Mantel tests on distance matrices derived from genetic, acoustic and morphological data sets (Mantel 1967). In addition, we conducted partial Mantel tests to evaluate correlations between genetic/phenotypic distances among populations of each study system and their separation by the river channel while controlling for effects of geographic distance between sampling sites (Smouse et al. 1986; Telles et al. 2001). We constructed a binary correspondence matrix, designating the values “0” and “1” for localities within the same and between opposite sides of the river, respectively.
Average genetic distances (16S) between sampling localities were calculated according to the Kimura 2-parameter (K2P) model in MEGA version 5. Acoustic distance matrices were obtained from the dataset of temperature-adjusted call measurements by calculating pairwise Euclidean distances between all possible pairs of populations by using the scores of the mean acoustic measurements on first and second components (which together explained 79.0 % of the total acoustic variation in A. paleovarzensis and 79.3 % in A. nidicola/masniger) produced by a principal components analysis (PCA). The principal components were obtained from the arithmetic means of the call traits among all sampled individuals for each of the localities, and were used in order to reduce the number of independent phenotypic variables. Morphological distance matrices were generated from the external measurements (except SVL) through the same procedure. The first two components explained 64.3 % of the total morphological variation in A. paleovarzensis and 71.1 % in A. nidicola/masniger. As body size was correlated with the scores of the first principal component of both acoustic (linear regression r 2 = 0.51, F = 9.39, p = 0.017 in A. paleovarzensis, and r 2 = 0.72, F = 23.78, p = 0.001 in A. nidicola/masniger) and morphological data sets (linear regression r 2 = 0.83, F = 43.65, p < 0.001 in A. paleovarzensis, and r 2 = 0.95, F = 174.09, p < 0.001 in A. nidicola/masniger), we regressed them against corresponding mean SVL measurements for each population, and used the residuals as new, size-independent acoustic variables from which new Euclidean distances were calculated. Body size adjustments are useful for controlling possible ontogenetic effects that might affect the outcome of comparative results. However, they can mask important call differentiation related to body size dissimilarities among populations and species, which were reported in the studied systems (Kaefer and Lima 2012; Kaefer et al. 2012). Therefore, we also calculated acoustic distance matrices without applying SVL corrections. Mantel tests were conducted in ZT (Bonnet and Van de Peer 2002) using permutation of the null models (Anderson and Legendre 1999), and applying 10,000 randomizations.
Results
Population Analyses
We obtained 276 16S rDNA sequences (Table 1), corresponding to 55 different haplotypes (Online Resource 5). The distribution of haplotypes among the sampled localities and the large number of mutations separating the genealogical groups showed a pronounced genetic structure among populations, with restricted haplotype sharing across riverbanks in A. paleovarzensis, while absent in the nidicola–masniger system (Figs. 1b, 2b). The AMOVA indicated that a small fraction of the overall genetic variation can be found within sampling localities, being concentrated among populations in the paleovarzensis system (60.37 %) and between opposite riverbanks in the nidicola–masniger system (59.74 %) (Table 2). The F ST values obtained for both study systems revealed an overall high and significant population structuring (Table 3). Genetic distances were more pronounced between populations of the nidicola–masniger system (reaching 5.2 %) than those observed in A. paleovarzensis (reaching 1.7 %), thus lending further support to the interspecific nature of the former.
Bayesian analysis of population structure supported the partition of the individuals of A. paleovarzensis into four genetic clusters (log ML value = −525.13; probability = 0.99), while seven groups were estimated for the nidicola–masniger system (log ML value = −1182.18; probability = 1.00). Co-occurrence of genetic clusters within sampling localities was nearly absent. The distribution of sampling localities exerted a clear effect on the determination of the genetic groupings, especially in A. nidicola and A. masniger, where six populations corresponded to completely segregated clusters (Fig. 3). The three neutrality tests indicated historical demographic changes in two clusters composed by individuals from almost the entire range of A. paleovarzensis. In contrast, in the nidicola–masniger system the cluster composed by the locality Road to Apuí was the only one to show significant departures from historical demographic equilibrium according to the three neutrality tests, including the R2, which is the most appropriate for small samples (Ramos–Onsins and Rozas 2002) (Table 4). The tests of demographic growth based on the sum of squared deviation (SSD) between observed and expected mismatch distributions, and Harpending’s raggedness index (Hri) did not reject the demographic expansion hypothesis, giving support for the significant values observed in the neutrality tests (Table 5, Online Resource 6).

Barplot from the Bayesian Analysis of Population Structure. a 142 individual 16S rDNA sequences of Allobates paleovarzensis from ten sampling localities. b 134 individual 16S rDNA sequences of Allobates nidicola and A. masniger from other ten sampling localities. Distinct colors represent each estimated genetic cluster. Individuals are displayed according to sampling localities, which are numbered as on Table 1
Divergence Time Estimation
The Bayesian evolutionary analysis indicated the time of the first divergence between the clades of A. paleovarzensis occupying the north and south sides of the middle Amazon River to be on Pleistocene (median = 1.24 million years (mya); HPD = 0.54, 2.23). The median time of the most recent common ancestor for the species was estimated at 2.29 mya (HPD = 1.23, 3.81), and most of the intraspecific cladogenetic events in A. paleovarzensis are indicated as having occurred during the last 1 million years (Fig. 4a). The split of the nidicola–masniger system between the left and right sides of the lower Madeira River and the time of the most recent common ancestor for this study system did not have occurred after Early Pliocene (HPD = 6.88, 15.33; median = 10.65 mya), with the subsequent cladogenetic events evenly distributed from the time of this divergence to the present (Fig. 4b).

Timetree of the a paleovarzensis and b nidicola–masniger systems based on fragments of the 16S rDNA gene. Only unique haplotypes were included. Age estimates and confidence intervals (horizontal bars) of the divergences were obtained via BEAST (Bayesian Evolutionary Analysis Sampling Trees). Posterior probabilities of the older nodes are presented. Terminals were colored according to their localities of origin, which are depicted in Figs. 1 and 2. Black and white terminals are outgroups. The black and grey vertical bars indicate the origin of each haplotype according to the riverside
Phenotypic Differentiation
Phenotypic traits showed wide intrapopulational ranges of variation, with values overlapping extensively among populations (see Online Resources 1–4). Discriminant function analyses using genetic clusters as grouping variables was able to correctly classify only 50.0 % of the individuals of Allobates paleovarzensis according to temperature-adjusted acoustic characters, and 43.0 % of the individuals with both temperature and body size-adjusted traits. Based on morphological characters, the percentage of correct membership assignment of A. paleovarzensis was even poorer: 38.0 % (Online Resource 7). Individuals of A. nidicola and A. masniger were more successfully classified than those of A. paleovarzensis in their respective genetic clusters based on acoustic characters adjusted for temperature (59.0 %), and both temperature and body size (48.0 %). Morphological characters also performed worse than acoustic traits within the nidicola–masniger system (41.0 % of individuals correctly classified; Online Resource 8).
Geographic Correlates of Phenotypic and Genetic Variation
The Mantel tests showed a significantly positive correlation between geographic and genetic distances in both study systems, indicating a strong role of stochastic processes in the process of evolutionary diversification of the studied molecular marker. In contrast, correlations between phenotypic (acoustic and morphological) and geographic distances were not significant (Table 6). We did not observe signal of the Amazon River as a barrier promoting differentiation in A. paleovarzensis in genetic or phenotypic distances. On the other hand, the Partial Mantel tests revealed a role of the lower Madeira River as a vicariant barrier on the nidicola–masniger system. Significant correlations were found between side of river and both overall genetic differentiation and uncorrected acoustic distances.
Discussion
Pronounced Population Genetic Structure
The high levels of genetic structure at the mitochondrial level found among populations, especially in A. nidicola and A. masniger, are notable even considering earlier reports on this subject, which indicate that panmictic scenarios are the exception among amphibian populations (Crawford 2003; Zeisset and Beebee 2008). Studies on other Amazonian species in similar and even wider geographic scales revealed lower levels of interpopulational genetic structure (e.g., Lougheed et al. 1999; Noonan and Gaucher 2006; Elmer et al. 2007; Simões 2010). The main life history characteristics pointed out to promote and maintain high genetic structure levels in anurans, such as small body size and strong site attachment (Vences and Wake 2007) are widely reported among Allobates (Pröhl 2005; Kaefer et al. in press). In addition, the studied species are reported to be territorial (Lima et al. 2010; Caldwell and Lima 2003), and to exhibit direct larval development in the nidicola–masniger system (Tsuji-Nishikido et al. 2012). Given that water bodies are believed to constitute the main extraterritorial resource for dendrobatoid species (Pröhl 2005), the absence of tadpole transport behavior may further restrict the use of space of adult frogs, as well as concentrate offspring close to parental individuals, contributing to the limited gene flow among lineages. The effects of this presumed high phylopatry in A. nidicola and A. masniger may also be observed from the results of the analysis of molecular variance, where a low within-population molecular variability was observed for this study system (only 2.02 % of the total variation).
The configuration of genetic clusters recovered from the Bayesian analyses showed significant genetic structuring over short geographic distances, resembling the pattern observed in the haplotype networks. The star-shaped network of A. paleovarzensis, together with the results of the neutrality tests, indicated that this species experienced recent demographic changes across most of its range. Such patterns were less pronounced in the molecular data of A. nidicola and A. masniger. Although the population growth tests did not reject the population expansion hypothesis, the highly structured haplotype network and the non-significant results in neutrality tests in most of the genetic clusters did not support past demographic expansion events within A. nidicola and A. masniger. A mixed result in neutrality tests was also observed through the range of A. femoralis along the Madeira River, and might be related to demographic expansion of determinate populations which experienced dispersal events, probably allowed by historical river course vagility (Simões 2010).
Spatio-Temporal Contexts of the Inferred Vicariant Events
Both study systems exhibited signatures in their genetic structures that are congruent with the transposition of large Amazonian rivers. The effectiveness of the lower Madeira River as a barrier to gene flow in Allobates was observed to be higher than that of the middle Amazon River. This observation is supported by the (1) absence of haplotype sharing among sides of the Madeira River (which was restricted to different banks of the Amazon); (2) accountability for most (60.37 %) of the genetic variability in A. nidicola and A. masniger (the river barrier was the second major factor explaining molecular variation within A. paleovarzensis); (3) effect on overall variability of acoustic characters (significant differences between interfluves were restricted to determinate call traits in A. paleovarzensis) (Kaefer and Lima 2012); (4) Existence of reciprocally monophyletic clades on opposite sides of the Madeira river (were paraphyletic between sides of the Amazon). This last characteristic satisfies even the most stringent predictions of the classic river barrier hypothesis (Gascon et al. 1996, 1998), indicating the completeness of the speciation process in the nidicola–masniger system according to both the evolutionary (sensu Wiley 1978) and phylogenetic (Rosen 1978) species concepts. Under neutrality, the usual chronology of evolutionary events leading towards speciation begins with the stage of polyphyly in a gene tree, with successive paraphyly stages before the eventual achievement of the status of reciprocal monophyly (de Queiroz 2007). According to this model, the monophyly observed in the gene tree of the nidicola–masniger system is expected to arise from old geographic barriers, in which diverging populations have experienced the time necessary to achieve such status. Nevertheless, non-monophyly (both paraphyly and polyphyly) can be observed in many of the currently recognized species (Funk and Omland 2003; McKay and Zink 2009).
Idiosyncratic characteristics of the sampling design and the history of the studied species can be related the differential roles of the rivers as barriers that have been reported (Colwell 2000). In this study, these system-specific characteristics may include: (1) Different sampling designs: while the effect of the Madeira River as a barrier was accessed in its lower portion, where it is believed to play a significant role as barrier to gene flow (Simões 2010), A. paleovarzensis occurs along the middle section of the Amazon River, where it was observed to exert intermediate effects as a vicariant barrier (Hayes and Sewlal 2004). (2) Distinct study models: although belonging to the same genus, aspects of the natural history of the studied species, such as dispersal abilities, might be responsible for distinct microevolutionary arrangements (see Discussion on genetic structure above). (3) Asynchronous divergence times: besides the large confidence intervals estimated by the Bayesian analyses, the median dates for the split of clades inhabiting opposite sides of the river in both study systems indicates that these events took place in different geological epochs. The cladogenic event separating haplotypes of A. paleovarzensis inhabiting the north and south of the Amazon River probably occurred during the Pleistocene, being younger that the divergence time estimated between A. nidicola and A. masniger, which might have not occurred after the early Pliocene. According to these findings, we infer that A. paleovarzensis might not have undergone the level and duration of genetic isolation required for population differentiation to cross the threshold necessary to met the criteria of speciation under monophyly- and phenotype-based species concepts (Coyne and Orr 2004).
Our timetree suggests that the lineages of A. paleovarzensis occupying opposite riverbanks of the middle Amazon River diverged during the late phase of its formation, when its alluvial fan experienced an expressive increase in sedimentation rates and the river had high vagility in the course of its drainage (Rossetti et al. 2005; Figueiredo et al. 2009). Therefore, although the Amazon River initiated as a transcontinental watercourse between 11.8 and 11.3 Mya ago, we believe that sedimentation-induced channel course dynamics, which are evidenced by Quaternary palaeochannels and meander relicts (Costa et al. 2001), allowed dispersal events across its middle section. The late Miocene split between A. nidicola and A. masniger at the portion of the Madeira River where it meets the Amazon seems to match the onset of the latter as a channel running from the Andes to the Atlantic Ocean (Hoorn 1994; Figueiredo et al. 2009). The estimated age of this evolutionary split is in accordance with studies that indicated that most of the speciation events in Amazonia largely predated the Pleistocene (Hoorn et al. 2010; Ribas et al. 2011). For amphibians, the origin of most of the current Amazonian species is estimated to have occurred during late Miocene (Santos et al. 2009; Funk et al. 2012; this study), with estimates reaching the late Oligocene (Elmer et al. 2007; Fouquet et al. 2012b).
The present appraisal of the effects of rivers as barriers must be interpreted cautiously as they represent byproducts of a multi-character approach conducted in species with restricted distributions. Investigation on this subject should involve species (or species groups) that can be found throughout the course of a river to be assessed, thus allowing for a multiple paired sampling design (e.g., Silva and Patton 1998; Simões 2010). In addition, nuclear markers should be considered in future genetic studies in order to access their potential in disentangle chronological aspects on the history of the studied populations (Zhang and Hewitt 2003). In spite of the limitations of our data to provide accurate details on the temporal and spatial mechanisms by which these vicariant barriers operate, the results presented here and elsewhere (Kaefer and Lima 2012; Kaefer et al. 2012; Tsuji-Nishikido et al. 2012) support the notion that the Amazon and Madeira Rivers do play a role in the distribution of the genetic and phenotypic variability of the studied systems. Our conclusions add to a growing body of studies that supported the effectiveness of the Amazon (e.g., Capparella 1988; Hayes and Sewlal 2004), Madeira (e.g., Cohn-Haft 2000; Simões et al. 2008; Fernandes et al. 2012) and other Amazonian rivers (e.g., Funk et al. 2007; Ribas et al. 2011; Fouquet et al. 2012a) as vicariant barriers promoting and maintaining evolutionary diversity. In this context, the various studies conducted at the Juruá River, another southern tributary of the Amazon, which rejected the river barrier hypothesis (e.g., Patton et al. 1994; Gascon et al. 1996, 1998, 2000; Lougheed et al. 1999) seem to reflect the ineffectiveness of this particular meandering river as a barrier to gene flow rather than reject the river barrier hypothesis for the entire biome.
Stochastic and Selective Forces in Evolutionary Differentiation
In addition to the genetic discontinuities associated with the transposition of the large Amazonian rivers, results from both study systems suggested that a large part of their genetic variation is correlated with geographic distance. This was more evident in A. paleovarzensis, for which Mantel tests indicated a pronounced correlation between genetic and geographic distances together with a non-significant role of the river barrier in genetic differentiation. Distance effects have been reported from phylogeographic studies involving Amazonian frogs (Funk et al. 2007; Amézquita et al. 2009; Simões 2010) and suggest an important role for isolation by distance and genetic drift on their evolutionary diversification. Divergent sexual selection was observed to occur in Amazonian frog species where genetic divergence and reduced gene flow were associated with differential female preferences (Boul et al. 2007; Guerra and Ron 2008). Therefore, a combination of stochastic and selective forces seems to have contributed to the richness of frog lineages observed in Amazonia.
The acoustic and morphological traits analyzed in this study did not respond to linear geographic distance among sampling localities, as observed with molecular characters. In addition, the discriminant function analyses revealed that a large fraction of the individuals could not be correctly classified in their respective genetic clusters according to their phenotypes. This misclassification may be attributed to a single marker-effect, given that mitochondrial genealogies may exhibit patterns of variation different that those obtained from the analysis of nuclear markers (e.g., Sequeira et al. 2011; Turmelle et al. 2011, but see Creer et al. 2004; Zink and Barrowclough 2008). Nonetheless, the significant correlations between our mitochondrial data and the main geographic barriers of the study areas (river and distance) suggest that the 16S marker was sensitive to the recent history of connectedness among these populations. Therefore, it is reasonable to explore other explanations to the relationship between the evolution of genetic and phenotypic characters.
The results of this study, together with the observation that there is low among-population variability in phenotypic characters (Kaefer and Lima 2012; Kaefer et al. 2012), indicate a prevailing role of stabilizing selective forces in phenotypic differentiation. In this case, evolutionary constraints may be preventing segregation in morphology and calls among populations. Additionally, the time elapsed since the allopatric condition may not have been sufficient for the accumulation of significant differences.
Supporting the first explanation is the observation that well diverged lineages of frogs generally exhibit low phenotypic differentiation (Wiens 2008; Cherry et al. 1978), and that the functional importance of these characters imposes evolutionary constraints (Lougheed et al. 2006). This evolutionary conservatism, in which diversification can occur with little phenotypic change (Adams et al. 2009) might be related, for example, to the historical failure of researchers in providing reasonable estimates of species richness for many anuran groups based on the classic morphological and biological species concepts (Camargo et al. 2006; Fouquet et al. 2007; Angulo and Reichle 2008; Funk et al. 2012). In both studied systems, bioacoustic characters did not echo fine-scale phylogeographic structure, supporting the notion that mate-recognition signals are subject to severe constraining or diversifying pressures and therefore are not closely related to geographic distance (Gerhardt and Huber 2002). In fact, behavioral traits are expected to be relatively evolutionarily malleable (Blomberg et al. 2003). However, exceptions to this pattern were reported (Ryan et al. 1996; Wycherley et al. 2002; Pröhl et al. 2007; Amézquita et al. 2009), depicting the large array of evolutionary forces underlying geographic divergence, as well as the idiosyncratic nature of their prevalence.
In favour of the second interpretation is the observation that the only the more differentiated system, composed by A. nidicola and A. masniger, exhibited significant differences in summarized acoustic parameters between riversides. It suggests that the diversifying effects of genetic drift assumed prominence in ancient, isolated lineages simply because of the accumulation of mutations over a longer time span. This interpretation also assumes that, under non-selective evolutionary forces, neutrally evolving genotypic characters diverge first, and that more time is needed for phenotypic differences to arise.
Conclusions
This study indicated that the distribution of genetic variability in three species of neotropical frogs is determined by a combination of the effects of isolation-by-distance and the transposition of large Amazonian rivers acting as vicariant barriers. Thus, we confirm our expectation that genetic drift plays a central role in the emergence of evolutionary differentiation that may lead to the formation of new species. The taxonomic validity of the three studied species is corroborated by our results according to both the evolutionary and phylogenetic species concepts. In addition, the current phenotypic divergence was loosely related with genetic differentiation and did not successfully predict assignment of individuals to genetic groups. These results argue against the expectation that the phenotypic differentiation is related to the genetic groupings that compose the study systems, and are surprising when considering the high levels of genetic structuring observed. Together with recent studies conducted on these model organisms (Kaefer and Lima 2012; Kaefer et al. 2012; Tsuji-Nishikido et al. 2012), we suggest a prevailing role of stabilizing selective forces in phenotypic differentiation. In this case, evolutionary constraints, which have no effect on neutrally evolving molecular markers, may be preventing phenotypic differentiation.
References
Adams, D. C., Berns, C. M., Kozak, K. H., & Wiens, J. (2009). Are rates of species diversification correlated with rates of morphological evolution? Proceedings of the Royal Society B, 276, 2729–2738.
Amézquita, A., Lima, A. P., Jehle, R., Castellanos, L., Ramos, O., Crawford, A. J., et al. (2009). Calls, colours, shapes, and genes: A multi–trait approach to the study of geographic variation in the Amazonian frog Allobates femoralis. Biological Journal of the Linnean Society, 98, 826–838.
Anderson, M. J., & Legendre, P. (1999). An empirical comparison of permutation methods for tests of partial regression coefficients in a linear model. Journal of Statistical Computation and Simulation, 62, 271–303.
Angulo, A., & Reichle, S. (2008). Acoustic signals, species diagnosis, and species concepts: The case of a new cryptic species of Leptodactylus (Amphibia, Anura, Leptodactylidae) from the Chapare region, Bolivia. Zoological Journal of the Linnean Society, 152, 58–77.
Antonelli, A., Quijada-Mascareñas, A., Crawford, A. J., Bates, J. M., Velazco, P. M., & Wüster, W. (2010). Molecular studies and phylogeography of Amazonian tetrapods and their relation to geological and climatic models. In C. Hoorn & F. P. Wesselingh (Eds.), Amazonia, landscape and species evolution (pp. 386–403). Oxford: Blackwell Publishing.
Avise, J. C. (2000). Phylogeography: The history and formation of species. Cambridge: Harvard University Press.
Avise, J. C. (2004). Molecular markers, natural history and evolution (2nd ed.). Sunderland: Sinauer Associates Inc.
Ayres, J. M., & Clutton-Brock, T. H. (1992). River boundaries and species range size in Amazonian primates. The American Naturalist, 140, 531–537.
Blomberg, S. P., Garland, T., Jr, & Ives, A. R. (2003). Testing for phylogenetic signal in comparative data: Behavioral traits are more labile. Evolution, 57, 717–745.
Bonnet, E., & Van de Peer, Y. (2002). ZT: A software tool for simple and partial Mantel tests. Journal of Statistical Software, 7, 1–12.
Boul, K. E., Funk, W. C., Darst, C. R., Cannatella, D. C., & Ryan, M. J. (2007). Sexual selection drives speciation in an Amazonian frog. Proceedings of the Royal Society B, 274, 399–406.
Caldwell, J. P., & Lima, A. P. (2003). A new Amazonian species of Colostethus (Anura: Dendrobatidae) with a nidicolous tadpole. Herpetologica, 59, 219–234.
Camargo, A., de Sá, R., & Heyer, W. R. (2006). Phylogenetic analyses of mtDNA sequences reveal three cryptic lineages in the widespread neotropical frog Leptodactylus fuscus (Schneider, 1799) (Anura, Leptodactylidae). Biological Journal of the Linnean Society, 87, 325–341.
Campbell, P., Pasch, B., Pino, J. L., Crino, O. L., Phillips, M., & Phelps, S. M. (2010). Geographic variation in the songs of neotropical singing mice: Testing the relative importance of drift and local adaptation. Evolution, 64, 1955–1972.
Capparella, A. P. (1988). Genetic variation in Neotropical birds: Implications for the speciation process. In H. Ouellet (Ed.), Acta XIX Congressus Internationalis Ornithologici (pp. 1658–1664). Ottawa: University of Ottawa Press.
Charif, R. A., Clark, C. W., & Fristrup, K. M. (2004). Raven 1.2 User’s manual. Ithaca: Cornell Laboratory of Ornithology.
Charif, R. A., Waack, A. M., & Strickman, L. M. (2008). Raven Pro 1.3 User’s manual. Ithaca: Cornell Laboratory of Ornithology.
Cherry, L. M., Case, S. M., & Wilson, A. C. (1978). Frog perspective on the morphological difference between humans and chimpanzees. Science, 200, 209–211.
Clement, M., Posada, D., & Crandall, K. A. (2000). TCS: A computer program to estimate gene genealogies. Molecular Ecology, 9, 1657–1659.
Cohn-Haft, M. (2000). A case study in amazonian biogeography: Vocal and DNA sequence variation in Hemitriccus flycatchers. PhD thesis. Luisiana State University.
Colwell, R. K. (2000). A barrier runs through it… or maybe just a river. Proceedings of the National Academy of Sciences, 97, 13470–13472.
Corander, J., Marttinen, P., Sirén, J., & Tang, J. (2008). Enhanced Bayesian modelling in BAPS software for learning genetic structures of populations. BMC Bioinformatics, 9, 539.
Costa, J. B. S., Bemerguy, R. L., Hasui, Y., & Borges, M. S. (2001). Tectonics and paleogeography along the Amazon River. Journal of South American Earth Sciences, 14, 335–347.
Coyne, J. A., & Orr, H. A. (2004). Speciation. Sunderland: Sinauer Associates Inc.
Crawford, A. J. (2003). Huge populations and old species of Costa Rican and Panamanian dirt frogs inferred from mitochondrial and nuclear gene sequences. Molecular Ecology, 12, 2525–2540.
Crawford, A. J., Lips, K. R., & Bermingham, E. (2010). Epidemic disease decimates amphibian abundance, species diversity, and evolutionary history in the highlands of central Panama. Proceedings of the National Academy of Sciences, 107, 13777–13782.
Creer, S., Thorpe, R. S., Malhotra, A., Chou, W. H., & Stenson, A. G. (2004). The utility of AFLPs for supporting mitochondrial DNA phylogeographical analyses in the Taiwanese bamboo viper, Trimeresurus stejnegeri. Journal of Evolutionary Biology, 17, 100–107.
Crisci, J. V., Katinas, L., & Posadas, P. (2003). Historical biogeography: An introduction. Cambridge: Harvard University Press.
Dall, S. R. X. (1997). Behaviour and speciation. Trends in Ecology & Evolution, 12, 209–210.
Dayrat, B. (2005). Towards integrative taxonomy. Biological Journal of the Linnean Society, 85, 407–415.
de Queiroz, K. (2007). Species concepts and species delineation. Systematic Biology, 56, 879–886.
Diniz-Filho, J. A. F., Telles, M. P. C., Bonatto, S. L., Eizirik, E., Freitas, T. R. O., de Marco, P., et al. (2008). Mapping the evolutionary twilight zone: Molecular markers, populations and geography. Journal of Biogeography, 35, 753–763.
Drummond, A. J., & Rambaut, A. (2007). BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evolutionary Biology, 7, 214.
Elmer, K. R., Dávila, J. A., & Lougheed, S. C. (2007). Cryptic diversity and deep divergence in an upper Amazonian leaflitter frog, Eleutherodactylus ockendeni. BMC Evolutionary Biology, 7, 247.
Erdtmann, L., & Amézquita, A. (2009). Differential evolution of advertisement call traits in Dart–Poison Frogs (Anura: Dendrobatidae). Ethology, 115, 801–811.
Excoffier, L., & Lischer, H. E. L. (2010). Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows. Molecular Ecology Research, 10, 564–567.
Excoffier, L., Smouse, P., & Quattro, J. (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics, 131, 479–491.
Fernandes, A. M., Wink, M., & Aleixo, A. (2012). Phylogeography of the chestnut tailed antbird (Myrmeciza hemimelaena) clarifies the role of rivers in Amazonian biogeography. Journal of Biogeography, 39, 1524–1535.
Figueiredo, J., Hoorn, C., van der Ven, P., & Soares, E. (2009). Late Miocene onset of the Amazon River and the Amazon deep-sea fan: Evidence from the Foz do Amazonas Basin. Geology, 37, 619–622.
Fouquet, A., Gilles, A., Vences, M., Marty, C., Blanc, M., & Gemmell, N. J. (2007). Underestimation of species richness in Neotropical frogs revealed by mtDNA analyses. PLoS ONE, 10, e1109.
Fouquet, A., Noonan, B. P., Rodrigues, M. T., Pech, N., Gilles, A., & Gemmell, N. J. (2012a). Multiple quaternary Refugia in the Eastern Guiana shield revealed by comparative phylogeography of 12 frog species. Systematic Biology, 61, 461–489.
Fouquet, A., Recoder, R., Teixeira, M., Jr, Cassimiro, J., Amaro, R. C., Camacho, A., et al. (2012b). Molecular phylogeny and morphometric analyses reveal deep divergence between Amazonia and Atlantic Forest species of Dendrophryniscus. Molecular Phylogenetics and Evolution, 62, 826–838.
Fu, Y. X. (1997). Statistical tests of neutrality of mutations against population growth, hitchhiking and background selection. Genetics, 147, 915–925.
Funk, W. C., Caldwell, J. P., Peden, C. E., Padial, J. M., de la Riva, I., & Cannatella, D. C. (2007). Tests of biogeographic hypotheses for diversification in the Amazonian forest frog, Physalaemus petersi. Molecular Phylogenetics and Evolution, 44, 825–837.
Funk, W. C., Caminer, M., & Ron, S. R. (2012). High levels of cryptic species diversity uncovered in Amazonian frogs. Proceedings of the Royal Society B, 279, 1806–1814.
Funk, D. J., & Omland, K. E. (2003). Species-level paraphyly and polyphyly: frequency, causes, and consequences, with insights from animal mitochondrial DNA. Annual Review of Ecology Evolution and Systematics, 34, 397–423.
Gascon, C., Lougheed, S. C., & Bogart, J. P. (1996). Genetic and morphological variation in Vanzolinius discodactylus: a direct test of the riverine barrier hypothesis. Biotropica, 28, 376–387.
Gascon, C., Lougheed, S. C., & Bogart, J. P. (1998). Patterns of genetic population differentiation in four species of Amazonian frogs: A test of the Riverine Barrier Hypothesis. Biotropica, 30, 104–119.
Gascon, C., Malcolm, J. R., Patton, J. L., Silva, M. N. F., Bogart, J. P., Lougheed, S. C., et al. (2000). Riverine barriers and the geographic distribution of Amazonian species. Proceedings of the National Academy of Sciences, 97, 13672–13677.
Gerhardt, H. C., & Huber, F. (2002). Acoustic communication in insects and anurans: Common problems and diverse solutions. Chicago: University of Chicago Press.
Goicochea, N., De La Riva, I., & Padial, J. M. (2009). Recovering phylogenetic signal from frog mating calls. Zoologica Scripta, 39, 141–154.
Grant, T., Frost, D. R., Caldwell, J. P., Gagliardo, R., Haddad, C. F. B., Kok, P. J. R., et al. (2006). Phylogenetic systematics of dart–poison frogs and their relatives (Anura: Athesphatanura: Dendrobatidae). Bulletin of the American Museum of Natural History, 299, 1–262.
Groeneveld, L. F., Weisrock, D. W., Rasoloarison, R. M., Yoder, A. D., & Kappeler, P. M. (2009). Species delimitation in lemurs: Multiple genetic loci reveal low levels of species diversity in the genus Cheirogaleus. BMC Evolutionary Biology, 9, 30.
Guerra, M. A., & Ron, S. R. (2008). Mate choice and courtship signal differentiation promotes speciation in an Amazonian frog. Behavioral Ecology, 19, 1128–1135.
Guillot, G., Renaud, S., Ledevin, R., Michaux, J., & Claude, J. (2012). A unifying model for the analysis of phenotypic, genetic and geographic data. Systematic Biology,. doi:10.1093/sysbio/sys038.
Haffer, J. (1969). Speciation in Amazonian Forest Birds. Science, 165, 131–137.
Hafner, J. C., & Upham, N. S. (2011). Phylogeography of the dark kangaroo mouse, Microdipodops megacephalus: cryptic lineages and dispersal routes in North America’s Great Basin. Journal of Biogeography, 38, 1077–1097.
Hall, T. A. (1999). BioEdit: A user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symposium Series, 41, 95–98.
Harpending, R. C. (1994). Signature of ancient population growth in a low–resolution mitochondrial DNA mismatch distribution. Human Biology, 66, 591–600.
Hayes, F. E., & Sewlal, J. N. (2004). The Amazon River as a dispersal barrier to passerine birds: Effects of river width, habitat and taxonomy. Journal of Biogeography, 31, 1809–1818.
Hoorn, C. (1994). An environmental reconstruction of the palaeo-Amazon River system (Middle-Late Miocene, NW Amazonia). Palaeogeography, Palaeoclimatology, Palaeoecology, 112, 187–238.
Hoorn, C., Wesselingh, F. P., ter Steege, H., Bermudez, M. A., Mora, A., Sevink, J., et al. (2010). Amazonia through time: Andean uplift, climate change, landscape evolution, and biodiversity. Science, 330, 927–931.
Hubert, N., Meyer, C. P., Bruggemann, H. J., Guérin, F., Komeno, R. J. L., Espiau, B., et al. (2012). Cryptic diversity in Indo–Pacific coral–reef fishes revealed by DNA–barcoding provides new support to the centre–of–overlap hypothesis. PLoS ONE, 7, e28987.
Hutchison, D. W., & Templeton, A. R. (1999). Correlation of pairwise genetic and geographic distance measures: Inferring the relative influences of gene flow and drift on the distribution of genetic variability. Evolution, 53, 1898–1914.
Irion, G., & Kalliola, R. (2010). Long–term landscape development processes in Amazonia. In C. Hoorn & F. P. Wesselingh (Eds.), Amazonia, landscape and species evolution (pp. 185–197). Oxford: Blackwell Publishing.
Kaefer, I. L., & Lima, A. P. (2012). Sexual signals of the Amazonian frog Allobates paleovarzensis: Geographic variation and stereotypy of acoustic traits. Behaviour, 149, 15–33.
Kaefer, I. L., Montanarin, A., Costa, R. S., & Lima, A. P. (in press). Temporal patterns of reproductive activity and site attachment of the Brilliant–thighed Frog Allobates femoralis from Central Amazonia. Journal of Herpetology.
Kaefer, I. L., Tsuji-Nishikido, B. M., & Lima, A. P. (2012). Beyond the river: underlying determinants of population acoustic signal variability in Amazonian direct–developing Allobates (Anura: Dendrobatoidea). Acta Ethologica, 15, 187–194.
Kimura, M. (1980). A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. Journal of Molecular Evolution, 16, 111–120.
Lemmon, E. M. (2009). Diversification of conspecific signals in sympatry: Geographic overlap drives multidimensional reproductive character displacement in frogs. Evolution, 63, 1155–1170.
Librado, P., & Rozas, J. (2009). DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics, 25, 1451–1452.
Lima, A. P., Caldwell, J. P., Biavati, G., & Montanarin, A. (2010). A new species of Allobates (Anura: Aromobatidae) from paleovárzea forest in Amazonas, Brazil. Zootaxa, 2337, 1–17.
Lougheed, S. C., Austin, J. D., Bogart, J. P., Boag, P. T., & Chek, A. A. (2006). Multi–character perspectives on the evolution of intraspecific differentiation in a neotropical hylid frog. BMC Evolutionary Biology, 6, 23.
Lougheed, S. C., Gascon, C., Jones, D. A., Bogart, J. P., & Boag, P. T. (1999). Ridges and rivers: a test of competing hypothesis of Amazonian diversification using a dart–poison frog (Epipedobates femoralis). Proceedings of the Royal Society B, 266, 1829–1835.
Lynch, M., & Crease, T. J. (1990). The analysis of population survey data on DNA sequence variation. Molecular Biology and Evolution, 7, 377–394.
Mantel, N. (1967). The detection of disease clustering and a generalized regression approach. Cancer Research, 27, 209–220.
McKay, B. D., & Zink, R. M. (2009). The causes of mitochondrial DNA gene tree paraphyly in birds. Molecuar Phylogenetics and Evolution, 54, 647–650.
Morales, V. R. (2002). Sistematica y biogeografía del grupo trilineatus (Amphibia, Anura, Dendrobatidae, Colostethus), con descripción de once nuevas especies. Publicación de la Asociación Amigos Doñana, 13, 1–59.
Moritz, C., Patton, J. L., Schneider, C. J., & Smith, T. B. (2000). Diversification of rainforest faunas: An integrated molecular approach. Annual Review of Ecology and Systematics, 31, 533–563.
Noonan, B. P., & Gaucher, P. (2006). Refugial isolation and secondary contact in the dyeing poison frog Dendrobates tinctorius. Molecular Ecology, 15, 4425–4435.
Nosil, P., Harmon, L. J., & Seehausen, O. (2009). Ecological explanations for (incomplete) speciation. Trends in Ecology & Evolution, 24, 145–156.
Palumbi, S. R. (1996). Nucleic acids II: the polymerase chain reaction. In D. M. Hillis, C. Moritz, & B. K. Mable (Eds.), Molecular systematics (pp. 205–247). Sunderland: Sinauer Associates Inc.
Panhuis, T. M., Butlin, R., Zuk, M., & Tregenza, T. (2001). Sexual selection and speciation. Trends in Ecology & Evolution, 16, 364–371.
Patton, J. L., Silva, M. N. F., & Malcolm, J. R. (1994). Gene genealogy and differentiation among arboreal spiny rats (Rodentia: Echymidae) of the Amazon Basin: a test of the riverine barrier hypothesis. Evolution, 48, 1314–1323.
Pröhl, H. (2005). Territorial behavior in dendrobatid frogs. Journal of Herpetology, 39, 354–365.
Pröhl, H., Hagemann, S., Karsch, J., & Höbel, G. (2007). Geographic variation in male sexual signals in Strawberry Poison Frogs (Dendrobates pumilio). Ethology, 113, 825–837.
Pröhl, H., Koshy, R. A., Mueller, U. G., Rand, A. S., & Ryan, M. J. (2006). Geographic variation of genetic and behavioral traits in northern and southern túngara frogs. Evolution, 60, 1669–1679.
Rambaut, A., & Drummond, A. J. (2007). Tracer v. 1.5. [http://beast.bio.ed.ac.uk/Tracer]. Accessed 04 May 2012.
Ramos–Onsins, S. E., & Rozas, J. (2002). Statistical properties of new neutrality tests against population growth. Molecular Biology and Evolution, 19, 2092–2100.
Ribas, C. C., Aleixo, A., Nogueira, A. C. R., Miyaki, C. Y., & Cracraft, J. (2011). A palaeobiogeographic model for biotic diversification within Amazonia over the past three million years. Proceedings of the Royal Society B, 279, 681–689.
Rosen, D. E. (1978). Vicariant patterns and historical explanation in biogeography. Systematic Zoology, 27, 159–188.
Rossetti, D. F., de Toledo, P. M., & Góes, A. M. (2005). New geological framework for Western Amazonia (Brazil) and implications for biogeography and evolution. Quaternary Research, 63, 78–89.
Ryan, M. J., Rand, A. S., & Weigt, L. A. (1996). Allozyme and advertisement call variation in the túngara frog, Physalaemus pustulosus. Evolution, 50, 2435–2453.
Santini, F., Miglietta, M. P., & Faucci, A. (2012). Where are we now? An introduction to a special issue on speciation. Evolutionary Biology, 39, 141–147.
Santos, J. C., Coloma, L. A., Summers, K., Caldwell, J. P., Ree, R., & Cannatella, D. C. (2009). Amazonian amphibian diversity is primarily derived from Late Miocene Andean lineages. PLoS Biology, 7, e1000056.
Santos, S., Hrbek, T., Farias, I. P., Schneider, H., & Sampaio, I. (2006). Population genetic structuring of the king weakfish, Macrodon ancylodon (Sciaenidae), in Atlantic coastal waters of South America: Deep genetic divergence without morphological change. Molecular Ecology, 15, 4361–4373.
Schneider, S., & Excoffier, L. (1999). Estimation of demographic parameters from the distribution of pairwise differences when the mutation rates vary among sites: Application to human mitochondrial DNA. Genetics, 152, 1079–1089.
Sequeira, F., Sodré, D., Ferrand, N., Bernardi, J., Sampaio, I., Schneider, H., et al. (2011). Hybridization and massive mtDNA unidirectional introgression between the closely related Neotropical toads Rhinella marina and R. schneideri inferred from mtDNA and nuclear markers. BMC Evolutionary Biology, 11, 264.
Silva, M. N. F., & Patton, J. L. (1998). Molecular phylogeography and the evolution and conservation of mammals. Molecular Ecology, 7, 475–486.
Simões, P. I. (2010). Diversificação do complexo Allobates femoralis (Anura, Dendrobatidae) em florestas da Amazônia brasileira: desvendando padrões atuais e históricos. PhD thesis. Manaus: Instituto Nacional de Pesquisas da Amazônia.
Simões, P. I., Lima, A. P., Magnusson, W. E., Hödl, W., & Amézquita, A. (2008). Acoustic and morphological differentiation in the frog Allobates femoralis: Relationships with the Upper Madeira River and other potential geological barriers. Biotropica, 40, 607–614.
Sioli, H. (1984). The Amazon: Limnology and landscape ecology of a mighty tropical river and its basin. Dordrecht: Dr W. Junk Publisher.
Smouse, P. E., Long, J. C., & Sokal, R. R. (1986). Multiple regression and correlation extensions of the Mantel test of matrix correspondence. Systematic Zoology, 35, 727–732.
Solomon, S. E., Bacci, M., Martins, J., Vinha, G. G., & Mueller, U. G. (2008). Paleodistributions and comparative molecular phylogeography of leafcutter ants (Atta spp.) provide new insight into the origins of Amazonian diversity. PLoS ONE, 3, e2738.
Tajima, F. (1989). Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics, 123, 585–595.
Tamura, K., Peterson, D., Peterson, N., Stecher, G., Nei, M., & Kumar, S. (2011). MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Molecular Biology and Evolution, 28, 2731–2739.
Telles, M. P. C., Silva, R. S. M., Chaves, L. J., Coelho, A. S. G., & Diniz–Filho, J. A. F. (2001). Divergência entre subpopulações de cagaiteira (Eugenia dysenterica) em resposta a padrões edáficos e distribuição espacial. Pesquisa Agropecuária Brasileira, 36, 1387–1394.
Templeton, A. R., Crandall, K. A., & Sing, C. F. (1992). A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram estimation. Genetics, 132, 619–633.
Thompson, J. D., Higgins, D. G., & Gibson, T. J. (1994). Improved sensitivity of profile searches through the use of sequence weights and gap excision. Computer Applications in the Biosciences, 10, 19–29.
Tsuji-Nishikido, B. M., Kaefer, I. L., Freitas, F. C., Menin, M., & Lima, A. P. (2012). Significant but not diagnostic: Differentiation through morphology and calls in the Amazonian frogs Allobates nidicola and A. masniger. Herpetological Journal, 22, 105–114.
Turmelle, A. S., Kunz, T. H., & Sorenson, M. D. (2011). A tale of two genomes: contrasting patterns of phylogeographic structure in a widely distributed bat. Molecular Ecology, 20, 357–375.
Vences, M., Kosuch, J., Lötters, S., Widmer, A., Jungfer, K., Köhler, J., et al. (2000). Phylogeny and classification of Poison Frogs (Amphibia: Dendrobatidae), based on mitochondrial 16S and 12S ribosomal RNA gene sequences. Molecular Phylogenetics and Evolution, 15, 34–40.
Vences, M., Thomas, M., Meijden, A., Chiari, Y., & Vieites, D. R. (2005). Comparative performance of the 16S rRNA gene in DNA barcoding of amphibians. Frontiers in Zoology, 2, 5.
Vences, M., & Wake, D. B. (2007). Speciation, species boundaries and phylogeography of amphibians. In H. H. Heatwole & M. Tyler (Eds.), Amphibian biology. Volume 6: Systematics (pp. 2613–2669). Chipping Norton: Surrey Beatty & Sons.
Verdade, V. K., & Rodrigues, M. T. (2007). Taxonomic review of Allobates (Anura, Aromobatidae) from the Atlantic Forest, Brazil. Journal of Herpetology, 41, 566–580.
Vieites, D. R., Wollenberg, K. C., Adreone, F., Köhler, J., Glaw, F., & Vences, M. (2009). Vast underestimation of Madagascar’s biodiversity evidenced by an integrative amphibian inventory. Proceedings of the National Academy of Sciences, 106, 8267–8272.
Wallace, A. R. (1852). On the monkeys of the Amazon. Proceedings of the Zoological Society of London, 20, 107–110.
West-Eberhard, M. J. (1989). Phenotypic plasticity and the origins of diversity. Annual Review of Ecology and Systematics, 20, 249–278.
Wiens, J. J. (2007). Species delimitation: New approaches for discovering diversity. Systematic Biology, 56, 875–878.
Wiens, J. J. (2008). Systematics and herpetology in the age of genomics. BioScience, 58, 297–307.
Wiley, E. O. (1978). The evolutionary species concept reconsidered. Systematic Zoology, 27, 17–26.
Wright, S. (1951). The genetical structure of populations. Annals of Human Genetics, 15, 323–354.
Wycherley, J., Doran, S., & Beebee, T. J. C. (2002). Male advertisement call characters as phylogeographical indicators in European water frogs. Biological Journal of the Linnean Society, 77, 355–365.
Zeisset, I., & Beebee, T. J. C. (2008). Amphibian phylogeography: a model for understanding historical aspects of species distributions. Heredity, 101, 109–119.
Zhang, D. X., & Hewitt, G. M. (2003). Nuclear DNA analyses in genetic studies of populations: Practice, problems and prospects. Molecular Ecology, 12, 563–584.
Zink, R. M., & Barrowclough, G. F. (2008). Mitochondrial DNA under siege in avian phylogeography. Molecular Ecology, 17, 2107–2121.
Zink, R. M., Blackwell-Rago, R. C., & Ronquist, F. (2000). The shifting roles of vicariance in biogeography. Proceedings of the Royal Society B, 267, 497–503.
Acknowledgments
We thank Anelise Montanarin, Francisco C. de Freitas, Irene da S. Melo, Maria A. Carvalho, Moisés da S. Melo and Raimundo N. Amorim for fieldwork assistance; Daniela Leroy e Waleska Gravena for help in laboratory procedures; Adolfo Amézquita, Adrian Garda, Andrew J. Crawford, Camila Ribas, Heike Pröhl, Janet W. Reid, Jeffrey Podos, Luciana K. Erdtmann, Marcelo Gordo, Marcelo Menin, Marina Anciães, Pedro Ivo Simões, Tomas Hrbek, Vanessa Verdade and Walter Hödl for valuable suggestions during this study. We extend thanks to two anonymous reviewers whose observations led to further improvements in the text. We also thank the Brazilian Conselho de Desenvolvimento Científico e Tecnológico for financial support (CNPq-CT Amazônia 553997/2006-8 and 575572/2008-6).
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical standards
This study complies with the current Brazilian laws and was allowed by RAN-ICMBio/IBAMA (licences 13777-2, 18516-1, 20065-2, and 21950-1).
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
11692_2012_9205_MOESM1_ESM.pdf
Online Resource 1 Advertisement call measurements of individuals of Allobates paleovarzensis in each study locality in Brazilian Amazonia. Values are presented as mean (above) and standard deviation (below). Call traits are: Note rate (NR, in notes/s); Note duration (ND, in s); Internote interval, as the silent interval between two consecutive notes of a call (InI, in s); Call rate (CR, in calls/s); Call duration (CD, in s); Intercall interval, as the silent interval between two consecutive calls (IcI, in s); Maximum (peak) frequency, as the frequency of higher intensity calculated for the entire note by a power spectrum function of Raven Pro 1.3 (MF, in Hz); Lowest frequency (LF, in Hz); Highest frequency (HF, in Hz); Note modulation, as the difference between the highest and the lowest frequencies of the call (NM, in Hz). Sampling locality codes, sample sizes and respective geographic coordinates are given on Table 1. (PDF 23 kb)
11692_2012_9205_MOESM2_ESM.pdf
Online Resource 2 Advertisement call measurements of individuals of Allobates nidicola and A. masniger in each study locality in Brazilian Amazonia. Values are presented as mean (above) and standard deviation (below). Call traits are: Note duration (ND, in s); Interval between notes (IN, in s); Lowest frequency (LF, in Hz); Highest frequency (HF, in Hz); Note modulation, calculated as the difference between the maximum and minimum frequencies (NM, in Hz); and Peak frequency (PF, in Hz). Sampling locality codes, sample sizes and respective geographic coordinates are given on Table 1. (PDF 18 kb)
11692_2012_9205_MOESM3_ESM.pdf
Online Resource 3 Morphometric measurements (in mm) of individuals of Allobates paleovarzensis in each study locality in Brazilian Amazonia. Values are presented as mean (above) and standard deviation (below). Morphometric traits are: Snout-vent length (SVL); Head length from angle of jaws to tip of snout (HL); Head width at level of angle of jaws (HW); Snout length from anterior corner of eye to tip of snout (SL); Eye to nostril distance from anterior corner of eye to centre of nostril (EN); Inter-nostril distance (IN); Eye length from anterior to posterior corner (EL); Inter-orbital distance (IO); Diameter of tympanum (TYM); Forearm length from proximal edge of palmar tubercle to outer edge of flexed elbow (FAL); Upper arm length from trunk insertion to outer edge of flexed elbow (UAL); Hand length from proximal edge of palmar tubercle to tip of fingers I, II, III and IV (HAND1, HAND2, HAND3 and HAND4); Width of disc on finger III (WFD); Tibia length from outer edge of flexed knee to heel (TL); Foot length from proximal edge of outer metatarsal tubercle to tip of toe IV (FL); Femur length (LL); Diameter of palmar tubercle (DPT); Width of tenar tubercle (WTT); Width of disc on toe IV (WTD); Width of finger III (WPF). Sampling locality codes, sample sizes and respective geographic coordinates are given on Table 1. (PDF 37 kb)
11692_2012_9205_MOESM4_ESM.pdf
Online Resource 4 Morphometric measurements (in mm) of individuals of Allobates nidicola and A. masniger in each study locality in Brazilian Amazonia. Values are presented as mean (above) and standard deviation (below). Morphometric traits are described in Online Resource 3. Sampling locality codes, sample sizes and respective geographic coordinates are given on Table 1. (PDF 37 kb)
11692_2012_9205_MOESM5_ESM.pdf
Online Resource 5 Distribution of 16S rDNA haplotypes of Allobates among 20 sampled localities in Brazilian Amazonia. Collection numbers of vouchers (INPA-H) and GenBank accession numbers are provided. Sampled localities are numbered according to Table 1. (PDF 24 kb)
11692_2012_9205_MOESM6_ESM.pdf
Online Resource 6 Mismatch distributions obtained from pairwise nucleotide site differences among 16S rDNA sequences composing the clusters of the (A) paleovarzensis and the (B) nidicola–masniger systems. Clusters were determined via Bayesian Analysis of Population Differentiation. Pairwise differences were not calculated for Cluster 4 of Allobates paleovarzensis due to the small number of samples that compose it. (PDF 334 kb)
11692_2012_9205_MOESM7_ESM.pdf
Online Resource 7 Classification matrix based on the discriminant function analysis, using phenotypic data from males of Allobates paleovazensis pertaining to four genetic clusters. Clusters are coded from C1 to C4, according to Table 4. Body-shape traits were used to discriminate individuals based on morphology. Acoustic measurements were adjusted for temperature and for both temperature and body size through linear regressions. The number and percentage of individuals correctly assigned to each genetic cluster are indicated. (PDF 18 kb)
11692_2012_9205_MOESM8_ESM.pdf
Online Resource 8 Classification matrix based on the discriminant function analysis, using phenotypic data from males of Allobates nidicola and A. masniger pertaining to seven genetic clusters. Clusters are coded from C1 to C7, according to Table 4. Body-shape traits were used to discriminate individuals based on morphology. Acoustic measurements were adjusted for temperature and for both temperature and body size through linear regressions. The number and percentage of individuals correctly assigned to each genetic cluster are indicated. (PDF 24 kb)
Rights and permissions
About this article
Cite this article
Kaefer, I.L., Tsuji-Nishikido, B.M., Mota, E.P. et al. The Early Stages of Speciation in Amazonian Forest Frogs: Phenotypic Conservatism Despite Strong Genetic Structure. Evol Biol 40, 228–245 (2013). https://doi.org/10.1007/s11692-012-9205-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11692-012-9205-4