
Abstract
Studies have reported lower striatal D2/D3 receptor availability in both alcoholics and cigarette smokers relative to healthy controls. These substances are commonly co-abused, yet the relationship between comorbid alcohol/tobacco abuse and striatal D2/D3 receptor availability has not been examined. We sought to determine the degree to which dual abuse of alcohol and tobacco is associated with lower D2/D3 receptor availability. Eighty-one subjects (34 nontreatment-seeking alcoholic smokers [NTS-S], 21 social-drinking smokers [SD-S], and 26 social-drinking non-smokers [SD-NS]) received baseline [11C]raclopride scans. D2/D3 binding potential (BPND ≡ Bavail/KD) was estimated for ten anatomically defined striatal regions of interest (ROIs). Significant group effects were detected in bilateral pre-commissural dorsal putamen, bilateral pre-commissural dorsal caudate; and bilateral post-commissural dorsal putamen. Post-hoc testing revealed that, regardless of drinking status, smokers had lower D2/D3 receptor availability than non-smoking controls. Chronic tobacco smokers have lower striatal D2/D3 receptor availability than non-smokers, independent of alcohol use. Additional studies are needed to identify the mechanisms by which chronic tobacco smoking is associated with striatal dopamine receptor availability.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alcohol and tobacco are the two most commonly abused substances in the United States. In people over the age of 12, the percentage reporting lifetime use of alcohol is 82.5%, and is 67.5% for tobacco (SAMHSA 2011). These two drugs interact in several domains. Specifically, tobacco cigarette-dependent individuals are approximately six times more likely to be alcohol dependent than non-tobacco cigarette-dependent individuals (Grant et al. 2004), and alcohol-dependent individuals are over five times more likely to be tobacco cigarette-dependent than non-alcohol-dependent individuals (Hasin et al. 2007). Comorbid abuse of alcohol and cigarettes has also been associated with higher rates of certain types of cancer than the abuse of either substance in isolation, including oral, laryngeal, esophageal, and liver cancer (Pelucchi et al. 2006). Evidence for additive effects of alcohol and cigarettes on cardiovascular disease is less conclusive (Mukamal 2006). Even so, abuse of either substance imparts increased risk for cardiovascular disease. Overall, the economic burden of alcohol and cigarette abuse in the U.S. is estimated at $185 billion (Harwood 2000) and $138 billion (Rice 1999), respectively.
Many factors likely contribute to the prevalence of comorbid alcohol and cigarette abuse (Drobes 2002), including the potential overlap of neurobiological mechanisms that subserve alcohol and cigarette dependence. One circuit implicated in most, if not all, addictive processes is the striatal dopamine (DA) system (Koob 1992; Leshner and Koob 1999; Robinson and Berridge 2003; Volkow et al. 2009; Di Chiara and Imperato 1988). A growing body of in vivo evidence suggests that striatal DA receptors may be altered in human addicts. PET and SPECT imaging studies have documented lower striatal D2/D3 receptor availability in several populations of abstinent and/or detoxified substance-dependent individuals, including users of cocaine (Martinez et al. 2009; Volkow et al. 1997), methamphetamine (Volkow et al. 2001), opiates (Wang et al. 1997), alcohol [(Hietala et al. 1994; Martinez et al. 2005; Volkow et al. 1996; Volkow et al. 2002; Heinz et al. 2004), although see (Guardia et al. 2000; Repo et al. 1999)], and cigarette-smoking subjects [(Busto et al. 2009; Fehr et al. 2008), although see (Yang et al. 2006)].
The high occurrence of comorbid alcohol and cigarette abuse, coupled with the association of abuse of both substances with deficits in the striatal dopaminergic (DAergic) system, are suggestive that alcohol and cigarettes act similarly on neurobiological circuits that underlie addiction. However, it is currently unknown if the individuals who abuse both alcohol and cigarettes have similar or greater deficits in D2/D3 availability compared to those who abuse only one substance. To begin to address this question, we conducted a retrospective analysis of baseline [11C]raclopride (RAC) PET data collected from several studies in the laboratory (RAC is a dopamine D2/D3 receptor antagonist used to estimate in vivo striatal receptor density). Baseline RAC PET data were compiled for three groups: nontreatment-seeking alcoholic smokers (NTS-S), social-drinking smokers (SD-S), and social-drinking non-smokers (SD-NS). We hypothesized that chronic, comorbid alcohol and cigarette abuse would be associated with greater deficits in D2/D3 receptor availability than abuse of cigarettes alone.
Methods
All study procedures were approved by the Indiana University Institutional Review Board and performed in accordance with the ethical standards of the Belmont Report. Subjects were recruited by local advertising in the greater Indianapolis area. After a complete description of the study to the subjects, written informed consent was obtained. Eighty-one right-hand dominant, adult subjects completed study procedures. Data from subsets of subjects have been published previously (Yoder et al. 2011a; Yoder et al. 2012; Albrecht et al. 2012). The presence or absence of alcohol abuse or dependence was assessed by either by the Semi-Structured Assessment for the Genetics of Alcoholism (Bucholz et al. 1994) (n = 73) or the Structured Clinical Diagnostic Interview for DSM-IV disorders (SCID) I Substance Use Disorders section (Module E) (n = 8). Subjects were excluded from participation if they endorsed recreational use of legal or illicit stimulants, pain medications, sedatives, and/or regular consumption of >2 marijuana cigarettes (or equivalent) per week. Urine toxicology screens (Q-10, Proxam) were administered during the screening visit, and on the day of PET imaging. Any positive result for an illicit substance on the screening visit was exclusionary (with the exception of THC when sporadic use was endorsed). Positive results on the day of scanning were recorded. NTS-S subjects had not received treatment for alcohol use disorders within the past year and were not actively seeking treatment. In cigarette smokers, tobacco dependence was assessed with the Fagerström Test for Nicotine Dependence (Pomerleau et al. 1994); these data were unavailable for three smoking subjects.
General scan day procedures
A BrAC measurement of 0.000 mg% was confirmed prior to scan day procedures for the majority of subjects (n = 73); BrAC was not measured in eight control subjects. Subjects received a structural MRI and a baseline [11C]raclopride (RAC) PET scan. All but seven smoking subjects received a transdermal nicotine patch, which has been shown to effectively control craving; variance in baseline RAC binding is also stable with patch placement (Yoder et al. 2011a; Yoder et al. 2012). Patch placement occurred approximately 5.5 h before RAC PET scanning (mean, 5.42 h; range, 1–7 h). Patch dose was based on subjects’ self-report of number of cigarettes smoked per day, per package instructions. Thirty-seven subjects received a 21 mg patch, 10 received a 14 mg patch, and one subject received a 7 mg patch. Cigarette craving was measured with the second dimension of the Cigarette Withdrawal Scale (CWS; (Etter 2005)), which specifically captures the individual’s current subjective state of cigarette craving. There are four items in this dimension, anchored by 1 (totally disagree) and 5 (totally agree). The final metric is a composite sum of the scores for each item; thus, the craving score range is 4–20. Cigarette craving data were available for 47 of the 55 smoking subjects. Forty-two of these subjects completed a paper version of the CWS prior to the rest scan; 5 subjects completed an electronic version immediately after RAC injection. On the day of scanning, two NTS subjects tested positive for cocaine, though both subjects denied recent cocaine use. One NTS and one SD-S subject tested positive for opiates on the scan day; both subjects reported that drugs had been prescribed for recent dental work. As previously described (Yoder et al. 2011a), NTS subjects were monitored for alcohol withdrawal with the Clinical Withdrawal Assessment for Alcohol, Revised (CIWA-Ar; (Sullivan et al. 1989)).
Image acquisition
A magnetized prepared rapid gradient echo (MP-RAGE) magnetic resonance image (MRI) was acquired using a Siemens 3T Trio for anatomic co-registration of PET data. RAC was synthesized as reported previously (Fei et al. 2004). RAC PET scans were acquired on an ECAT HR+ (3D mode; septa retracted). Prior to each PET scan, a 10-min transmission scan using three internal rod sources was acquired for attenuation correction. RAC PET scans were initiated with an IV infusion of 522.4 ± 55.6 MBq RAC over the course of 1.5 min. Injected mass was 0.14 ± 0.07 nmol/kg. Dynamic data acquisition lasted 50 min.
Image processing
Image processing was similar to that described previously (Yoder et al. 2011a; Yoder et al. 2012). MRI and PET images were converted to Neuroimaging Informatics Technology Initiative (NIfTI) format (http://nifti.nimh.nih.gov/) and processed with SPM5 (http://www.fil.ion.ucl.ac.uk/spm/). For each subject, an early mean PET image was coregistered to the anatomic MRI using the mutual information (MI) algorithm in SPM5. To facilitate motion-correction and to place all data into MRI space, all PET frames were coregistered to the MRI-registered mean PET. Each subject’s MRI was spatially normalized to Montreal Neurological Institute (MNI) space. The transformation matrix obtained from the spatial normalization step was then applied to the motion-corrected, MRI-registered PET data from each subject.
Region of interest analysis
Regions of interest (ROIs) were drawn on individual subjects’ spatially normalized MRIs using MRIcron (http://www.cabiatl.com/mricro/mricron/). Striatal ROIs were drawn according to specific anatomic landmarks (Martinez et al. 2003; Mawlawi et al. 2001), and consisted of the left and right ventral striatum (VST), pre- and postcommissural dorsal caudate (pre-/post-DCA), and pre- and postcommissural dorsal putamen (pre/post-DPU). Cerebellar ROIs were used as the reference region (tissue that contains little to no D2/D3 receptor density). Individual gray matter cerebellar ROIs were created for each subject; the vermis was excluded. For each subject and each ROI, the number of voxels in the ROI was recorded and converted to volume (cc). Time-activity curves for all ROIs were extracted from the dynamic RAC data using the MarsBaR toolbox (http://marsbar.sourceforge.net/). The RAC binding potential for each ROI ((defined as bound tracer concentration relative to nondisplaceable tracer concentration; BPND (Innis et al. 2007)) was estimated with the multilinear reference tissue method model (MRTM; Ichise et al. 2003). One subject had substantial atrophy of the caudate; caudate data from this individual were excluded from analyses.
Statistical analysis
One-way analysis of variance (ANOVA) was used to test for mean differences in outcome variables across the three groups. To identify sources of significant group effects, post-hoc testing was conducted using the Least Square Difference (LSD) method. Bonferroni corrections were applied to account for multiple comparisons. To test for effects of nicotine patch on BPND, one-way ANOVA was conducted in subsets of age-matched smokers, with nicotine patch dose as a fixed factor (no patch; 7/14 mg; 21 mg). Statistical tests were performed in Microsoft Excel 2007 or SPSS 19. Unless otherwise specified (e.g., in the case of Bonferroni correction), statistical significance was set at p < 0.05.
Results
Subject characteristics
Subject demographics and drinking characteristics are shown in Table 1. Groups were balanced for handedness, race, ethnicity, and education. Fagerström scores were not significantly different between smoking groups (Table 1). There was a main effect of group for number of drinks per week (p < 1.0 × 10-15). Post-hoc testing showed that NTS drank significantly more than both social-drinking groups (Table 1). SD-S and SD-NS did not differ in amount of alcohol consumed per week (Table 1). One-way ANOVA revealed a main effect of age (p < 0.05): SD-NS subjects were significantly younger than both SD-S and NTS-S subjects (Table 1). There was a main effect of injected radioactivity (p < 0.05). Post-hoc testing revealed that injected radioactivity in SD-S subjects was significantly higher than NTS-S subjects (Table 1). Mass dose was not significantly different across the three groups.
Striatal BPND: ROI analysis
There was a main effect of group for BPND in the L-pre-DCA, L-pre-DPU, L-post-DPU , R-pre-DCA, R-pre-DPU, and R-post-DPU (Table 2)
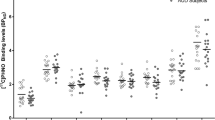
. Figure 1 illustrates the distribution of BPND in the R-pre-DPU for all three groups. Although only the R-pre-DPU and L-post-DPU survived Bonferroni correction, we observed that the mean BPND values for the smoking groups were both lower relative to the non-smokers (Table 2). To test the hypothesis that BPND is lower in the smokers compared to non-smokers, we performed an orthogonal planned contrast within the general linear model framework to compare the mean of the SD-NS group to the combined means of the NTS-S and SD-S groups. Applying Bonferroni correction to account for multiple comparisons lowered the threshold for significance to p < 0.005. At this corrected significance level, smokers had significantly lower striatal BPND values in six regions (Table 3).

Individual BPND data from the right pre-commissural dorsal putamen (R-pre-DPU), by group. Black diamonds: Nontreatment-seeking alcoholic smokers, NTS-S: Gray squares: Social-drinking smokers, SD-S. White triangles: Social-drinking non-smokers, SD-NS. Horizontal lines indicate group means
Striatal BPND: effect of nicotine patch dose
To determine if transdermal nicotine patches have an effect on striatal BPND, we examined data from three age-matched subsets (n = 7 each) of cigarette smokers. This approach is analogous to that recently reported by Weerts et al. (2013) with [11C]carfentanil. These subsets included: (1) smokers who did not receive a nicotine patch during the scan day (37.6 ± 7.2 years old), (2) smokers who received a patch dose of 7 or 14 mg nicotine (one subject received a 7 mg patch, the rest received 14 mg; 38.1 ± 10.5 y.o.), and (3) smokers who received a patch dose of 21 mg nicotine (37.0 ± 6.4 y.o.). Groups were also balanced for drinking status: each group contained four SD-S subjects and three NTS-S subjects. One-way ANOVA did not reveal any main effects of patch dose on BPND in any of the ten striatal regions, indicating that there was no effect of nicotine on BPND (data not shown).
ROI volumes: group differences
There was a main effect of group on ROI volume in the L-pre-DCA, L-pre-DPU, L-post-DCA, R-pre-DCA, R-pre-DPU, and R-post-DCA (Table 4). Post-hoc testing revealed that, regardless of smoking status, ROI volumes for both social-drinking groups (SD-S and SD-NS) were significantly greater than those for the NTS-S group in several regions (Table 4).
Discussion
The current study investigated whether chronic abuse of both alcohol and tobacco cigarettes has a differential effect on D2/D3 receptor availability compared to what has been previously reported for alcohol or tobacco-cigarette dependence alone (Busto et al. 2009; Fehr et al. 2008; Martinez et al. 2005; Volkow et al. 1996). The major result of this study was that cigarette smoking was associated with lower RAC BPND, independent of drinking status.
The finding that cigarette smoking is associated with low RAC BPND is in line with previous studies that reported lower D2/D3 receptor availability in chronic cigarette smokers relative to non-smokers (Busto et al. 2009; Fehr et al. 2008; Stokes et al. 2011). However, the current results are inconsistent with data from Martinez et al. (Martinez et al. 2005), which documented reduced striatal D2/D3 receptor availability in detoxified alcoholic subjects, even with matching control subjects for smoking status, as we did in the present study. In other studies of D2/D3 receptor availability in alcoholics, imbalances in smoking status between alcoholics and controls may have accounted for some reported lower D2/D3 receptor availability in alcoholics (Heinz et al. 2004; Hietala et al. 1994; Volkow et al. 1996; Volkow et al. 2002). An important difference between the current and previous studies is that we sampled nontreatment-seeking alcoholics from the local community, whereas prior work studied abstinent alcoholics after inpatient detoxification. These are likely two distinct populations of alcohol-dependent subjects. Treatment-seeking alcoholics have more severe alcoholism than nontreatment-seekers (Fein and Landman 2005), have a higher comorbidity of psychiatric disorders (Di Sclafani et al. 2008), and a greater degree of gray and white matter abnormalities (Gazdzinski et al. 2008). Thus, it is possible that individuals from a community-based, currently heavy-drinking (and smoking) population may not have apparent deficits in striatal D2/D3 receptor availability when compared to social-drinking controls matched for cigarette smoking status.
Because BPND is a compound index (Bavail/KD), lower striatal BPND in smokers relative to non-smokers could be interpreted as either lower numbers of D2/D3 receptors, or higher synaptic/extrasynaptic DA concentration. We speculate that cigarette smoking produces an apparently lower D2/D3 availability via increased striatal DA concentration. This would be consistent with a post-mortem study of chronic cigarette smokers, which reported that DA levels in smokers’ striatal tissue were significantly higher compared to non-smokers, whereas D2 and D3 receptor levels were not different between groups (Court et al. 1998). It is possible that smoking-induced increases in striatal DA occur through inhibition of monoamine oxidase (MAO). Dopamine is a substrate for both MAO isoforms (MAO-A and -B), and chronic treatment with either MAO-A or MAO-B inhibitors increases basal striatal DA (Lamensdorf et al. 1996; Kaseda et al. 1999; Lakshmana et al. 1998). Several studies established that platelet MAO activity is lower in current cigarette smokers relative to non-smokers and former smokers (Berlin et al. 1995; Norman et al. 1987; Oreland et al. 1981). Multiple PET studies have demonstrated inhibition of both MAO isoforms (MAO-A and -B) in the brains of smokers (Leroy et al. 2009; Fowler et al. 1996a; Fowler et al. 1996b). Cigarette smoke itself is a potent inhibitor of both MAO isoforms (Yu and Boulton 1987), and several of the inhibitory compounds in cigarette smoke extract have been identified, including the β-carbolines harman and norharman (Herraiz and Chaparro 2005). Collectively, this body of evidence strongly supports the possibility that cigarette smoke increases DA levels, resulting in apparent lower striatal RAC D2/D3 availability in smokers.
Alternatively, it is possible that the nicotine patches worn by a majority of subjects induced DA release. All but seven smokers were administered nicotine patches on the scan day, and studies in the animal literature suggest that nicotine itself may cause measurable DA release (Nisell et al. 1994; Schiffer et al. 2001). Three human RAC studies found similar results (Brody et al. 2006; Brody et al. 2004; Takahashi et al. 2008). However, in the two Brody et al. studies, the designs included subjects physically smoking while inside the scanner, and it is possible that the chemosensory properties of smoking a cigarette are central factors contributing to the DA release observed in these studies. In fact, recent data support this possibility: Domino et al. (2012) showed that smoking denicotinized cigarettes causes measurable DA release, indicating that the presence of nicotine may not be a necessary condition for increased DA during the act of smoking. Although Takahashi et al. (2008) reported that chewing nicotinized gum increased striatal DA in cigarette smokers, the design included a placebo condition as the “baseline”. If the placebo condition induced a negative prediction error (nicotine expected from the gum, but not delivered), this could have induced a decrease in striatal DA (Yoder et al. 2009) during the placebo condition, producing results that show an apparent “increase” in DA during the nicotinized gum condition (see discussion of design confounds in Yoder et al. (2011b)). There is also strong pharmacological evidence that nicotine itself does not induce DA release measurable by RAC PET. When nicotine was administered intranasally to humans and intravenously to unanesthetized monkeys, there were no significant reductions in RAC binding (Montgomery et al. 2007; Tsukada et al. 2002). Finally, our data did not show any evidence of a measurable effect of nicotine patches on BPND. Therefore, we suggest that it is unlikely that the use of nicotine patches was a significant confound in this study.
There are limitations to this retrospective study. As all of our alcoholic subjects were also cigarette smokers, we could not assess the specific contributions of alcohol and cigarette use to D2/D3 availability in this sample. Inclusion of a group of non-smoking alcoholics would be needed to confirm the conclusion that the group differences observed in the current study were due solely to chronic smoking. Also, although every effort was made to screen for use of illicit substances, some subjects tested positive for drugs besides marijuana (2 for cocaine, 2 for opiates) on the day of scanning. Examination of these subjects’ data indicated that the BPND values were well within two standard deviations of the group mean, indicating that they were not outliers skewing the results. A history of substance abuse or dependence was an exclusion criteria; however, we cannot preclude the possibility that prior recreational use of other illicit substances contributed variance to the data. The reduced striatal volumes in the NTS-S group introduces another potential confound, via the risk of partial volume effects on BPND (Morris et al. 2004). NTS-S subjects had smaller striatal volumes (Table 4) than the other two social drinking groups, but there were no differences in striatal RAC BPND between the alcoholic smokers and social drinking smokers. If striatal atrophy (and hence, partial volume effect) was the sole source of apparent lower BPND between smokers and non-smokers, then we might expect similar levels of atrophy in both smoking groups. However, this was not the case. Therefore, we suggest that striatal atrophy in the alcoholic smokers is an unlikely source of significant variance in our data. Another potential concern is the significantly younger age of the SD-NS subjects compared to the SD-S and NTS-S subjects (Table 1). Age-related decline of striatal D2/D3 receptor availability is well-documented, with estimates ranging from 4 % to 8 % decrease per decade (for comprehensive review, see Ishibashi et al. 2009). However, the majority of such studies were conducted with a very wide age range (e.g., from 20 to 80 years of age) in healthy individuals. In the present work, we did not observe a correlation between BPND and age in either social drinking group, perhaps because of the limited age range of our samples. However, we did observe a correlation between age and BPND in the smoking alcoholic group (data not shown). Because age was not uniformly related to BPND across our samples, we believed it was not appropriate to use age as a covariate in the data analyses. Our observations of age-related decline in BPND in NTS-S (especially in a sample with a restricted age range) is consistent with recent evidence, which suggests that this decline may be accelerated in certain populations such as schizophrenia (Kegeles et al. 2010) and alcoholism (Rominger et al. 2012).
In conclusion, the primary finding in the present study is that, regardless of drinking status, cigarette smokers have lower striatal D2/D3 receptor availability compared to non-smokers. This is important, as it suggests that some component(s) of cigarette smoke may act on the dopamine system independently of alcohol abuse. This adds to the growing literature demonstrating the adverse effects of cigarette smoking on brain structure and chemistry (for review, see Durazzo et al. 2007). Additionally, recent findings indicate that continued cigarette use during treatment for substance abuse may be detrimental to clinical outcome (for review, see Kalman et al. 2010). Although it is tempting to speculate that the effects of cigarette smoking on brain dopamine may interfere with treatment for alcoholism and other addictions, more research is needed to explore this possibility. In addition to studying the clinical implications of smoking on D2/D3 availability, future studies must address the molecular ramifications of chronic cigarette abuse on the dopamine system.
References
Albrecht, D. S., Skosnik, P. D., Vollmer, J. M., Brumbaugh, M. S., Perry, K. M., Mock, B. H., Zheng, Q. H., Federici, L. A., Patton, E. A., Herring, C. M., & Yoder, K. K. (2012). Striatal D(2)/D(3) receptor availability is inversely correlated with cannabis consumption in chronic marijuana users. Drug and Alcohol Dependence. doi:10.1016/j.drugalcdep.2012.07.016.
Berlin, I., Said, S., Spreux-Varoquaux, O., Olivares, R., Launay, J. M., & Puech, A. J. (1995). Monoamine oxidase A and B activities in heavy smokers. Biological Psychiatry, 38(11), 756–761. doi:10.1016/0006-3223(95)00084-4.
Brody, A. L., Mandelkern, M. A., Olmstead, R. E., Scheibal, D., Hahn, E., Shiraga, S., Zamora-Paja, E., Farahi, J., Saxena, S., London, E. D., & McCracken, J. T. (2006). Gene variants of brain dopamine pathways and smoking-induced dopamine release in the ventral caudate/nucleus accumbens. Archives of General Psychiatry, 63(7), 808–816. doi:10.1001/archpsyc.63.7.808.
Brody, A. L., Olmstead, R. E., London, E. D., Farahi, J., Meyer, J. H., Grossman, P., Lee, G. S., Huang, J., Hahn, E. L., & Mandelkern, M. A. (2004). Smoking-induced ventral striatum dopamine release. The American Journal of Psychiatry, 161(7), 1211–1218.
Bucholz, K. K., Cadoret, R., Cloninger, C. R., Dinwiddie, S. H., Hesselbrock, V. M., Nurnberger, J. I., Jr., Reich, T., Schmidt, I., & Schuckit, M. A. (1994). A new, semi-structured psychiatric interview for use in genetic linkage studies: a report on the reliability of the SSAGA. Journal of Studies on Alcohol, 55(2), 149–158.
Busto, U. E., Redden, L., Mayberg, H., Kapur, S., Houle, S., & Zawertailo, L. A. (2009). Dopaminergic activity in depressed smokers: a positron emission tomography study. Synapse, 63(8), 681–689. doi:10.1002/syn.20646.
Court, J. A., Lloyd, S., Thomas, N., Piggott, M. A., Marshall, E. F., Morris, C. M., Lamb, H., Perry, R. H., Johnson, M., & Perry, E. K. (1998). Dopamine and nicotinic receptor binding and the levels of dopamine and homovanillic acid in human brain related to tobacco use. Neuroscience, 87(1), 63–78.
Di Chiara, G., & Imperato, A. (1988). Drugs abused by humans preferentially increase synaptic dopamine concentrations in the mesolimbic system of freely moving rats. Proceedings of the National Academy of Sciences of the United States of America, 85(14), 5274–5278.
Di Sclafani, V., Finn, P., & Fein, G. (2008). Treatment-naive active alcoholics have greater psychiatric comorbidity than normal controls but less than treated abstinent alcoholics. Drug and Alcohol Dependence, 98(1–2), 115–122. doi:10.1016/j.drugalcdep.2008.04.019.
Domino, E. F., Ni, L., Domino, J. S., Yang, W., Evans, C., Guthrie, S., Wang, H., Koeppe, R. A., & Zubieta, J. K. (2012). Denicotinized versus average nicotine tobacco cigarette smoking differentially releases striatal dopamine. Nicotine Tob Res. doi:10.1093/ntr/nts029.
Drobes, D. J. (2002). Concurrent alcohol and tobacco dependence: mechanisms and treatment. Alcohol Research & Health, 26(2), 136–142.
Durazzo, T. C., Gazdzinski, S., & Meyerhoff, D. J. (2007). The neurobiological and neurocognitive consequences of chronic cigarette smoking in alcohol use disorders. Alcohol and Alcoholism, 42(3), 174–185. doi:10.1093/alcalc/agm020.
Etter, J. F. (2005). A self-administered questionnaire to measure cigarette withdrawal symptoms: the cigarette withdrawal scale. Nicotine & Tobacco Research, 7(1), 47–57.
Fehr, C., Yakushev, I., Hohmann, N., Buchholz, H. G., Landvogt, C., Deckers, H., Eberhardt, A., Klager, M., Smolka, M. N., Scheurich, A., Dielentheis, T., Schmidt, L. G., Rosch, F., Bartenstein, P., Grunder, G., & Schreckenberger, M. (2008). Association of low striatal dopamine d2 receptor availability with nicotine dependence similar to that seen with other drugs of abuse. The American Journal of Psychiatry, 165(4), 507–514. doi:10.1176/appi.ajp.2007.07020352.
Fei, X., Mock, B. H., DeGrado, T. R., Wang, J. Q., Glick-Wilson, B. E., Sullivan, M. L., Hutchins, G. D., & Zheng, Q. H. (2004). An improved synthesis of PET dopamine D2 receptors radioligand [11C]raclopride. Synthetic Communications, 34, 1897–1907.
Fein, G., & Landman, B. (2005). Treated and treatment-naive alcoholics come from different populations. Alcohol, 36(2), 19–26.
Fowler, J. S., Volkow, N. D., Wang, G. J., Pappas, N., Logan, J., MacGregor, R., Alexoff, D., Shea, C., Schlyer, D., Wolf, A. P., Warner, D., Zezulkova, I., & Cilento, R. (1996a). Inhibition of monoamine oxidase B in the brains of smokers. Nature, 379(6567), 733–736. doi:10.1038/379733a0.
Fowler, J. S., Volkow, N. D., Wang, G. J., Pappas, N., Logan, J., Shea, C., Alexoff, D., MacGregor, R. R., Schlyer, D. J., Zezulkova, I., & Wolf, A. P. (1996b). Brain monoamine oxidase A inhibition in cigarette smokers. Proceedings of the National Academy of Sciences of the United States of America, 93(24), 14065–14069.
Gazdzinski, S., Durazzo, T. C., Weiner, M. W., & Meyerhoff, D. J. (2008). Are treated alcoholics representative of the entire population with alcohol use disorders? A magnetic resonance study of brain injury. Alcohol, 42(2), 67–76. doi:10.1016/j.alcohol.2008.01.002.
Grant, B. F., Hasin, D. S., Chou, S. P., Stinson, F. S., & Dawson, D. A. (2004). Nicotine dependence and psychiatric disorders in the United States: results from the national epidemiologic survey on alcohol and related conditions. Archives of General Psychiatry, 61(11), 1107–1115. doi:10.1001/archpsyc.61.11.1107.
Guardia, J., Catafau, A. M., Batlle, F., Martin, J. C., Segura, L., Gonzalvo, B., Prat, G., Carrio, I., & Casas, M. (2000). Striatal dopaminergic D(2) receptor density measured by [(123)I]iodobenzamide SPECT in the prediction of treatment outcome of alcohol-dependent patients. The American Journal of Psychiatry, 157(1), 127–129.
Harwood, H. J. (2000). Updating estimates of the economic costs of alcohol abuse in the United States: Estimates, update methods, and data. Rockville: National Institute on Alcohol Abuse and Alcoholism.
Hasin, D. S., Stinson, F. S., Ogburn, E., & Grant, B. F. (2007). Prevalence, correlates, disability, and comorbidity of DSM-IV alcohol abuse and dependence in the United States: results from the National Epidemiologic Survey on Alcohol and Related Conditions. Archives of General Psychiatry, 64(7), 830–842. doi:10.1001/archpsyc.64.7.830.
Heinz, A., Siessmeier, T., Wrase, J., Hermann, D., Klein, S., Grusser, S. M., Flor, H., Braus, D. F., Buchholz, H. G., Grunder, G., Schreckenberger, M., Smolka, M. N., Rosch, F., Mann, K., & Bartenstein, P. (2004). Correlation between dopamine D(2) receptors in the ventral striatum and central processing of alcohol cues and craving. The American Journal of Psychiatry, 161(10), 1783–1789. doi:10.1176/appi.ajp.161.10.1783.
Herraiz, T., & Chaparro, C. (2005). Human monoamine oxidase is inhibited by tobacco smoke: beta-carboline alkaloids act as potent and reversible inhibitors. Biochemical and Biophysical Research Communications, 326(2), 378–386. doi:10.1016/j.bbrc.2004.11.033.
Hietala, J., West, C., Syvalahti, E., Nagren, K., Lehikoinen, P., Sonninen, P., & Ruotsalainen, U. (1994). Striatal D2 dopamine receptor binding characteristics in vivo in patients with alcohol dependence. Psychopharmacology, 116(3), 285–290.
Ichise, M., Liow, J. S., Lu, J. Q., Takano, A., Model, K., Toyama, H., Suhara, T., Suzuki, K., Innis, R. B., & Carson, R. E. (2003). Linearized reference tissue parametric imaging methods: application to [11C]DASB positron emission tomography studies of the serotonin transporter in human brain. Journal of Cerebral Blood Flow and Metabolism, 23(9), 1096–1112. doi:10.1097/01.WCB.0000085441.37552.CA.
Innis, R. B., Cunningham, V. J., Delforge, J., Fujita, M., Gjedde, A., Gunn, R. N., Holden, J., Houle, S., Huang, S. C., Ichise, M., Iida, H., Ito, H., Kimura, Y., Koeppe, R. A., Knudsen, G. M., Knuuti, J., Lammertsma, A. A., Laruelle, M., Logan, J., Maguire, R. P., Mintun, M. A., Morris, E. D., Parsey, R., Price, J. C., Slifstein, M., Sossi, V., Suhara, T., Votaw, J. R., Wong, D. F., & Carson, R. E. (2007). Consensus nomenclature for in vivo imaging of reversibly binding radioligands. Journal of Cerebral Blood Flow and Metabolism, 27(9), 1533–1539. doi:10.1038/sj.jcbfm.9600493.
Ishibashi, K., Ishii, K., Oda, K., Kawasaki, K., Mizusawa, H., & Ishiwata, K. (2009). Regional analysis of age-related decline in dopamine transporters and dopamine D2-like receptors in human striatum. Synapse, 63(4), 282–290. doi:10.1002/syn.20603.
Kalman, D., Kim, S., DiGirolamo, G., Smelson, D., & Ziedonis, D. (2010). Addressing tobacco use disorder in smokers in early remission from alcohol dependence: the case for integrating smoking cessation services in substance use disorder treatment programs. Clinical Psychology Review, 30(1), 12–24. doi:10.1016/j.cpr.2009.08.009.
Kaseda, S., Nomoto, M., & Iwata, S. (1999). Effect of selegiline on dopamine concentration in the striatum of a primate. Brain Research, 815(1), 44–50.
Kegeles, L. S., Slifstein, M., Xu, X., Urban, N., Thompson, J. L., Moadel, T., Harkavy-Friedman, J. M., Gil, R., Laruelle, M., & Abi-Dargham, A. (2010). Striatal and extrastriatal dopamine D2/D3 receptors in schizophrenia evaluated with [18F]fallypride positron emission tomography. Biological Psychiatry, 68(7), 634–641. doi:10.1016/j.biopsych.2010.05.027.
Koob, G. F. (1992). Drugs of abuse: anatomy, pharmacology and function of reward pathways. Trends in Pharmacological Sciences, 13(5), 177–184.
Lakshmana, M. K., Rao, B. S., Dhingra, N. K., Ravikumar, R., Govindaiah, Sudha, S., Meti, B. L., & Raju, T. R. (1998). Role of monoamine oxidase type A and B on the dopamine metabolism in discrete regions of the primate brain. Neurochemical Research, 23(8), 1031–1037.
Lamensdorf, I., Youdim, M. B., & Finberg, J. P. (1996). Effect of long-term treatment with selective monoamine oxidase A and B inhibitors on dopamine release from rat striatum in vivo. Journal of Neurochemistry, 67(4), 1532–1539.
Leroy, C., Bragulat, V., Berlin, I., Gregoire, M. C., Bottlaender, M., Roumenov, D., Dolle, F., Bourgeois, S., Penttila, J., Artiges, E., Martinot, J. L., & Trichard, C. (2009). Cerebral monoamine oxidase A inhibition in tobacco smokers confirmed with PET and [11C]befloxatone. Journal of Clinical Psychopharmacology, 29(1), 86–88. doi:10.1097/JCP.0b013e31819e98f.
Leshner, A. I., & Koob, G. F. (1999). Drugs of abuse and the brain. Proceedings of the Association of American Physicians, 111(2), 99–108.
Martinez, D., Gil, R., Slifstein, M., Hwang, D. R., Huang, Y., Perez, A., Kegeles, L., Talbot, P., Evans, S., Krystal, J., Laruelle, M., & Abi-Dargham, A. (2005). Alcohol dependence is associated with blunted dopamine transmission in the ventral striatum. Biological Psychiatry, 58(10), 779–786. doi:10.1016/j.biopsych.2005.04.044.
Martinez, D., Greene, K., Broft, A., Kumar, D., Liu, F., Narendran, R., Slifstein, M., Van Heertum, R., & Kleber, H. D. (2009). Lower level of endogenous dopamine in patients with cocaine dependence: findings from PET imaging of D(2)/D(3) receptors following acute dopamine depletion. The American Journal of Psychiatry, 166(10), 1170–1177. doi:10.1176/appi.ajp.2009.08121801.
Martinez, D., Slifstein, M., Broft, A., Mawlawi, O., Hwang, D. R., Huang, Y., Cooper, T., Kegeles, L., Zarahn, E., Abi-Dargham, A., Haber, S. N., & Laruelle, M. (2003). Imaging human mesolimbic dopamine transmission with positron emission tomography. Part II: amphetamine-induced dopamine release in the functional subdivisions of the striatum. Journal of Cerebral Blood Flow and Metabolism, 23(3), 285–300.
Mawlawi, O., Martinez, D., Slifstein, M., Broft, A., Chatterjee, R., Hwang, D. R., Huang, Y., Simpson, N., Ngo, K., & Van Heertum, R. (2001). Imaging human mesolimbic dopamine transmission with positron emission tomography: I. Accuracy and precision of D2 receptor parameter measurements in ventral striatum. Journal of Cerebral Blood Flow and Metabolism, 21(9), 1034–1057.
Montgomery, A. J., Lingford-Hughes, A. R., Egerton, A., Nutt, D. J., & Grasby, P. M. (2007). The effect of nicotine on striatal dopamine release in man: a [11C]raclopride PET study. Synapse, 61(8), 637–645. doi:10.1002/syn.20419.
Morris, E. D., Endres, C. J., Schmidt, K. C., Christian, B. T., Muzik, J. R. F., & Fisher, R. E. (2004). Kinetic modeling in positron emission tomography. In Emission tomography: The fundamentals of PET and SPECT (pp. 499–540): Academic Press.
Mukamal, K. J. (2006). The effects of smoking and drinking on cardiovascular disease and risk factors. Alcohol Research & Health, 29(3), 199–202.
Nisell, M., Nomikos, G. G., & Svensson, T. H. (1994). Infusion of nicotine in the ventral tegmental area or the nucleus accumbens of the rat differentially affects accumbal dopamine release. Pharmacology & Toxicology, 75(6), 348–352.
Norman, T. R., Chamberlain, K. G., & French, M. A. (1987). Platelet monoamine oxidase: low activity in cigarette smokers. Psychiatry Research, 20(3), 199–205.
Oreland, L., Fowler, C. J., & Schalling, D. (1981). Low platelet monoamine oxidase activity in cigarette smokers. Life Sciences, 29(24), 2511–2518.
Pelucchi, C., Gallus, S., Garavello, W., Bosetti, C., & La Vecchia, C. (2006). Cancer risk associated with alcohol and tobacco use: focus on upper aero-digestive tract and liver. Alcohol Research & Health, 29(3), 193–198.
Pomerleau, C. S., Carton, S. M., Lutzke, M. L., Flessland, K. A., & Pomerleau, O. F. (1994). Reliability of the fagerstrom tolerance questionnaire and the fagerstrom test for nicotine dependence. Addictive Behaviors, 19(1), 33–39.
Repo, E., Kuikka, J. T., Bergstrom, K. A., Karhu, J., Hiltunen, J., & Tiihonen, J. (1999). Dopamine transporter and D2-receptor density in late-onset alcoholism. Psychopharmacology, 147(3), 314–318.
Rice, D. P. (1999). Economic costs of substance abuse, 1995. Proceedings of the Association of American Physicians, 111(2), 119–125.
Robinson, T. E., & Berridge, K. C. (2003). Addiction. Annual Review of Psychology, 54, 25–53. doi:10.1146/annurev.psych.54.101601.145237.
Rominger, A., Cumming, P., Xiong, G., Koller, G., Boning, G., Wulff, M., Zwergal, A., Forster, S., Reilhac, A., Munk, O., Soyka, M., Wangler, B., Bartenstein, P., la Fougere, C., & Pogarell, O. (2012). [(18) F]fallypride PET measurement of striatal and extrastriatal dopamine D(2/3) receptor availability in recently abstinent alcoholics. Addiction Biology, 17(2), 490–503. doi:10.1111/j.1369-1600.2011.00355.x.
SAMHSA (2011). Results from the 2011 national survey on drug use and health: National findings. In O. o. A. Studies (Ed.). Rockville, MD.
Schiffer, W. K., Gerasimov, M. R., Marsteller, D. A., Geiger, J., Barnett, C., Alexoff, D. L., & Dewey, S. L. (2001). Topiramate selectively attenuates nicotine-induced increases in monoamine release. Synapse, 42(3), 196–198. doi:10.1002/syn.10000.
Stokes, P. R., Egerton, A., Watson, B., Reid, A., Lappin, J., Howes, O. D., Nutt, D. J., & Lingford-Hughes, A. R. (2011). History of cannabis use is not associated with alterations in striatal dopamine D2/D3 receptor availability. Journal of Psychopharmacology. doi:10.1177/0269881111414090.
Sullivan, J. T., Sykora, K., Schneiderman, J., Naranjo, C. A., & Sellers, E. M. (1989). Assessment of alcohol withdrawal: the revised clinical institute withdrawal assessment for alcohol scale (CIWA-Ar). British Journal of Addiction, 84(11), 1353–1357.
Takahashi, H., Fujimura, Y., Hayashi, M., Takano, H., Kato, M., Okubo, Y., Kanno, I., Ito, H., & Suhara, T. (2008). Enhanced dopamine release by nicotine in cigarette smokers: a double-blind, randomized, placebo-controlled pilot study. The International Journal of Neuropsychopharmacology, 11(3), 413–417. doi:10.1017/S1461145707008103.
Tsukada, H., Miyasato, K., Kakiuchi, T., Nishiyama, S., Harada, N., & Domino, E. F. (2002). Comparative effects of methamphetamine and nicotine on the striatal [(11)C]raclopride binding in unanesthetized monkeys. Synapse, 45(4), 207–212. doi:10.1002/syn.10102.
Volkow, N. D., Chang, L., Wang, G. J., Fowler, J. S., Ding, Y. S., Sedler, M., Logan, J., Franceschi, D., Gatley, J., Hitzemann, R., Gifford, A., Wong, C., & Pappas, N. (2001). Low level of brain dopamine D2 receptors in methamphetamine abusers: association with metabolism in the orbitofrontal cortex. The American Journal of Psychiatry, 158(12), 2015–2021.
Volkow, N. D., Fowler, J. S., Wang, G. J., Baler, R., & Telang, F. (2009). Imaging dopamine’s role in drug abuse and addiction. Neuropharmacology, 56(Suppl 1), 3–8. doi:10.1016/j.neuropharm.2008.05.022.
Volkow, N. D., Wang, G. J., Fowler, J. S., Logan, J., Gatley, S. J., Hitzemann, R., Chen, A. D., Dewey, S. L., & Pappas, N. (1997). Decreased striatal dopaminergic responsiveness in detoxified cocaine-dependent subjects. Nature, 386(6627), 830–833. doi:10.1038/386830a0.
Volkow, N. D., Wang, G. J., Fowler, J. S., Logan, J., Hitzemann, R., Ding, Y. S., Pappas, N., Shea, C., & Piscani, K. (1996). Decreases in dopamine receptors but not in dopamine transporters in alcoholics. Alcoholism, Clinical and Experimental Research, 20(9), 1594–1598.
Volkow, N. D., Wang, G. J., Maynard, L., Fowler, J. S., Jayne, B., Telang, F., Logan, J., Ding, Y. S., Gatley, S. J., Hitzemann, R., Wong, C., & Pappas, N. (2002). Effects of alcohol detoxification on dopamine D2 receptors in alcoholics: a preliminary study. Psychiatry Research, 116(3), 163–172.
Wang, G. J., Volkow, N. D., Fowler, J. S., Logan, J., Abumrad, N. N., Hitzemann, R. J., Pappas, N. S., & Pascani, K. (1997). Dopamine D2 receptor availability in opiate-dependent subjects before and after naloxone-precipitated withdrawal. Neuropsychopharmacology, 16(2), 174–182. doi:10.1016/S0893-133X(96)00184-4.
Weerts, E. M., Wand, G. S., Kuwabara, H., Xu, X., Frost, J. J., Wong, D. F., & McCaul, M. E. (2013). Association of smoking with μ-opioid receptor availability before and during naltrexone blockade in alcohol-dependent subjects. Addiction Biology.
Yang, Y. K., Yao, W. J., McEvoy, J. P., Chu, C. L., Lee, I. H., Chen, P. S., Yeh, T. L., & Chiu, N. T. (2006). Striatal dopamine D2/D3 receptor availability in male smokers. Psychiatry Research, 146(1), 87–90. doi:10.1016/j.pscychresns.2005.09.008.
Yoder, K. K., Morris, E. D., Constantinescu, C. C., Cheng, T. E., Normandin, M. D., O'Connor, S. J., Kareken, D. A. (2009). When what you see isn't what you get: alcohol cues, alcohol administration, prediction error, and human striatal dopamine. Alcohol Clin Exp Res 33(1):139–149.
Yoder, K. K., Albrecht, D. S., Kareken, D. A., Federici, L. M., Perry, K. M., Patton, E. A., Zheng, Q. H., Mock, B. H., O’Connor, S., & Herring, C. M. (2011a). Test-retest variability of [(11)C]raclopride-binding potential in nontreatment-seeking alcoholics. Synapse, 65(7), 553–561. doi:10.1002/syn.20874.
Yoder, K. K., Albrecht, D. S., Kareken, D. A., Federici, L. M., Perry, K. M., Patton, E. A., Zheng, Q. H., Mock, B. H., O’Connor, S. J., & Herring, C. M. (2012). Reliability of striatal [(11)C]raclopride binding in smokers wearing transdermal nicotine patches. European Journal of Nuclear Medicine and Molecular Imaging, 39(2), 220–225. doi:10.1007/s00259-011-1965-z.
Yoder, K. K., Kareken, D. A., & Morris, E. D. (2011b). Assessing dopaminergic neurotransmission with PET: basic theory and applications in alcohol research. Current Medical Imaging Reviews, 7, 118–124.
Yu, P. H., & Boulton, A. A. (1987). Irreversible inhibition of monoamine oxidase by some components of cigarette smoke. Life Sciences, 41(6), 675–682.
Acknowledgments
Supported by ABMRF/The Foundation for Alcohol Research (KKY), NIAAA 5P60AA007611-25 (pilot P50 to KKY), NIAAA R21AA016901 (KKY), NIAAA R01AA018354 (KKY) and the Indiana Clinical and Translational Sciences Institute (NIH TR000006, Indiana Clinical Research Center).
The authors would like to thank Christine Herring, Lauren Federici, Cari Cox Lehigh, and Elizabeth Patton for assistance with recruitment and data collection; Kevin Perry for acquisition of PET data; Michele Beal and Courtney Robbins for assistance with MR scanning; and Dr. Bruce Mock, Dr. Clive Brown-Proctor, Dr. Qi-Huang Zheng, Barbara Glick-Wilson and Brandon Steele for [11C]raclopride synthesis. We also thank Dr. Andrew Saykin, Dr. Gary Hutchins, and Dr. Nicholas Grahame for valuable discussion regarding data analysis.
Conflict of Interest
The authors declare that they have no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Albrecht, D.S., Kareken, D.A. & Yoder, K.K. Effects of smoking on D2/D3 striatal receptor availability in alcoholics and social drinkers. Brain Imaging and Behavior 7, 326–334 (2013). https://doi.org/10.1007/s11682-013-9233-4
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11682-013-9233-4