
Abstract
To elucidate the reoxidation mechanism of Al-killed ultra-low C steel by FetO-containing slag, the kinetics of a reaction 2Al + 3(FetO) = (Al2O3) + 3Fe between the steel and CaO–Al2O3–FetO–MgOsat. slag was investigated mostly at 1823 K. Al contents (total and soluble), and total O content in steel samples were measured during the reactions under various initial compositions of slag ((pct CaO)\(_0\)/(pct Al2O3)\(_0\), (Fe\(_t\)O)\(_0\)), and the reaction temperature. The experimental results were analyzed using the reaction rate model developed in the present study, which is based on probable rate controlling step and employing CALPHAD thermodynamics using FactSage thermochemical software and databases. When the (pct Fe\(_t\)O)\(_0\) was higher than 10, the rate model could explain the measured data with an assumption that the rate was solely controlled by mass transport of Al in the steel. However, mixed transport control theory should be used to interpret the reaction rate when the (pct Fe\(_t\)O)\(_0\) was lower than 10. Decreasing (pct Fe\(_t\)O) during the reoxidation reaction changes the reaction mechanism in terms of the mode of rate-controlling step. The mass transport coefficient of Al in the steel (k MAl was \(5\times 10^{-4}\) m s\(^{-1}\) at 1823 K (1550 °C), which is in favorable agreement with those in the literature. The mass transport coefficients of Al\(_2\)O\(_3\) was formulated to depend on the viscosity of the slag. In the mixed transport control regime, the apparent mass transport coefficient gradually decreased due to the slow mass transport of Al\(_2\)O\(_3\) as a resistance to the overall mass transport. This was also additionally supported by evaluating the activation energy of the apparent mass transport coefficient, which turned out to increase as the reoxidation reaction proceeds. Therefore, it can be concluded that the reaction mechanism gradually changes during the reoxidation reaction. (pct CaO)\(_0\)/(pct Al\(_2\)O\(_3\))\(_0\) ratio affects the reoxidation rate only when (Fe\(_t\)O)\(_0\) was low.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Ultra-Low Carbon (ULC) steel is one of the clean steels showing outstanding formability and ductility.[1,2,3,4] It has been applied for producing various structural parts of automotive, which require sophisticated shaping and forming. However, due to the low C content (typically in a range of 10 to 30 mass ppm), this steel grade is susceptible to oxidation during steelmaking and casting processes. While this steel is produced via BF (Blast Furnace)—BOF (Basic Oxygen Furnace)—RH (Ruhrstahl Heraeus)—CC (Continuous Casting) processes, reoxidation is likely to occur in the RH and the CC processes, and it is a major problem generating non-metal inclusion (NMI).[5,6,7]
ULC steel is deoxidized by aluminum alloys during the RH process. Once the liquid steel is well refined, it is sent to a casting machine through a tundish. Between the RH process and the casting process, the reoxidation may occur by several reasons (reaction with the RH slag/tundish flux,[8] open eye formation in the tundish,[9,10,11] reaction with refractories of tundish[12] and submerged entry nozzle,[13,14,15] aspiration through sliding gate,[16] etc. Therefore, Al in the liquid steel is consumed, then NMI is produced such as alumina. This deteriorates not only the process efficiency,[13,17,18] but also quality of final products.[19,20,21]
If the scope of the present study is confined to the reoxidation due to RH slag, Fe\(_t\)O, SiO\(_2\), and MnO may be responsible for the reoxidation.[6,22] Higher contents of those weak oxides would accelerate the reoxidation reaction. On the other hand, since the slag absorbs NMI at the steel–slag interface, a faster dissolution rate of NMI is also required.[23,24,25,26] The dissolution rate of alumina inclusion in various slags has been reported as summarized elsewhere.[27] In particular, the present authors investigated the dissolution rate of alumina inclusion in Fe\(_t\)O-containing RH slag,[27] utilizing high-temperature dissolution experimental technique coupled with the modified invariant interface approximation.[24] They found that decreasing viscosity of slag is one of the key factors to increase the dissolution rate, which is consistent with a proposal for various types of slags.[28] Since major components of RH slag are CaO, Al\(_2\)O\(_3\), and Fe\(_t\)O, increasing Fe\(_t\)O content ((pct Fe\(_t\)O)) would be favorable in increasing the dissolution rate. However, cleanliness of the liquid steel would deteriorate by increasing the (pct FetO), at the same time. Therefore, it is required that understanding both phenomena (dissolution of alumina into the RH slag and reoxidation of the liquid steel by the RH slag) is essential in terms of (pct Fe\(_t\)O) in the RH slag. The role of (pct Fe\(_t\)O) in the dissolution point of view was recently discussed by the present authors.[27]
There have been several reports about kinetics and mechanism of the chemical reaction between Al in liquid steel and oxide components in molten slags or fluxes.[22,29,30,31,32,33,34,35,36,37,38,39,40] Many of these reports focused on reaction kinetic between Al and SiO\(_2\).[22,29,30,31,32,33,34,39] Mass transport of Al in the liquid steel was the rate-controlling step in the majority of the previous reports.[22,29,30,31,40] However, under certain conditions, mass transport of oxide components in slag took part in the overall rate-controlling step,[33,37,38,39] thereby suggesting a mixed rate-controlling regime. The reactions involving Fe\(_t\)O occur more vigorously.[41,42,43,44] The reaction mechanism of the reoxidation reaction involving Fe\(_t\)O is relatively less clear than that involving SiO\(_2\). It was reported that the rate-controlling step of the reaction involving Fe\(_t\)O is either the mass transport of Al in liquid steel[22] or the mass transport of Fe\(_t\)O.[35] This vague understanding needs to be further resolved.
In the present study, the reaction kinetics between ULC-Al killed liquid steel and Fe\(_t\)O-containing slag representing RH slag was investigated by high-temperature chemical reaction experiment and reaction rate model analysis. Several possible rate-controlling steps were considered, and these were compared with the experimental data measured in the present study to draw the rate controlling step. A gradual change of the reaction rate-controlling step was emphasized. Finally, a suggestion for the practical RH operation is provided based on the conclusion found in the present study.
Experimental
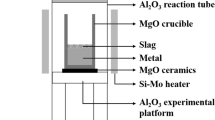
A series of high-temperature reactions between Fe–Al alloy and CaO–Al\(_2\)O\(_3\)–MgOsat.–Fe\(_t\)O slag were carried out, mostly at 1823 K under well-controlled atmosphere. The reaction represents an interfacial reaction between Al-killed ULC steel and RH slag. The initial Al content in the liquid steel ([pct Al]\(_0\)) was about 0.1.
Several master slags were preliminarily synthesized by mixing desired proportion of CaO (calcined from CaCO\(_3\) powder, > 99.5 pct, Kanto, Japan), Al\(_2\)O\(_3\) (> 99.0 pct, Samchun chemical Ltd., Korea), and MgO (> 98.0 pct, Kanto, Japan). Those were melted in graphite crucibles charged in an induction melting furnace above 1773 K (1550 °C). After the melting, the slags were poured on a water-cooled stainless steel plate, followed by crushing in a disk-milling machine. The slag was then burned under air for two hours at 1173 K (900 °C) to remove any residual carbon. Afterward, these were mixed with FeO reagent (> 99.5 pct, Kojundo, Japan) to set the initial composition of the slags, listed in Table I. The slag compositions were designed to have various initial C/A ratio ((pct CaO)\(_0\)/(pct Al\(_2\)O\(_3\))\(_0\)) and initial Fe\(_t\)O content ((pct FeO)\(_0\)). The initial MgO content ((pct MgO)\(_0\)) was set to its saturation content, which was obtained by phase diagram calculation using FactSage 7.3 with FTOxid database.[45,46] In the Table I, letters “A”, “B”, and “C” mean C/A = 1.2, 1.0, and 0.8, respectively. Numbers following the letters mean the (pct FeO)\(_0\). In the present article, “pct” means mass percent, unless others are stated.
500 g of electrolytic iron (> 99.99 pct, \(\sim \) 0.01 pct O) was charged in a magnesia crucible (OD 60 × ID 50 × H 100 (mm)). The crucible was then located in a graphite susceptor in a quartz reaction tube sealed by brass endcap on both sides. The temperature at the bottom of the crucible was monitored by a B-type thermocouple, which was connected to a proportional–integral–derivative (PID) controller. Ar gas purified by passing through Drierite and Mg chips heated at 773 K (500 °C) flowed in the reaction chamber while the crucible was heated. After the temperature reached the desired temperature (mostly 1823 K (1550 °C), some of runs at 1873 K (1600 °C) and 1923 K (1650 °C)), the atmosphere in the quartz tube was replaced with Ar-5 vol pct H\(_2\) at a flow rate of 1 L min\(^{-1}\) to remove the O in the steel down to approximately 0.002 pct. It took about 4 hours. Subsequently, Al pellets wrapped by an iron foil were dropped on the liquid steel surface. The liquid steel was homogenized for 30 minutes. Then, 40 g of the master slag prepared as above was charged onto the molten steel surface using an alumina guide tube through a hole available in the upper endcap. This moment was set to the beginning of the reaction. After the pre-determined time, a small portion of the liquid steel was sampled using a quartz tube, followed by quenching in water.
The composition of the liquid steel was analyzed by Inductively Coupled Plasma - Atomic Emission Spectroscopy (Thermo-Fisher Scientific ICAP 6500, Waltham, MA) for soluble Al content ([pct. S. Al]) and total Al content ([pct. T. Al]), although only [pct. S. Al] was further considered in the kinetic analysis. The steel specimen (1 g) was dissolved in a mixture of 1:1 HCl (20 mL) and 1:1 HNO\(_3\) (10 mL) on a hot plate at 403 K (130 °C). The solution was filtered by a 5B filter, which was additionally washed by distilled water. The filter was carefully dried in a Pt crucible at 373 K (100 °C), then was ignited thereafter. The residue was fused with 1 g of a flux (Na\(_2\)CO\(_3\):H\(_3\)BO\(_3\) = 3:1) and the melt was dissolved in 1:1 HCl 5 mL with 10 mL of the distilled water. The solution was mixed with the other solution composed of the filtrate and the washings. This was subject to the ICP analysis for [pct. T. Al]. In case of [pct. S. Al], only the filtrate and the washings were used. Total O content in the steel ([pct. T. O]) was analyzed by inert gas fusion infrared absorptiometry (LECO ON836, St. Joseph, MI). Distribution of NMI was analyzed using a field-emission scanning electron microscopy with an energy-dispersive X-ray spectroscopy (ULTRA-55, ZEISS, Oberkochen, Germany), to obtain inclusion number density for some samples. It was not attempted to sample the slag during the reaction.
Results
Figures 1 (a) through (c) show typical results obtained in the present study: [pct. T. Al] and [pct. S. Al] vs reaction time t (seconds) of liquid steel reacted with slags (A5, A10, and A15) at 1823 K. Both decreased continuously, and the decreasing rate was higher when (pct Fe\(_t\)O)\(_0\) was higher. The following chemical reaction occurred:
where underline and parenthesis refer to states dissolved in liquid steel and in liquid slag, respectively. [pct. S. Al] decreased below 0.001 after 300 seconds, when A10 and A15 slags were used. Difference between [pct. T. Al] and [pct. S. Al] may be proportional to the content of alumina inclusions suspending in the liquid steel, in some cases.

Composition change of the liquid steel ([pct. Al]\(_0\) = 0.1 reacted with CaO–Al\(_2\)O\(_3\)–MgO\(_{\text{sat.}}\)–Fe\(_t\)O slag): (a) to (c) [pct. T. Al] and [pct. S. Al], (d) to (f) [pct. T. O] of the liquid steel reacted with the slag A5, A10, and A15, respectively, measured in the present study. Closed circles in (f) are the measured inclusion number density NA, mm\(^{-2}\)). (color online)
Figures 1(d) through (f) show [pct. T. O] varied during the reaction. When (pct. Fe\(_t\)O)\(_0\) = 5, [pct. T. O] first increased until 120 seconds, then decreased continuously (Figure 1(d)). On the other hand, when (pct. Fe\(_t\)O)\(_0\) = 10, [pct. T. O] first rapidly increased, showed a maximum, then decreased. After 300 sec, [pct. T. O] did not vary noticeably (Figure 1(e)). When (pct. Fe\(_t\)O)\(_0\) = 15, [pct. T. O] showed a maximum, followed by a minimum, then increased again until 1800 seconds (Figure 1(f)), which was the last sampling time in the present study. The major source of [pct. T. O] should be Fe\(_t\)O in the slag through the Reaction [1].
Figure 2 shows a calculated Al deoxidation equilibria in liquid steel (Fe–Al–O system) at 1823 K using the Modified Quasichemical Model applied for deoxidation[47,48,49] assuming activity of Al\(_2\)O\(_3\)(s) being unity (\(a_{{\text{Al}_2}\text{O}_3(\text{s})} = 1\)). Therefore, the curve represents [pct. S. Al] vs [pct. S. O] in equilibrium with the pure solid alumina. The experimental data ([pct. S. Al] vs [pct. T. O]) in Figure 1 were shown together. It can be seen that all the [pct. T. O] data measured in the experiment are higher than the calculated equilibrium [pct. S. O]. Therefore, the measured [pct. T. O] consists of not only the soluble O but also O from suspending alumina inclusions.[22] However, according to an inclusion number density (\(N_A\) (mm\(^{-2}\))) measurement shown in Figure 1(f) for the A15 sample, the \(N_A\) first rapidly increased (t = 60 seconds), then gradually decreased. This was due to the floating up of the alumina inclusions in the condition of the present study (liquid steel in a small crucible). The moment of the maximum \(N_A\) coincides with that of [pct. T. O]. The analyzed [pct. T. O] is a sum of [pct. S. O] and [pct. I. O], and the \(N_A\) should be proportional to [pct. I. O].[50] Therefore, it is reasonable that the first rapid increase in [pct. T. O] was due to suspending alumina inclusions. Then, it is obvious that apart from the Reaction [1], the following reaction occurred:
Some of the alumina inclusions were entrapped in the steel, followed by floating up again toward the steel–slag interface. Subsequently, the alumina inclusions must have been dissolved in the slag. It was reported that dissolution of alumina inclusion into Fe\(_t\)O-containing RH slag is very fast (within a second for 20 \(\mu \)m size alumina for the slag containing 10 pct Fe\(_t\)O).[27]

This suggests that the latter increase of [pct. T. O] was due to soluble O by
where [pct. S. Al] is almost negligible. This might happen for the A10 and A15 samples. Therefore, the observed change of [pct. T. O] in the samples shown in Figure 1 can be explained as follows. When the slag comes into contact with the steel, the Reaction [2] takes place rapidly. It increases the [pct. T. O] due to the increase of [pct. I. O]. The generated alumina inclusions then float up and were absorbed in the slag, thereby decreasing [pct. T. O], as can be seen in Figure 1(f). And as seen in the Figures 1(a) through (c), [pct. S. Al] decreased. This lowers the driving force of the Reaction [2]. When (pct. Fe\(_t\)O) was lower, there was no additional source for the reoxidation, and [pct. T. O] gradually decreased as seen in Figure 1(a). However, when (pct. Fe\(_t\)O) was high, the additional reoxidation reaction (Reaction [3]) can take place. This increases [pct. T. O] due to an increase of [pct. S. O] as seen in Figures 1(e) and (f). Higher (pct. Fe\(_t\)O)\(_0\) resulted in higher [pct. T. O] as the reaction proceeded.
The analyzed [pct. T. O] did not approach the calculated deoxidation curve, suggesting a possibility of supersaturation during the reoxidation.[51,52,53,54]
Experiments using other slags of different C/A ratios also showed a similar trend in [pct. S. Al], [pct. T. O], and the \(N_A\).
From the above observation, the analyzed [pct. T. O] cannot be used to represent the extent of reoxidation by Al. Regardless of the Reactions [1] or [2], soluble Al and Fe\(_t\)O reacted and this is the main reoxidation reaction. Therefore, chasing [pct. S. Al] is useful to interpret the reaction rate. In the present study, [pct. S. Al] data were used in a series of kinetic analyses, which will be discussed in Section IV.
Discussions
In this section, the measured experimental data ([pct. S. Al] in liquid steels reacted with various C/A ratios and (pct. Fe\(_t\)O)\(_0\)) were used to interpret the reoxidation reaction mechanism by analyzing the rate of the reoxidation reaction. The stoichiometry of Fe\(_t\)O was assumed to be “FeO” for the sake of simplicity during the present kinetic analysis.
Basic Conditions and Assumptions
In the present study, the reoxidation reaction rate is represented by d[pct. S. Al]/dt due to the Reactions [1] and [2]. Since reaction rate of most high-temperature metallurgical processes is controlled by mass transport of reaction species, it was decided that (1) chemical reaction occurred rapidly,[55] (2) mass transport of Fe did not limit the rate, and therefore, (3) mass transport of Al, Al\(_2\)O\(_3\), and FeO only limit the reaction rate, exclusively or simultaneously.
Mass Balance
Only Al and O contents in the steel samples were analyzed. Therefore, composition of the slag was estimated by a mass balance between the steel and the slag by the reaction stoichiometry as follows:
where \(w_i\) and M\(_i\) are the mass and atomic (or molecular) mass of i (g and g mol\(^{-1}\)), Wsteel is the mass of steel (g), [pct. S. Al] is mass pct of the soluble Al at time t. “\(_0\)” means the beginning of the reaction (\(t = 0\) sec). Therefore, the content of Al\(_2\)O\(_3\) and FeO in the slag are expressed as:
It was assumed that mass balance was kept within the system (the steel and the slag), and CaO and MgO did not react during the reaction. Loss of soluble Al was partly due to the Reaction [2], but its contribution to the above mass balance equations was not significant compared to the initial contents of Al\(_2\)O\(_3\) and FeO. Therefore, its contribution was neglected for the sake of simplicity.
Local Equilibrium at the Interface
It was additionally assumed that the Reaction [1] arrived at equilibrium at the steel–slag interface. The standard Gibbs-energy change of the Reaction [1] (\(\Delta G^{\circ }_{1}\)) is − 639, − 631, and − 622 kJ at 1823 K (1550 °C), 1873 K (1600 °C), and 1923 K (1650 °C), respectively, where the standard states are liquid Al, liquid FeO, solid Al\(_2\)O\(_3\) (corundum), and liquid Fe, respectively. Those were obtained from FactSage FactPS database.[45,46] The equilibrium constant of the Reaction [1] is
where \(^i\) refers to “interface”. Since the steel samples contain a maximum 0.2 pct of Al and a few hundred ppm of O, aFe can be reasonably set to unity. In addition to this, the Reaction [2] is likely to occur preferentially at the interface. Some of the alumina inclusions entrap into the liquid steel, responsible for the insoluble O. The other part immediately dissolves into the liquid slag. Therefore, it may be assumed that the alumina inclusion formed by the reoxidation at the interface is indeed pure solid alumina and \(a^i_{{{\text{Al}_2}}{\text{O}_3}} = 1\). (pct. FeO)\(^i\) is then assumed to be that of the slag saturated by the alumina. In the present kinetic analysis, these assumptions were employed:
where \(a^i_{\text{FeO(sat.)}}\) is the activity of FeO in the slag saturated by alumina. This was obtained by allowing a slag of its bulk composition to be saturated by Al\(_2\)O\(_3\) using FactSage EQUILIB module.[45,46]
Thermodynamic data for liquid steel and molten slag
The activity of Al in the liquid steel (simplified to the system Fe–Al–O) was calculated using FactSage FSStel database, which employed the Modified Quasichemical Model for deoxidation in liquid steel.[47,48,49] The actual calculation was carried out using FactSage EQUILIB module. The activity of FeO in the slag composed of CaO–Al\(_2\)O\(_3\)–MgO–FeO(–Fe\(_2\)O\(_3\)) was also calculated using FactSage FTOxid database, which also employed the Modified Quasichemical Model for liquid oxide.[47]
Reaction Rate Model: Rate-controlling due to Mass Transport of Al in Liquid Steel (Model A)
Mass transport of Al in liquid steel often limits or controls the reaction rate between Al and reducible oxide components in molten slag or flux.[22,29,30,31,40] The molar flux of Al in a boundary layer of the liquid steel, JAl (mol m\(^{-2}\) s\(^{-1}\)), can be expressed as:
where A, \(n_i\), \(c_i\), and k MAl are the reaction area (m\(^2\)), the number of moles of i (mol), the concentration of i (mol m\(^{-3}\)), and the mass transport coefficient of Al (m s\(^{-1}\)), respectively. The molar flux equation can be transformed into:
where \(\rho _{\text{steel}}\) is the density of the liquid steel (= 7120 kg m\(^{-3}\)). A was assumed to be the same as the cross-sectional area of the crucible (\(= 1.96\times 10^{-3}\) m\(^2\)). [pct. S. Al]\(^i\) was obtained as follows. At a time t, both the steel and the slag are characterized by their bulk contents ([pct. S. Al], [pct. S. O], (pct CaO), (pct. Al\(_2\)O\(_3\)), (pct. MgO), (pct. FeO)). As described in Section IV–A–2, the slag at the interface was assumed be saturated by Al\(_2\)O\(_3\). \(a^i_{\text{FeO(sat.)}}\) was obtained in the saturation condition. The \(a^i_{\text{FeO(sat.)}}\) was substituted in the Eq. [8] to get \(a_{\text{Al}}^i\) at the time t. Then, for the obtained \(a_{\text{Al}}^i\), [pct. S. Al]\(^i\) was obtained by calculating a constrained equilibrium where two conditions are imposed: \(a_{{\text{Al}_2}\text{O}_3} = 1\) and the \(a_{\text{Al}}^i\) just obtained. This is equivalent to read x-coordinate of the point on the deoxidation equilibria curve for a given \(a_{\text{Al}}^i\), as marked by small filled circles in Figure 2. The calculated [pct. S. Al]\(^i\) is then substituted in the Eq. [10], and the reaction rate at t is calculated along with A, \(\rho _{\text{steel}}\), \(W_{\text{steel}}\), and an initial guess of k MAl . This procedure is repeated by a finite element approach:
where \(\Delta t\) was set to 1 sec in the present study. Substituting the updated [pct S. Al] in the Eqs. [4] through [6] updates the slag composition, followed by updating \(a^i_{\text{FeO(sat.)}}\), \(a^i_{\text{Al}}\), and [pct. S. Al]\(^i\) in the order. These steps are repeated, yielding the [pct. S. Al] during the reoxidation reaction. The calculated [pct. S. Al] was fitted to the experimental data obtained in the present study, which yields the mass transport coefficient of Al in the liquid steel k MAl . The calculation procedure is schematically shown in Figure 3.

Flowchart of the model calculation when the rate is controlled by the Al mass transport (color online)
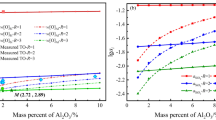
The model calculations are shown in Figure 4 with the experimental data. Closed circles are the experimental data of the present study, dotted curves were calculated using the rate model of Al mass transport control (Model A). The k MAl were fitted to the experimental data in Figures 4(a) through (c) for which (pct FeO)\(_0\) = 15. \(k_{\text{Al}}^{\text{M}} = 5 \times 10^{-4} ms^{-1}\) resulted in favorable agreement with the experimental data, and it is close to the previously reported \(k_{\text{Al}}^{\text{M}}\).[32,33] With the same \(k_{\text{Al}}^{\text{M}}\), the calculations were repeated for other cases as seen in Figures 4(d) through (i). As seen in the figure, the agreement becomes worse, as (pct FeO)\(_0\) decreased. The calculated \(\log \frac{\text{[pct. S. Al]}}{\text{[pct. S. Al]}}_0\) was lower than the experimental data. It means that the model calculation overestimated the reoxidation rate when (pct FeO)\(_0\) = 10 and 5, and decreasing the (pct FeO)\(_0\) resulted in additional resistance to the reoxidation reaction: the rate controlled not only by the mass transport of Al in the liquid steel but also components in the slag. In a series of previous investigations on reaction between Al-containing steel and CaO–SiO\(_2\) based molten slags[29,33] or molten flux,[31,32] it was reported that a rate of Al decrease in the steel was controlled by the mass transport of Al for some cases. However, the single rate-controlling step was no more valid when the molten flux becomes more viscous due to the accumulation of Al\(_2\)O\(_3\) in the consumption of SiO\(_2\).[32] In such a case, a general mixed control model was developed in which mass transport of components in molten flux was simultaneously considered.[38] In the light of this fact, decreasing (pct. FeO)\(_0\) increases the viscosity of the slag employed in the present study, and this may cause retardation of mass transport in the slag phase. This suggests an additional consideration of rate-controlling in the slag phase. Mass transport of Al\(_2\)O\(_3\) or FeO may be responsible for the discrepancy seen in Figures 4(d) through (i).

Comparison between the experimental data and the model calculations for \(\log \frac{\text{[pct. S. Al]}}{\text{[pct. S. Al]}}_0\): (a) A15, (b) B15, (c) C15, (d) A10, (e) B10, (f) C10, (g) A5, (h) B5, and (i) C5. Closed circles are the experimental data of the present study, dotted curves were calculated using the rate model of Al mass transport control (Model A), solid curve were calculated using the rate model of the mixed control by Al and Al\(_2\)O\(_3\) (Model B) (color online)
Reaction Rate Model: Mixed Transport Control by the Involvement of Slag Components
The additional consideration of rate-controlling step may be done for mass transport of Al\(_2\)O\(_3\) or that of FeO, since these two are involved in the reoxidation reaction. It is unlikely that the mass transport of the slag component solely controls the reaction rate, but the mass transport of the slag component would gradually increase resistance to the overall reaction rate. Therefore, it is reasonable to consider a mixed reaction rate model.
Consideration of Mass Transport of Al\(_2\)O\(_3\) in Liquid Slag (Model B)
When Al\(_2\)O\(_3\) accumulates in the boundary layer of the slag phase during the reaction, the diffusion of Al2O3 slows down. This would retard the reaction rate. The following analysis is for the case where mass transport of Al2O3 participates in rate-controlling step along with the mass transport of Al2O3. The molar flux of Al in the boundary layer of the molten slag, \(J_{{{\text{Al}_2}}{\text{O}_3}}\) (mol m−2 s−1), can be expressed as:
where \(k_{{{\text{Al}_2}}\text{O}_3}^{\text{S}}\) is the mass transport coefficient of Al2O3 in the slag. The minus sign was added since \(c_{{{\text{Al}_2}}\text{O}_3}^i \ge c_{{{\text{Al}_2}}\text{O}_3}\). Assuming there is no accumulation of Al\(_2\)O\(_3\) at the interface, \(J_{\text{Al}} = 2J_{{{\text{Al}_2}}\text{O}_3}\). This leads to the following relationship:
where an apparent rate constant for this mixed rate model by Al–Al\(_2\)O\(_3\) mass transport is
and
Equation [14] is then transformed into:
It should be noted that the Eq. [16] contains the interfacial concentration terms via \(L_{{\text{Al-Al}_2}\text{O}_3}\), which varies during the reaction. At a time t, \(c_{\text{Al}}^i\) was obtained as:
\(c_{{\text{Al}_2}\text{O}_3}^i\) was obtained similarly:
where \(\rho _{\text{slag}}\) is the density of the slag (kg m\(^{-3}\)). \(\rho _{\text{slag}}\) was calculated using the partial vol data.[56] Therefore,
The interfacial contents (\([\text{pct. S. Al}]^i\) and \((\text{pct. Al}_2{\text{O}}_3)^i\)) were calculated as described in Section IV–B and Figure 3. At a time t, the interfacial contents were calculated using FactSage EQUILIB module along with FSStel and FTOxid databases, thereby giving \(L_{{\text{Al-Al}_2}\text{O}_3}\). The obtained \(L_{{\text{Al-Al}_2}\text{O}_3}\) was inserted in the Eq. [14], and the \(k_{{\text{Al-Al}_2}\text{O}_3}^{\text{app}}\) was calculated along with \(k^{\text{M}}_{\text{Al}}\) (already determined in Section IV–B) and \(k^{\text{S}}_{{\text{Al}_2}\text{O}_3}\), which has been unknown yet. Then, the \(k_{{\text{Al-Al}_2}\text{O}_3}^{\text{app}}\) was substituted into the Eq. [16] in order to get the reoxidation rate, along with the \(\rho _{\text{slag}}\). [pct. S. Al] and (pct. Al\(_2\)O\(_3\)) can be updated as was done in Section IV–B. The unknown \(k^{\text{S}}_{{\text{Al}_2}\text{O}_3}\) was fitted to the present experimental data well.
During the present study, it was found that a constant \(k^{\text{S}}_{{\text{Al}_2}\text{O}_3}\) value was not satisfactory. A constant \(k^{\text{S}}_{{\text{Al}_2}\text{O}_3}\) value, which improved the fitting in one case, did not show additional good agreement with the experimental data in the other cases. In general, setting \(k^{\text{S}}_{{\text{Al}_2}\text{O}_3}\) to be a constant resulted in overestimating the reoxidation rate at lower (pct. FeO)\(_0\). This suggests that \(k^{\text{S}}_{{\text{Al}_2}\text{O}_3}\) would depend on the slag composition, partly due to changing fluidity of the slag. The fluidity of the slag should be linked to the viscosity of the slag, thereby the mass transport coefficient of a slag component being inversely proportional to the viscosity (\(\eta \)) as was pointed out previously.[30,38] In the present study, a similar approach was used.[38] The \(k^{\text{S}}_{{\text{Al}_2}\text{O}_3}\) was assumed to be inversely proportional to viscosity:
where \(k^{\text{S},\circ }_{{\text{Al}_2}\text{O}_3}\) and \(\eta ^\circ \) are the mass transport coefficient of Al\(_2\)O\(_3\) and viscosity of a reference slag. The A15 slag was chosen as the reference slag (\(k^{\text{S},\circ }_{{\text{Al}_2}\text{O}_3} = 2.5\times 10^{-9}\;\;{\text{m s}}^{-1} \), \(\eta ^\circ = 0.0735\) Pa.s at 1823 K (1550 °C)). The viscosity of the slag was calculated by a viscosity model proposed by Grundy et al.[57] using FactSage viscosity module.
The calculated results using the mixed rate control model by Al and Al\(_2\)O\(_3\) (Model B) are shown in Figure 4 by solid curves. It is seen that the agreement with the experimental data was improved, in particular for the cases of lower (pct. FeO)\(_0\). The calculation results in this mixed model become similar to those of the Model A as (pct. FeO)\(_0\) increased. Therefore, it is likely that higher (pct. FeO)\(_0\) eliminates the resistance to the reaction in the slag phase by lowering the viscosity of the slag.
Consideration of Mass Transport of FeO in Liquid Slag (Model C)
Delivery of FeO to the reaction interface may be slow and may limit the reaction rate. The following analysis is for the case where mass transport of FeO participates in rate controlling step along with the mass transport of Al. The molar flux of FeO in the boundary layer of the molten slag, \(J_{\text{FeO}}\) (mol m\(^{-2}\) s\(^{-1}\)), can be expressed as:
where \(k_{\text{FeO}}^{\text{S}}\) is the mass transport coefficient of FeO in the slag. According to the stoichiometric relationship, \(J_{\text{Al}} = \frac{2}{3}J_{\text{FeO}}\). This leads to the following relationship:
where an apparent rate constant for this mixed rate model by Al–FeO mass transport is
and
Equation [22] is then transformed into:
In this model, the apparent mass transport coefficient \(k_{\text{Al-FeO}}^{\text{app}}\) becomes higher than \(k^{\text{M}}_{\text{Al}}\), which can increase the overall rate. The term in the parenthesis in the Eq. [25] may be small, thereby decreasing the rate even though the increase of \(k^{\text{M}}_{\text{Al}}\). However, since the term \(L_{\text{Al-FeO}}\) appears in both places (one in the \(k_{\text{Al-FeO}}^{\text{app}}\) and the other in the parenthesis) and its effect cancels out, it is unlikely. Indeed, the present case is similar to enhancement of diffusion-limited rates of vaporization of metals where vaporization rate is accelerated by the counter flux of two reactants.[58] Thus, this consideration ruled out a possibility of retarding the reoxidation rate by FeO mass transport in the slag, and this Model C is not applicable.
From the analysis using the present models (Model B and Model C), it is clear that the mass transport of Al\(_2\)O\(_3\) indeed participates as a part of rate controlling step.
Effect of Temperature on the Reaction Rate
Figure 5 shows the change of [pct. S. Al] in the steel reacted with A5 slag (C/A = 1.2, (pct. FeO)\(_0\) = 5) at three different temperature. The increasing temperature increased the reaction rate. Model calculations using the Model B show good agreement with the experimental data. For this calculation, mass transport coefficients of Al and Al\(_2\)O\(_3\) were obtained as follows:
-
\(k^{\text{M}}_{\text{Al}}\): the value obtained at 1823 K (1550 °C) (\(5 \times 10^{-4}\) m s\(^{-1}\)) was extrapolated to 1873 K (1600 °C) and 1923 K (1650 °C) using an Arrhenius equation (\(k^{\text{M}}_{\text{Al}} = C \exp (-E_{\text{A(Al)}}/\text{R}T)\)), along with the activation energy \(E_{\text{A(Al)}}\) (= 119 kJ mol\(^{-1}\)) reported previously[32] (a solid line in Figure 6).
-
\(k_{{\text{Al}_2}\text{O}_3}^{\text{S}}\): fitted to the experimental data in Figure 6, after the extrapolation of the \(k^{\text{M}}_{\text{Al}}\).
Using the fitted \(k_{{\text{Al}_2}\text{O}_3}^{\text{S}}\), the activation energy for the \(k_{{\text{Al}_2}\text{O}_3}^{\text{S}}\) (\(E_{{\text{A(Al}_2}\text{O}_3)}\)) was obtained by the Arrhenius plot, as seen in Figure 6(a). The obtained \(E_{{\text{A(Al}_2}\text{O}_3)}\) was 1147 kJ mol\(^{-1}\), respectively, which are much greater than \(E_{\text{A(Al)}}\). It should be noted that such high \(E_{{{\text{A(Al}_2}\text{O}_3})}\) was not obtained by a rigorous analysis of Al\(_2\)O\(_3\) mass transport in the slag. Therefore, this value should be considered to be valid only in the present mixed transport case.

Decrease of [pct. S. Al] in liquid steel reacted with A5 slag (C/A = 1.2, (pct. FeO)\(_0\)) at 1823 K (1550 °C), 1873 K (1600 °C), and 1923 K (1650 °C) (color online)

Arrhenius plot of (a) \(k^{\text{M}}_{\text{Al}}\) and \(k^{\text{S}}_{{\text{Al}_2}\text{O}_3}\), and (b) \(k_{{\text{Al-Al}_2}\text{O}_3}^{\text{app}}\). A solid line in (a) was taken from Ref. [32] (color online)
Gradual Variation of Rate-Controlling Step
It was seen that the reoxidation rate was controlled by mass transport of Al in liquid steel when (pct. FeO)\(_0\) was high (e.g. 15). On the other hand, the rate was additionally limited by mass transport of the slag components (Al\(_2\)O\(_3\)) when (pct. FeO)\(_0\) was low (e.g. 5). Since the reoxidation reaction accompanies with the continuous reduction of the FeO and increase of Al\(_2\)O\(_3\) in the slag, the rate-controlling step would have to change gradually, i.e., mass transport of Al in liquid steel \(\rightarrow \) mixed mass transport control of Al in liquid steel and Al\(_2\)O\(_3\) in the slag. This gradual change was examined by the reaction rate model developed in the present study (the Model B). As was seen in the relative size of the activation energy of individual mass transport coefficient (\(E_{\text{A(Al)}}\) and \(E_{{\text{A(Al}_2}\text{O}_3)}\)), the gradual change should increase activation energy of the apparent rate constant (\(k_{{\text{Al-Al}_2}\text{O}_3}^{\text{app}}\)). Figure 6(b) shows the other Arrhenius plot of \(k_{{\text{Al-Al}_2}\text{O}_3}^{\text{app}}\) for the reoxidation reaction by the A5 slag. The \(k_{{\text{Al-Al}_2}\text{O}_3}^{\text{app}}\) values were collected and used to extract the activation energy: (1) at the beginning of the reaction, (2) in the middle of the reaction ([pct. S. Al] becomes a half of [pct. S. Al]\(_0\)), and (3) in the other middle of the reaction (the reoxidation rate \(d \text{[pct. S. Al]}/dt\) decreased to half of its initial value (\(d \text{[pct. S. Al]}/dt|_{t=0}\))) . The apparent activation energy of \(k_{{\text{Al-Al}_2}\text{O}_3}^{\text{app}}\) (\(k_{{\text{Al-Al}_2}\text{O}_3}\)) increased from 250 to 337 kJ mol\(^{-1}\) when the “half-concentration” was reached, and it also increased to 291 kJ mol\(^{-1}\) when the “half-rate” was reached. This gives an evidence that the reoxidation reaction is governed not by a simple manner, but by gradual mechanism change during the reaction. It should be noted that the choices of the “half-concentration” and the “half-rate” do not represent any specific physical meaning, but were only arbitrary.
Practical Application
It was shown that the present experimental data could be explained by the Model B. Mass transport of Al in the liquid steel cannot be a sole rate-controlling step, and mass transport of the slag component is involved as a part of the rate-controlling step.
As was already seen in Figure 4, decreasing (pct. FeO)\(_0\) decreased the apparent reoxidation rate by the decreasing source of the reoxidation (FeO) and increasing viscosity of the slag. Therefore, (pct FeO)\(_0\) in the slag is the most critical factor in the control of the reoxidation rate. C/A ratio in the slag does not look as critical as the (pct FeO)\(_0\) in the control of the reoxidation rate.
In this section, the Model B was used for further analysis for the reoxidation reaction. Figure 7 shows calculated [pct. S. Al]\(^i\), \(L{_{\text{Al-Al}_2}\text{O}_3}\), and \(k^{\text{app}}_{{\text{Al-Al}_2}\text{O}_3}\) (m s\(^{-1}\)) at 1823 K, respectively. Numbers with right brace mean (pct. FeO)\(_0\). Solid, dashed, and dotted lines are the calculated results of different C/A ratio: 1.2, 1.0, and 0.8, respectively. Decreasing (pct. FeO)\(_0\) increased [pct. S. Al]\(^i\), therefore decreased the driving force of mass transport of Al in the liquid steel. It also decreased \(L_{{\text{Al-Al}_2}\text{O}_3}\) and \(k^{\text{app}}_{{\text{Al-Al}_2}\text{O}_3}\), which also decreased the reoxidation rate. When (pct. FeO)\(_0\) was 10 and 15, C/A ratio does not influence the three terms ([pct. S. Al]\(^i\), \(L_{{\text{Al-Al}_2}\text{O}_3}\), and \(k^{\text{app}}_{{\text{Al-Al}_2}\text{O}_3}\)) noticeably. Therefore, C/A is not a critical factor to the reoxidation rate in this case. On the other hand, when (pct FeO)\(_0\) was as low as 5, C/A ratio does influence the three terms: increasing C/A ratio increased [pct. S. Al]\(^i\) and decreased \(L_{{\text{Al-Al}_2}\text{O}_3}\) and \(k^{\text{app}}_{{\text{Al-Al}_2}\text{O}_3}\). This tells that increasing the C/A ratio would decrease the reoxidation rate. In Figures 4(g) through (i), initial reoxidation rate looks similar each other, but the [pct. S. Al]/[pct. S. Al]\(_0\) at the last moment (600 seconds) are seen to be different: the higher C/A ratio is, the higher the [pct. S. Al]/[pct. S. Al]\(_0\) is at 600 seconds. High C/A ratio decreased the reoxidation rate because \(L_{{\text{Al-Al}_2}\text{O}_3}\) is low, which is indeed due to the high [pct. S. Al]\(^i\). At the steel–slag interface, the [pct. S. Al]\(^i\) is determined by the equilibrium of the Reaction [1]. For a given steel and slag composition, the equilibrium [pct. S. Al]\(^i\) is governed by the activity of FeO in the slag (see the Eq. [8]). If slags of a similar level of (pct. FeO)\(_0\) with different C/A0 ratios are considered, [pct. S. Al]\(^i\) would be higher if the activity coefficient of FeO (\(\gamma _{\text{FeO}}\)) is lower in the slag. According to the available literature, increasing C/A0 ratio in the CaO–Al\(_2\)O\(_3\)–Fe\(_t\)O slag lowers \(\gamma _{\text{FeO}}\).[59,60] Although the slag used in the present study and practical RH slags contain some other minor components such as MgO, increasing C/A0 ratio in RH slag of low (pct. FeO)\(_0\) (i.e. \(\sim \) 5) can lower the reoxidation rate. This conclusion is in agreement with recent reports.[61,62]

Calculated various data from the Model B during the reoxidation reaction at 1823 K: (a) Al content at the interface, [pct. S. Al]\(^i\), (b) local equilibrium condition, \(L_{{\text{Al-Al}_2}\text{O}_3}\), and (c) apparent mass transport coefficient, \(k^{\text{app}}_{{\text{Al-Al}_2}\text{O}_3}\) (m s\(^{-1}\))(color online)
A recent work on viscosity (\(\eta \)) of similar slag system (CaO–Al\(_2\)O\(_3\)–Fe\(_t\)O–SiO\(_2\)–MgO) by Kim and Park showed that the \(\eta \) of low C/A ratio depends on (pct Fe\(_t\)O) more significantly than that of high C/A ratio, in the range of C/A ratio from 1.0 to 2.0.[63] This observation and the present results may be summarized as follows (see Table II). Upon increasing (pct. Fe\(_t\)O) in RH slags, [pct. S. Al]\(^i\) decreases. This increases \(L_{{\text{Al-Al}_2}\text{O}_3}\), and consequently increases \(k^{\text{app}}_{{\text{Al-Al}_2}\text{O}_3}\). These phenomena become more significant when the C/A ratio is high. At the same time, \(\eta \) of the slag decreases upon increasing the (pct. Fe\(_t\)O). However, the decrease of \(\eta \) was reported to be more significant when the C/A ratio is low. Since the \(\eta \) is thought to be inversely proportional to the reaction rate constant (\(k^{\text{app}}_{{\text{Al-Al}_2}\text{O}_3}\) in the present study or as was reported in Reference 38), the more significant effect of the (pct. Fe\(_t\)O) on the \(\eta \) at low C/A ratio is not fully consistent with the kinetic analysis presented in the present study, although the effect of \(\eta \) was indeed taken into account in the present reaction rate model. This implies that the reoxidation rate is governed partly by the \(\eta \) of the slag involved, but it is not a major determining factor. Clearly, thermodynamics at the steel-slag interface play more significant role.
In practical operation, FetO in the RH slag comes from BOF slag due to partial entrapment of the slag during tapping to a ladle.[64] The FetO is one of significant sources of reoxidation during RH process. Deoxidizing the RH slag by killing FetO before or during the process by adding Al or C source is one of the possible ways to suppress the reoxidation. However, it is generally not easy to reduce the FetO component in the slag because the top surface of the slag is partially solidified and a substantial amount of fume is generated. This lowers workability during the practical operation. On the other hand, higher FetO content in the slag lowers the viscosity of the slag, which is beneficial to absorb floating alumina inclusions into the slag, as was mentioned in Section I.[27] This suggests that having FetO in the RH slag may be utilized to absorb the inclusions floating up toward the steel-slag interface if reoxidation by the FetO is not concerned. A compromise for the content of FetO may be thought between removing a source of reoxidation to suppress the formation of alumina inclusions and keeping a source of lowering the viscosity of the slag to absorb inclusions rapidly. This depends on the conditions of each RH facility and workability in the plant. As an alternative, killing FetO and adding a non-reducible flux that lowers the viscosity of RH slag can suppress the reoxidation and increase the dissolution rate of inclusion in the slag. Of course, it adds then one more concern regarding production cost and refractory life.[65,66]
As was mentioned in Section III, the measured [pct. T. O] shown in Figures 1(e) through (f) at a later stage of the reaction seems due to soluble O. This is likely to happen beneath the slag–steel interface. When this soluble O reacts with soluble Al, this will also be another source of alumina inclusion due to the reoxidation reaction indirectly. A new method to analyze the soluble O in steel specimen using conventional inert gas fusion infrared absorptiometry was recently developed by the present author.[67] It is suggested that the cleanliness of liquid steel in terms of O content should consider the presence of soluble O, which is generated due to FetO in slag.
Conclusion
In view of reoxidation of ULC steel during RH process, reaction rate and mechanism of the following reaction was investigated by a series of laboratory-scale experiment and reaction rate model analyses coupling CALPHAD thermodynamics and mass transport theory:
(pct. FetO)\(_0\), C/A ratio in slag, and reaction temperature were varied to find critical factors influencing the reaction rate and mechanism. From the experiment and the model analysis, the followings were concluded:
-
1.
Increasing (pct. FetO)\(_0\) increased the reoxidation rate, and varying C/A ratio was important when (pct. FetO)\(_0\) was low (5 in the present study).
-
2.
Analyzed [pct. T. O] in steel was partly contributed by soluble O, and a possibility of supersaturation was observed.
-
3.
Reoxidation rate was nearly controlled by mass transport of Al in liquid steel, but decreasing (pct. FetO)\(_0\) alters the rate-controlling step toward mixed transport control involving Al\(_2\)O\(_3\) transport.
-
4.
A new reaction rate model was developed which takes into account a local equilibrium by CALPHAD method, mixed transport rate in both phases, effect of viscosity on mass transport coefficients of Al\(_2\)O\(_3\) or FetO) in the slag, and was shown to be in good agreement with the present experimental data.
-
5.
Activation energy of the mass transport process gradually increased as the reaction proceeded. This suggests that the rate-controlling step gradually changes during the reaction. Therefore, the reaction mechanism was changed during the reaction.
-
6.
At high (pct FetO)\(_0\), C/A0 ratio in the slag hardly affects the reoxidation rate. At low (pct FetO)\(_0\), increasing C/A0 lowers the reoxidation rate, thus is beneficial to suppress the reoxidation.
References
B. Jiang, X. Hu, G. He, H. Peng, H. Wang, Y. Liu, Met. Mater. Int. 26, 1295–1305 (2020)
V.J. Martinez, J.I. Verdeja, J.A. Pero-Sanz, Mater. Charact. 46, 45–53 (2001)
J. Choi, J. Kim, POSCO Tech Rep 20, 7–11 (2017)
G. Yin, C. Yang, Y. Lu, J. Mater. Sci. Technol. 26, 433–38 (2010)
C.J. Treadgold, Ironmak. Steelmaking 30, 120–24 (2003)
M. Wang, Y.-P. Bao, H. Cui, H.-J. Wu, W.-S. Wu, ISIJ Int. 50, 1606–11 (2010)
Q. Ren, Y. Zhang, Y. Ren, L. Zhang, J. Wang, Y. Wang, J. Mater. Sci. Technol. 61, 147–58 (2021)
T.S. Kim, Y. Chung, L. Holappa, J.H. Park, Metall. Mater. Trans. B 48B, 1736–47 (2017)
S. Chatterjee, D. Li, K. Chattopadhyay, ISIJ Int. 56, 1889–1892 (2016)
C. Chen, L.T.I. Jonsson, A. Tilliander, G. Cheng, P.G. Jönsson, Chem. Eng. Sci. 137, 914–937 (2015)
R. Singla, S. Tripathy, S.K. Das, Chem. Eng. Sci. 203, 391–401 (2019)
E. Zinngrebe, J. Small, S. van der Laan, A. Westendorp, Metall. Mater. Trans. B 51, 2321–2338 (2020)
J.-H. Lee, M.-H. Kang, S.-K. Kim, Y.-B. Kang, ISIJ Int. 58, 1257–1266 (2018)
J.-H. Lee, S.-K. Kim, M.-H. Kang, Y.-B. Kang, BHM Berg- und Hüttenmännische Monatshefte 163, 18–22 (2018)
J.-H. Lee, M.-H. Kang, S.-K. Kim, J. Kim, M.-S. Kim, Y.-B. Kang, ISIJ Int. 59, 749–758 (2019)
B. Thomas ,H. Bai, 18rd Process Technology Division Conference Proceedings, vol. 18, 2001, pp. 895–912
Y. Vermeulen, B. Coletti, B. Blanpain, P. Wollants, J. Vleugels, ISIJ Int. 42, 1234–1240 (2002)
S. Basu, S.K. Choudhary, N.U. Girase, ISIJ Int. 44, 1653–1660 (2004)
S.-M. Lee, S.-J. Kim, Y.-B. Kang, H.-G. Lee, ISIJ Int. 52, 1730–1739 (2012)
X. Deng, C. Ji, W. Dong, L. Li, X. Yin, Y. Yang, A. McLean, Ironmak. Steelmaking 45, 592–602 (2018)
H. Yasunaka, R. Yamanaka, T. Inoue, T. Saito, Tetsu-to-Hagane 81, 529–534 (1995)
H. Sun, K. Mori, ISIJ Int. 36, S34–S37 (1996)
J.-Y. Choi, H.-G. Lee, J.-S. Kim, ISIJ Int. 42, 852–860 (2002)
S. Feichtinger, S.K. Michelic, Y.-B. Kang, C. Bernhard, J. Am. Ceram. Soc. 97, 316–325 (2014)
A.-H. Bui, H.-M. Ha, I.-S. Chung, H.-G. Lee, Met. Mater. Int. 11, 319–326 (2005)
A.-H. Bui, H.-M. Ha, Y.-B. Kang, I.-S. Chung, H.-G. Lee, Met. Mater. Int. 11, 183–190 (2005)
Y.-J. Park, Y.-M. Cho, W.-Y. Cha, Y.-B. Kang, J. Am. Ceram. Soc. 103, 2210–2224 (2020)
M. Valdez, G.S. Shannon, S. Sridhar, ISIJ Int. 46, 450–457 (2006)
M.A. Rhamdhani, K.S. Coley, G.A. Brooks, Metall. Mater. Trans. B 36B, 219–227 (2005)
D.J. Kim, J.H. Park, Metall. Mater. Trans. B 43B, 875–886 (2012)
M.-S. Kim, S.-W. Lee, J.-W. Cho, M.-S. Park, H.-G. Lee, Y.-B. Kang, Metall. Mater. Trans. B 44B, 299–308 (2013)
Y.-B. Kang, M.-S. Kim, S.-W. Lee, J.-W. Cho, M.-S. Park, H.-G. Lee, Metall. Mater. Trans. B 44B, 309–316 (2013)
J. Park, S. Sridhar, R.J. Fruehan, Metall. Mater. Trans. B 45B, 1380–1388 (2014)
P. Ni, T. Tanaka, M. Suzuki, M. Nakamoto, M. Ersson, P.G. Jönsson, ISIJ Int. 59, 2024–2035 (2019)
A.-J. Deng, D.-D. Fan, H.-C. Wang, C.-H. Li, J. Iron. Steel Res. Int. 27, 409–419 (2020)
M.-S. Kim, M.-S. Park, Y.-B. Kang, Metall. Mater. Trans. B 50B, 2077–2082 (2019)
M.-S. Kim, M.-S. Park, S.-E. Kang, J.-K. Park, Y.-B. Kang, ISIJ Int. 58, 686–695 (2018)
M.-S. Kim, Y.-B. Kang, Calphad 61, 105–115 (2018)
M.-S. Kim, M.-S. Park, S.-H. Jung, S.-Y. Kim, Y.-B. Kang, Metall. Mater. Trans. B 51B, 3067–3078 (2020)
T. Isono, K. Ohnuki, K. Umezawa, 4th international conference on Molten Slags and Fluxes, (1992), 493–498
T. Ogura, R. Fujiwara, R. Mochizuki, Y. Kawamoto, T. Oishi, M. Iwase, Metall. Trans. B 23, 459–466 (1992)
H. Ohta, H. Suito, Metall. Mater. Trans. B 29B, 119–129 (1998)
S. Basu, A.K. Lahiri, S. Seetharaman, Metall. Mater. Trans. B 41B, 414–419 (2010)
S. Basu, A.K. Lahiri, S. Seetharaman, Metall. Mater. Trans. B 39B, 447–456 (2008)
C.W. Bale, E. Bélisle, P. Chartrand, S.A. Decterov, G. Eriksson, K. Hack, I.-H. Jung, Y.-B. Kang, J. Melançon, A.D. Pelton, C. Robelin, S. Petersen, Calphad 33, 295–311 (2009)
C.W. Bale, E. Bélisle, P. Chartrand, S.A. Decterov, G. Eriksson, A.E. Gheribi, K. Hack, I.H. Jung, Y.B. Kang, J. Melançon, A.D. Pelton, S. Petersen, C. Robelin, J. Sangster, P. Spencer, M.-A. Van Ende, Calphad 54, 35–53 (2016)
A.D. Pelton, P. Chartrand, Metall. Mater. Trans. A 32A, 1355–1360 (2001)
M.-K. Paek, J.-J. Pak, Y.-B. Kang, Metall. Mater. Trans. B 46B, 2224–2233 (2015)
Y.-B. Kang, ISIJ Int. 60, 2717–2730 (2020)
A. Karasev, H. Suito, Metall. Mater. Trans. B 30B, 259–270 (1999)
H. Suito, Proc. of the Ethem Turkdogan Symposium 141–152 (1994)
K. R. Lee, H. Suito ,Metallurgical and Materials Transactions B, vol. 27, (1996), p. 423
M.L. Turpin, J.F. Elliott, J. Iron Steel Inst. Lond. 204, 217–225 (1966)
G. Forward, J.F. Elliott, J. Metals 19, 54–59 (1967)
B. Larsen, in “Basic Open Hearth Steelmaking With Supplement on Oxygen in Steelmaking”, chapter 19. ed. by G. Derge, (AIME, 1964), pp. 583–611
R.A. Lange, I.S. Carmichael, Geochim. Cosmochim. Acta 51, 2931–2946 (1987)
A.N. Grundy, H. Liu, I.-H. Jung, S.A. Decterov, A.D. Pelton, Int. J. Mater. Res. 99, 1185–1194 (2008)
E. Turkdogan, P. Grieveson, J. Beisler, Trans. Metall. Soc. AIME 227, 1258–1264 (1963)
K.R. Lee, H. Suito, Metall. Mater. Trans. B 25B, 893–902 (1994)
V. Espejo, M. Iwase, Metall. Mater. Trans. B 26B, 257–264 (1995)
Y.-Q. Ji, C.-Y. Liu, Y. Lu, H.-X. Yu, F.-X. Huang, X.-H. Wang, Metall. Mater. Trans. B 49B, 3127–3136 (2018)
Y.-Q. Ji, C.-Y. Liu, H.-X. Yu, X.-X. Deng, F.-X. Huang, X.-H. Wang, J. Iron. Steel Res. Int. 27, 402–408 (2020)
T.-S. Kim, J.-H. Park, ISIJ Int. 61, 724–733 (2021)
M. Martín, M. Rendueles, M. Díaz, Chem. Eng. Sci. 60, 5781–5791 (2005)
J.S. Han, Y. Chung, J.H. Park, Met. Mater. Int. 25, 1360–1365 (2019)
B. Ma, Q. Zhu, Y. Sun, J. Yu, Y. Li, J. Mater. Sci. Technol. 26, 715–720 (2010)
H.-M. Hong ,Y.-B. Kang, ISIJ International, vol. 61, (2021), No. 9, https://doi.org/10.2355/isijinternational.ISIJINT-2020-596
Acknowledgments
This work was financially supported by POSCO, Rep. of Korea.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Manuscript submitted February 25, 2021, accepted May 9, 2021.
Rights and permissions
About this article

Cite this article
Cho, YM., Cha, WY. & Kang, YB. Reoxidation of Al-Killed Ultra-Low C Steel by FetO in RH Slag: Experiment, Reaction Rate Model Development, and Mechanism Analysis. Metall Mater Trans B 52, 3032–3044 (2021). https://doi.org/10.1007/s11663-021-02220-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-021-02220-8