
Abstract
In this study, kinetics of decarburization and manganese losses from Fe–Mn–C steels containing 10, 15, and 25 pct Mn and 0.18 and 0.42 pct C during Ar–O2 bubbling in the temperature range of 1823 to 1913 K were studied. The results showed that higher temperature resulted in higher rate of decarburization because of more oxygen partitioned to carbon oxidation than to manganese. Manganese loss was explained by considering multiple mechanisms: oxide formation and vapor formation, and evaporation-condensation. Manganese loss increased at higher temperatures which has been attributed to an increase in vapor pressure. Changing the depth of nozzle submergence did not make any difference in the decarburization and manganese loss, the reactions occurred well within the time the bubble was present in the melt. Prolonged time after reaction did not lead to a repartitioning of the species. Comparison of thermodynamic calculations with experimental observations showed that manganese and carbon in the bulk metal were not in equilibrium with the gas species in the bubble. After the bubble reacted near the tip of nozzle, the system did not proceed to true equilibrium.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The automotive industry is facing increasing global demand to comply with environmental regulations and fuel efficiency. This has driven steelmakers to fulfill these requirements by the development of new high strength steels with improved mechanical properties.[1,2,3,4] Manganese is a critical alloying element for advanced high strength steels due to its ability to improve mechanical properties such as toughness and ductility. The manganese concentration in TWIP and TRIP steels varies between 5 and 30 pct,[5,6,7] while the carbon concentration is required to be less than 0.1 pct.[8] Numerous research has been conducted regarding the mechanical properties and microstructure of these alloys.[9,10] However, published data about the processing of these steels are scarce.
Decarburization of liquid iron-manganese alloys is challenging due to the high vapor pressure of manganese and its affinity for oxygen. There has been more work on ferromanganese alloys that are used as alloy additions to make manganese-containing steels. Manganese oxygen refining (MOR) with a similar concept of basic oxygen furnace (BOF) allows the production of medium and low carbon ferromanganese by blowing oxygen through a lance or bottom tuyeres into high carbon ferromanganese.[11] The carbon concentration of 0.5 to 1 pct can be achieved if the temperature rises to above 2023 K (1750 °C). However, this leads to undesirable manganese losses by evaporation and oxidation which can be as high as 5 pct.[12] To achieve the decarburization to meet metallurgical requirements, it is necessary to use argon to dilute the blown oxygen.[13] Hence, argon-oxygen decarburization (AOD) is a vital step after melting the materials in the electric arc furnace (EAF), (the so-called EAF-AOD route) to economically produce high manganese steels with rapid decarburization while avoiding excessive loss of manganese. The manganese losses could be compensated by the addition of ferrosilicon during the reduction stage of AOD.[14] The AOD was first commercialized for stainless, which has been well researched,[15,16,17,18,19,20] whereas there are only a few published works on the argon-oxygen processing of ferromanganese alloys.[21,22,23,24] Yamamoto et al.[25] decarburized molten Fe-76Mn–7C alloys to 0.5 to 1 pct C with Ar–O2 blowing in a combined converter with a top lance for O2 blowing and a bottom plug for Ar stirring or Ar–O2 injection. The total manganese loss was reported to be about 3 to 4 pct due to oxidation and evaporation. As reported by Lee et al.[26] decarburization of high carbon ferromanganese proceeds in a series of sequential stages. According to these workers, in the first of three stages, the rate is controlled by the chemical reaction between oxygen and carbon at the interface between the gas and the metal. The reaction is controlled by the rate of supply of oxygen in the middle stage. In the final stage, at low carbon levels, the rate of mass transfer of carbon in the liquid to the gas/liquid interface was controlling. These researchers proposed that a portion of oxygen which is not used for the decarburization will react with manganese vapor and form MnO mist beyond the surface of the metal according to the fuming mechanism proposed by Turkdogan et al.[27] You et al.[21,22,23,24] have studied the decarburization of high carbon ferromanganese both in laboratory scale and pilot converters. These workers reported that efficiency of oxygen for carbon removal improved with decreasing concentration of oxygen in Ar-O2 gas mixtures and with increasing temperature. Furthermore, the evaporative and oxidative losses of manganese were reported to be in the range of 2–25 pct in the combined blowing converter. These workers did not determine the contribution of each of these mechanisms for manganese loss. Liu et al.[28] carried out a laboratory study of the decarburization and manganese loss from Fe–16Mn–3C alloy by injection of CO2–O2 showing that the introduction of CO2 to oxygen increased the rate of decarburization while helping manganese retention in the melt.
The current paper is a part of a larger experimental study that seeks to understand the kinetics and mechanisms of the decarburization and manganese losses from high manganese steels during Ar–O2 bubbling. A previous paper by authors[29] investigated the effect of alloy chemistry on decarburization and manganese losses. It has been shown that the total manganese loss in the processing of Fe–Mn–C alloys containing 10 to 25 pct Mn and 0.05 to 0.42 pct C was higher than being explained only as vapor and oxide. An evaporation-condensation mechanism was proposed wherein manganese evaporates in equilibrium with manganese in the metal and condenses at a lower temperature inside the bubble. This mechanism can work only if there is sufficient temperature gradient between the bubble surface and its interior. It was demonstrated that this mechanism works both from a thermodynamic and kinetic point of view. In a further study by current authors,[30] it has been shown that increasing the gas flow rate and oxygen fraction in the gas mixture led to higher rates of decarburization and manganese losses from Fe–15Mn–1C alloy. Moreover, it was shown that the ratio of manganese loss to decarburization was controlled by the relative mass transport of manganese and carbon in the metal. Unlike the previous study for lower range carbon alloys, assuming that products of oxidation were CO and MnO, there was a considerable amount of unconsumed oxygen. Based on mass balance, it has been shown how oxygen is partitioned among CO, CO2, MnO, and FeO. The objective of current paper is to examine the effect of melt temperature, alloy composition, and nozzle submergence depth on decarburization and manganese losses.
Experimental Methods
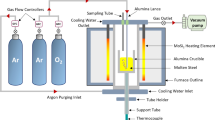
The experimental procedure in this paper is the same as previous work by authors,[29,30] but have repeated the description here for the convenience of the reader. The experiments were conducted in a resistant heated vertical tube furnace (Figure 1) in the temperature range of 1823 to 1913 K (1550 to 1640 °C). The temperature was controlled using a type-B platinum-rhodium thermocouple within ±8 K. There was an alumina working tube with a diameter of 0.079 m which was sealed using O-rings and water-cooled stainless steel caps at both ends allowing to evacuate the furnace using a vacuum pump (at the top end) or argon for purging being introduced to the furnace from bottom end. Six alloy compositions were used in this work as listed in Table I. Fe–Mn–C alloys containing 10, 15, and 25 pct Mn and 0.18 and 0.42 pct C. The desired amount of manganese flakes (99.990 pct purity), electrolytic iron powder (99.977 pct purity), and graphite rods (99.9999 pct purity) were mixed to prepare the alloys. Prior to mixing alloys, manganese flakes were acid pickled with 5 pct HCl solution to remove the surface oxide layer. The mass of alloys was 0.33 kilograms in all experiments. The alloy was placed in an alumina crucible with 0.038 m inner diameter, 0.04 m outer diameter, and height of 0.09 m and raised to the hot zone of the furnace. Before each experiment, furnace was sealed and evacuated to the range of 80 to 150 millitorr and backfilled with argon. Ar purging was continued until the end of the experiment to avoid oxidation of the liquid metal. When the target temperature for each experiment was reached, one hour was given to ensure that melt is homogenized. Then, a single-bore alumina tube with an outer diameter of 4.8 × 10−3 m and an inner diameter of 1.6 × 10−3 m was lowered into the melt so that submergence depth was reached the desired one (depending on the experiment, either 0.01, 0.02, 0.025 or 0.03 m). Then bubbling of Ar–O2 mixture started at the total flow rate of 5 × 10−6 Nm3/s (300 Nml/min) and composition of Ar–6.7 pct O2 for all experiments. The metal samples were taken frequently and analyzed by ICP-OES for manganese content and by LECO for carbon content. Also, at the end of the experiments fume samples were collected from the wall and cap of the furnace and analyzed by ICP-OES for their manganese and iron content.

Schematic diagram of the experimental setup, not to scale
Results
Effect of Temperature on Manganese Loss and Decarburization
To study the effect of temperature on the rate of manganese loss, experiments were carried at 1823, 1893, and 1913 K. The total gas flow rate and gas composition were 5 × 10−6 Nm3/s and Ar–6.7 pct O2 and nozzle submergence depth was 0.03 m. Figure 2 shows the three-stage pattern of manganese concentration in the bath vs time for Fe–25Mn–0.42C alloy at 1823 K. It should be noted that all manganese data in this work conform to this three-stage behavior. In stage 1, there was a rapid manganese loss at an almost constant rate, followed by a plateau in stage 2. In stage 3, manganese concentration decreased, again at a constant rate. The corresponding carbon behavior is also studied in these three stages. In order to make graphs clear to read, three stages are not delineated in each of Figures 3 through 10. The authors analyzed the final slag composition in previous work[29,30] but periodic slag sampling was challenging because of insufficient slag volume. Therefore, it was not possible to use the slag to investigate the middle stage of the reaction. The authors are not able to offer an explanation for this stage although it is interesting to note that this stage is also found in pilot-scale refining of ferromanganese.[23,26]

Three-stage pattern of manganese concentration vs time for Fe–25Mn–0.42C alloy at 1823 K, gas flow rate: \( 5 \times 10^{ - 6} \)Nm3/s and gas composition: Ar–6.7 pct O2, typical of all experiments. Ref. [29]

Manganese concentration in the bath vs time for (a) Fe–25Mn–0.18C, (b) Fe–15Mn–0.18C, and (c) Fe–10Mn–0.18C alloys, Gas flow rate: \( 5 \times 10^{ - 6} \)Nm3/s and gas composition: Ar–6.7 pct O2, variable: temperature, nozzle submergence depth: 0.03 m. Data at 1823 K are from Ref. [29]
Figures 3((a) through (c)) and 4((a) through (c)) show the change of manganese concentration in the bath vs time for Fe–0.18C and Fe–0.42C alloys, respectively. Figures a, b, and c correspond to the manganese concentrations of 25, 15, and 10 pct, respectively. The pattern of manganese behavior is similar for all temperatures and compositions. Initially, there was a manganese loss at a constant rate, followed by a reversion or plateau in the middle stage and another manganese loss at a constant rate in the final stage. Increasing temperature led to a faster rate of manganese loss and a shorter middle stage. Consequently, at 1893 K and 1913 K, manganese loss in stage 3 started earlier compared to that at 1823 K. You et al.[22] did not present the manganese vs time data for various temperatures in Ar–O2 bubbling to Fe–75Mn–6C alloys, they reported that the rate of manganese loss increased with temperature. Liu et al.[28] also observed higher rates of manganese loss at higher temperatures during CO2-O2 injection into Fe–16Mn–3C alloy.

Manganese concentration in the bath vs time for (a) Fe–25Mn–0.42C, (b) Fe–15Mn–0.42C, and (c) Fe–10Mn–0.42C alloys, Gas flow rate: \( 5 \times 10^{ - 6} \)Nm3/s and gas composition: Ar–6.7 pct O2 variable: temperature, nozzle submergence depth: 0.03 m. Data at 1823 K are from Ref. [29]
Figures 5((a) through (c)) illustrate the decarburization of the Fe–25Mn–0.18C, Fe–15Mn–0.18C, and Fe–10Mn–0.18C alloys for the various temperatures. Similarly, Figures 6((a) through (c)) show the decarburization of the Fe–25Mn–0.42C, Fe–15Mn–0.42C, and Fe–10Mn–0.42C alloys. Increasing temperature resulted in higher rates of decarburization. Depending on the initial manganese concentration, if enough bubbling time is given, alloys Fe–0.42C alloys can decarburize to 0.18 pct or lower carbon concentrations. For example, the final carbon content of Fe–15Mn–0.42C alloy at 1913 K in is 0.17 pct (Figure 6(b)). Also, the final carbon content of Fe–Fe–10Mn–042C in 1823 and 1913 K was 0.165 and 0.128 pct (Figure 6(c)). The effect of temperature on the rate of decarburization is consistent with You et al.[22] and Liu et al.[28] work.

Carbon concentration in the bath vs time for (a) Fe–25Mn–0.18C, (b) Fe–15Mn–0.18C, and (c) Fe–10Mn–0.18C alloys, variable: temperature, nozzle submergence depth: 0.03 m. Data at 1823 K are from Ref. [29]

Carbon concentration in the bath vs time for (a) Fe–25Mn–0.42C, (b) Fe–15Mn–0.42C, and (c) Fe–10Mn–0.42C alloys, variable: temperature, nozzle submergence depth: 0.03 m. Data at 1823K are from Ref. [29]
Effect of Alloy Composition
Figures 7((a) through (g)) show the effect of initial manganese content on the rate of decarburization of Fe–Mn–C alloys containing 10, 15, and 25 pct Mn and 0.18 and 0.42 pct C. In each case, the temperature was maintained constant, and the gas flow rate and composition were 5 × 10−6 Nm3/s and Ar–6.7 pct O2. The depth of submergence in all these experiments was 0.03 m. As indicated, in all cases the rate of decarburization increased with decreasing initial manganese concentration. As observed in Figures 7((a) through (c)), decarburization of alloys Fe–0.18C at all temperatures followed a curve shape and the rate of decarburization slowed down with the carbon concentration in the bath. However, alloys Fe–0.42C exhibited a linear pattern with a constant rate for a given manganese concentration at 1893 and 1913 K (Figures 7(e) and (f)). Alloys Fe–0.42C at 1823 K were the exception, where the rate of decarburization was slow in the first stage. Then, the rate increased in the middle stage and finally decreased with the carbon content in the bath (Figure 7(d)).

Effect of the initial Mn concentration on decarburization of Fe–0.18C alloys at (a) 1823 K, (b) 1893 K, (c) 1913 K, and Fe–0.42C alloys at (d) 1823 K, (e) 1893 K, (f) 1913 K, gas flow rate \( 5 \times 10^{ - 6} \)Nm3/s and gas composition: Ar–6.7 pct O2, nozzle submergence depth: 0.03 m. Data at 1823 K are from Ref. [29]
Figures 8((a) through (i)) compares the effect of initial carbon concentration on manganese loss of alloys at each temperature. In all cases, there is more manganese loss in Fe–0.18C alloys compared to the Fe–0.42C alloys. At 1823K, stage 3 starts at the same time for both carbon concentrations (Figures 8(a), (d), (g)). In contrast, with increasing temperature to 1893 and 1913 K, stage 3 started earlier. Furthermore, for Fe–0.18C alloys duration of the middle stage was shorter and stage 3 started earlier than for Fe–0.42C alloys.

Effect of the initial carbon concentration on manganese loss of Fe–25Mn alloys at (a) 1823 K, (b) 1893 K, (c) 1913 K, and Fe–15Mn alloys at (d) 1823 K, (e) 1893 K, (f) 1913 K, and Fe–10Mn alloys at (g) 1823 K, (h) 1893 K, (i) 1913 K, gas flow rate \( 5 \times 10^{ - 6} \)Nm3/s and gas composition: Ar–6.7 pct O2, nozzle submergence depth: 0.03 m. Data at 1823 K are from Ref. [29]
Effect of Nozzle Submergence Depth
To investigate the effect of nozzle submergence depth on the rate of decarburization and demanganization, several experiments were carried out at depths of 0.01, 0.02, 0.025, and 0.03 m for Fe–25Mn–0.18C and Fe–25Mn–0.42C alloys. These depths refer to the height of metal above the nozzle tip. The gas flow rate, gas composition, and temperature were maintained constant at, respectively, 5 × 10−6 Nm3/s, and Ar–6.7 pct O2, and 1893 K. Figures 9(a) and (b) show the change in carbon concentration in the metal with time for alloys with 0.18 pct and 0.42 pct C. The corresponding manganese concentrations in the metal are given in Figures 10(a) and (b). Carbon and manganese concentrations were not influenced by the submergence depth. In the previous work by the authors,[30] it was shown that the reaction times are of the order of 0.001 seconds which is about 1 pct of the residence time of bubble in the metal. Hence, before a bubble leaves the tip of the nozzle, reactions have occurred and completed so that change in the residence time of the bubble by increasing the depth of the nozzle will not affect the rate of reactions. The authors’ previous work also showed that although the overall reaction time was very short in comparison to the bubble residence time, the competition for oxygen between carbon and manganese was largely dictated by their calculated relative rates of mass transfer in the metal.

Carbon concentration in bath vs time for (a) Fe–25Mn–0.18C, (b) Fe–25Mn–0.42C, variable: submergence depth

Manganese concentration in bath vs time for (a) Fe–25Mn–0.18C, (b) Fe–25Mn–0.42C, variable: nozzle submergence depth
Chemical Composition of Collected Fume
There was no fume on the crucible wall. However, the fume deposited on the furnace wall and cap was collected. It was not possible to collect all of it, but it was clear that the amount of fume generated increased with the initial manganese concentration of the alloy due to the increase in the vapor pressure of manganese. This observation is in agreement with the work of Chu et al.[31] on the processing of Fe–Mn–C steels containing 2 to 30 pct Mn and 0.004 pct C under vacuum. In this work, the concentration of manganese and iron in fume was measured using ICP-OES. According to X-ray diffraction, these elements were present primarily as MnO and Fe2O3. However, because the fine powder in the deposit oxidized as soon as the furnace was opened the authors are not able to determine in which form the fume deposited. For this reason, we have presented the composition of fume as the ratio of Mn/Fe. This ratio is plotted in Figure 11(a) for various temperatures and alloy compositions. There was no appreciable difference between the composition of the fume at the wall and the cap of the furnace. For this reason, one composition for each experiment is plotted in this figure. As observed in Figure 11(a), the Mn/Fe ratio of the fume increased with the initial manganese concentration of the alloy and decreasing temperature. Alloys with lower carbon contents (0.18 pct C) had higher ratios of Mn/Fe in fume. With increasing temperature, the fraction of Fe in the fume increased. The Mn/Fe ratio of the fume is consistent with the trend of the ratio of equilibrium pressures of Mn/Fe as shown in Figure 11(b).

Variations of (a) the measured Mn/Fe ratio in the collected fume vs initial Mn in alloy and (b) ratio of equilibrium pressures of Mn/Fe vs initial Mn in the alloy, nozzle submergence depth: 0.03 m, variable: temperature
The equilibrium vapor pressures of Mn and Fe (\( {\text{p}}_{\text{Mn}} \) and \( {\text{p}}_{\text{Fe}} \)) are calculated for the bulk alloy composition using Equations [1] and [2].[32] \( {\text{p}}_{\text{Mn }}^{0} \) and \( {\text{p}}_{\text{Fe }}^{0} \)are the vapor pressure of the pure metals. The activities of Mn and Fe in the bulk are calculated using the data from the thermodynamic model by Lee[33] for Fe–Mn–C alloys.
The Mn/Fe ratio of the fume is consistent with the trend of the ratio of equilibrium pressures of Mn/Fe as shown in Figure 11(b). However, from experimental data, Mn/Fe ratios are lower than predicted from the data in Figure 11(b). For all except one alloy, the slope of the lines in Figure 11(a) are almost identical which would be consistent with a controlling role for mass transport of manganese in the metal. Furthermore, the rates increase with decreasing carbon concentration which would suggest competition for oxygen between manganese and carbon.
Discussion
Thermodynamic Analysis
In order to compare the experimental measurements with thermodynamic predictions, the equilibrium manganese and carbon concentrations in the melt were calculated for Fe–25Mn–0.42C alloy as an example with FactSage 8.0 using FactPS, FToxid, and FSstel databases in the Equilib module. FactSage has the ability to consider the incremental addition of species to a system over time; in this case, oxygen is added at a rate consistent with the flow rate. FactSage assumes instantaneous equilibration of the increment with the melt and calculates the new melt composition. By entering the gas flow rate, gas and melt compositions as well as temperature, FactSage will calculate the predicted change in melt composition with time. Figure 12((a) and (b)) shows the predicted equilibrium carbon and manganese concentration vs time for Fe–25Mn–0.42C alloy at temperatures of 1823 K to 1913 K. As was shown by similar calculations by the authors for alloy Fe–15Mn–1C,[30] above a critical carbon content, the predicted rate of decarburization is fast, and below the critical carbon content slows significantly (Figure 12(a)). The FactSage calculations predict that regardless of temperature, rates of decarburization above the critical carbon content are 1×10−4 pct C/s because they will be entirely controlled by the supply of oxygen, whereas below the critical carbon content rates decrease with temperature. Furthermore, above the critical carbon content, no loss of manganese is predicted. The experimental data do not agree with these predictions. For Fe–25Mn–0.42 C (Figure 6(a)) the experimental rates of decarburization are 1.8 to 2.3 times slower than the FactSage prediction for temperatures of 1823 to 1913 K (Figure 12(a)). Moreover, manganese behavior in the experiments (Figure 4(a)) does not follow the thermodynamic prediction by FactSage (Figure 12(b)). In stage 1 of the experiments, there is a rapid manganese loss which is not predicted by FactSage. Furthermore, rates of manganese losses in the experiment do not agree with the predicted rates by FactSage.

FactSage prediction for variation of (a) carbon and (b) manganese concentrations in the bath for Fe–25Mn–0.42C at various temperatures
It should be noted that the predicted fraction of iron in the fume, whether present as FeO or Fe is negligible and much less than that observed experimentally (Figure 11(a)). It should also be noted that FeO is unstable relative to MnO and CO under the current experimental conditions. Comparison of thermodynamic calculations with experimental observations shows that manganese and carbon in the bubble are not in equilibrium with the melt. This is consistent with observations from a previous publication by the authors[30] which showed that carbon and manganese oxidation occurred in proportion to the mass transfer rates of the species in the melt. Moreover, as indicated in Figures 9 and 10, changing the depth of submergence did not make any difference in the decarburization and manganese loss, the reactions occur well within the time the bubble is present the melt and that prolonged time after reaction does not lead to a repartitioning of the species. Therefore, it is clear that the bubble, having reacted, does not go through a transition toward true equilibrium.
Rate of Manganese Loss
Figure 13 shows the measured rates of manganese loss vs the initial manganese concentration of alloys. Increasing temperature resulted in higher rates of manganese loss in both stages 1 and 3 for Fe–0.18C alloys (Figure 13(a)). For stage 3, the effect of temperature on manganese loss was considerably smaller than for stage 1. Similarly, in Figure 13(b), the increasing temperature is seen to have a bigger effect on stage 1. Furthermore, in all cases, alloys with higher initial manganese content exhibited higher rates of manganese loss, particularly in stage 1 of the process. Increasing temperature from 1823 to 1913 K led to 20, 37, and 47 pct increase in the rate of manganese loss in stage 1 of the process for Fe–0.18C alloys. For Fe–42C alloys rate of manganese loss increased by 16, 67, and 65 pct for 10, 15, and 25 pct Mn alloys.

Rates of total manganese loss with the initial manganese concentration for (a) Fe–0.18C, and (b) Fe–0.42C alloys, Variable: temperature
Oxygen Utilization
Based on the measured decarburization and demanganization rates we know the ratio of manganese and carbon removed to oxygen supplied. If it is assumed that the products of oxidation are primarily CO and MnO, the oxygen utilization for decarburization and demanganization are the fraction of oxygen consumed for the formation of CO and MnO to the total supplied oxygen. Table II shows the oxygen utilization, calculated on this basis, for decarburization for each alloy and temperature. For Fe–0.42C alloys at 1893 and 1913 K, the oxygen utilizations were very similar in all three stages. However, for Fe–0.18C alloys, oxygen utilization decreased with carbon content in the bath (from stage 1 to 3). For alloys at 1823 K, oxygen utilization was low in stage 1, higher in stage 2, and low in stage 3 again. The available oxygen after CO formation is not sufficient to account for total manganese loss shown in Figure 13. Therefore, in addition to oxidation to MnO, there must be another mechanism involved in manganese loss. This observation agrees with studies by Lee et al.[26] and You et al.[21] for oxygen refining of high carbon ferromanganese. It was reported by these authors that the difference between the total manganese loss and oxidation was due to the evaporation of manganese. In a previous publication by the current authors, where the experimental temperature was confined to 1823K, two possible mechanisms were assessed. In that work, it was found the straight-forward loss by evaporation could not explain all the manganese loss. However, an additional mechanism, involving local heating of the bubble surface by the oxidation reactions to enhance manganese vaporization, followed by condensation of a fine mist of liquid manganese in the bubble, explained the additional loss over the loss by straight-forward evaporation. The additional mechanism is called the evaporation-condensation mechanism. In the following sections of this paper, the viability of these potential mechanisms extended the temperature range from 1823 to 1913K is discussed.
Potential Mechanisms for the Excess Manganese Loss
Manganese loss as Mn (g) by evaporation
If the difference between total manganese loss and loss as MnO is due to evaporative loss, the required vapor pressure of Mn (g) can be calculated readily, as detailed in Appendix A. For evaporative loss to be a viable mechanism, the vapor pressure of manganese would have to be very high, which requires a substantial temperature rise of up to 557 K (Table A.I). The required temperature for the excess manganese loss to be due to evaporation as Mn (g) is plotted vs the initial Mn of alloys in Figure 14. In both Figures 14(a) and (b), temperatures required to support evaporation as the source of excess manganese loss increased with increasing experimental temperature. For Fe–0.18C alloys, these required temperatures for stage 3 were considerably lower than for stage 1 (Figure 14(a)). Liu et al.[28] observed similar excess manganese loss from Fe–16Mn–3C alloy by injection of pure O2 at a flow rate of 8.3 × 10−6 Nm3/s into the melt. These workers did not present mass balance calculations, nor did they suggest a mechanism for manganese loss. However, these researchers have referred to the effect of CO2 introduction in decreasing evaporation loss of Mn, most likely due to the endothermic nature of decarburization with CO2. The mass of liquid steel was not given in their experiments; however, based on the crucible dimensions (inner diameter: 0.06 m and height 0.125 m, 20 pct of the crucible is filled) it is possible to make some estimate of the mass of steel to be at least 500 grams. With this assumption and knowing how much total manganese and carbon were removed based on their data, the vapor pressure of manganese required to support evaporative loss can be estimated to be 0.052 atm which would require a temperature rise of about 400 K which seems high. The current authors were not able to detect a temperature increase on the outer wall of the crucible and do not believe it is conceivable that a 400K temperature increase in the melt would not be detected on the crucible wall; however, it might be possible to have very high local temperature increases in the vicinity of the bubble.

The required temperature for the excess manganese loss being as vapor for (a) Fe–Mn–0.18C alloys, and (b) Fe–Mn–0.42C alloys, variable: temperature
Manganese loss as droplet inside bubble by evaporation-condensation
Given the difficulty in justifying the necessary temperature rise for evaporative loss, the authors previously proposed a mechanism based on evaporation-condensation.[29] The proposed mechanism assumes that in addition to loss as oxide and vapor, manganese may form a mist of liquid droplets inside the bubble as follows: the temperature of the bubble surface is increased by exothermic reactions (MnO and CO formation) the local temperature rise leads to Mn evaporation at a high vapor pressure. The manganese vapor then condenses as fine droplets inside the bubble. This mechanism is viable under conditions where manganese vapor is generated in equilibrium with manganese in the alloy and condenses at a lower temperature inside the bubble. This mechanism can work only if the temperature difference is enough for the manganese vapor pressure in equilibrium with the bubble surface \( {\text{p}}_{\text{Mn}}^{\text{S}} \) is higher than that in equilibrium with pure Mn (l) in the bubble interior \( {\text{p}}_{\text{Mn}}^{\text{mist}} \). Manganese condensation will continue as long as the temperature gradient between the bubble surface and its interior remains sufficiently high. This mechanism is equivalent to the concept of fume formation proposed by Turkdogan et al.[27] except in the current case, the metal condenses as a mist of liquid droplets rather than as oxide particles observed by Turkdogan et al..[27] The detailed calculation procedure for the evaporation-condensation mechanism is given in Appendix B. The vapor pressures and temperatures of manganese at the bubble surface for each alloy at various experimental conditions are provided in Table III. As indicated, the temperature at the surface of the bubble (TS) required for the proposed evaporation-condensation mechanism increased with the starting temperature. For a given manganese concentration TS is independent of carbon concentration and it is higher for alloys with lower manganese content.
It is mentioned earlier that the exothermic oxidation reactions generate the required heat for this mechanism to work. Here, the viability of this mechanism is explored from a heat balance point of view. Given the short time over which the manganese loss to the bubble occurs, it is assumed that there is no heat loss, and because the gas enters the melt through a heated ceramic tube, all the supplied oxygen is consumed to produce CO and MnO at the starting temperature. The generated heat from the oxidation reaction must be sufficient to heat the layer of metal surrounding the bubble to the required higher temperatures TS. The thickness of this layer can be calculated to determine the number of moles of steel and manganese in that layer. The details of these calculations are provided in Appendix B. Table III shows the thickness of the heated layer for each stage as well as the number of moles of manganese contained in that layer which is compared with the total number of moles of manganese removed per bubble for various alloys and temperatures. In most cases, the amount of manganese in the heated layer is more than sufficient to support the manganese loss and for the cases where this is not true, the amount is close. This would suggest that the mechanism is viable given the fact that manganese can transport into the heated layer and it is only the manganese at the surface that needs to be at the evaporation temperature. It should be noted that all these experiments were done in the same way. However, these inconsistencies may arise for several reasons. As mentioned earlier reactions are so fast that as the bubble is going to melt, the reaction is completed. This leads to some variations. The tip of the alumina nozzle could be eroded, and the bubble could be different in some cases. Furthermore, there might be some buildup on the nozzle. These possibilities make it difficult to interpret the result definitively. The authors attempted to measure the temperature rise for a small number of experiments, but no measurable temperature rise was detected using a thermocouple in contact with the outside of the crucible. However, it does not exclude the possibility of a localized temperature rise at the surface of the bubble, in fact, badly eroded nozzle tips are strong evidence for such temperature rise.
Figure 15 shows the temperature at the surface of the bubble required for the evaporation-condensation mechanism to operate. In this mechanism, the temperature requirement only depends on the manganese concentration of alloy and temperature of the melt. Comparing with Figure 14, temperatures for this mechanism are lower between 55 K and 270 K in stage 1, and between 28 K and 170 K in stage 3. Alloy Fe–10Mn–0.18C was an exception where the required temperature for the simple evaporation mechanism was about 50 K lower than the evaporation-condensation mechanism. On balance it seems more likely that evaporation-condensation operates driven by a local temperature increase at the bubble/metal interface. It is also worth noting that the mechanisms of evaporation-condensation and simple evaporation are not mutually exclusive, and it is likely that both operated in parallel.

The temperature at the surface of the bubble vs the initial Mn concentration of the alloy
The contribution of each mechanism of Mn loss is shown in Figures 16 and 17 for Fe–0.18C and Fe–0.42C alloys, respectively. Manganese losses are as oxide, vapor, and mist droplets inside the bubble. These different contributions were calculated by assuming all oxygen remaining after the carbon loss was accounted for, reacted with manganese, any additional losses of manganese were first attributed to evaporation and then evaporation-condensation. Based on the much greater stability of MnO and CO, it is further assumed that amount of FeO compared with MnO is negligible. As shown in Figures 16((a) through (i)), the total rate of manganese loss for alloy Fe–25Mn–0.18C and Fe–15Mn–0.18C, Fe–10Mn–0.18C in both stages 1 and 3 increases with temperature. In both stages, the contribution of oxidation loss of manganese decreased with temperature, while its loss as vapor increased. In Figure 16(c), loss as oxide, vapor, and mist overlapped in stage 3. The contribution of manganese loss as liquid droplets followed the same trend as vapor, it increased with temperature. However, for alloy Fe–10Mn–0.18C in stage 3, the contribution of loss as droplets was zero (Figures 16(g) through (i))). Although according to heat balance calculations there is more than enough manganese in the heated layer as shown in Table III, the total manganese loss in stage 3 was not that high to reach that high vapor pressure the manganese vapor inside the bubble in equilibrium with mist must be at the activity of 1. Therefore, the contribution of manganese mist is zero in stage 3. Likewise, in Figures 17((a) through (i)), manganese loss as oxide decreased with temperature, whereas loss as vapor increased in all alloys at both stages 1 and 3 for Fe–25Mn–0.42C alloys. The contribution of manganese loss as droplet increased with temperature in stage 1 for all alloys; however, in stage 3, it remained constant (Figures 17(d) and (f)) or decreased slightly with temperature (Figures 17(a) compared with (b), (c)).

Rates of Mn losses as vapor, oxide, and liquid droplet inside the bubble for Fe–25Mn–0.18C at (a) 1823 K, (b) 1893 K, (c) 1913 K, for Fe–15Mn–0.18C at (d) 1823 K, (e) 1893 K, (f) 1913 K, and for Fe–10Mn–0.18C at (g) 1823 K, (h) 1893 K, (i) 1913 K

Rates of Mn losses as vapor, oxide, and liquid droplet inside the bubble for Fe–25Mn–0.42C at (a) 1823 K, (b) 1893 K, (c) 1913 K, for Fe–15Mn–0.42C at (d) 1823 K, (e) 1893 K, (f) 1913 K, and for Fe–10Mn–0.42C at (g) 1823 K, (h) 1893 K, (i) 1913 K
Conclusions
In this paper, the decarburization and manganese losses from Fe–Mn–C alloys with different manganese and carbon concentrations in the temperature range of 1823 K to 1913 K were studied.
-
1.
The decarburization and total manganese loss increased with increasing the temperature. In the case of carbon; this is because more oxygen is partitioned to carbon oxidation than to manganese. The increase in manganese loss at higher temperatures is because of the large increase in vapor pressure leading to more loss as mist and by simple vaporization.
-
2.
Based on the observation that changing the depth of nozzle submergence did not make any difference in the decarburization and manganese loss, the reactions occur well within the time the bubble is present in the melt and that prolonged time after reaction does not lead to repartitioning of the species.
-
3.
Total manganese loss increased with manganese concentration in alloy due to the higher vapor pressure of manganese. Furthermore, due to the competition between carbon and manganese for oxygen, the observed rate of decarburization increased with lower carbon content in the melt.
-
4.
Comparison of thermodynamic calculations with experimental observations shows that manganese and carbon in the bubble are not in equilibrium with the melt. This is consistent with observations from a previous publication by the authors which showed that carbon and manganese oxidation occurred in proportion to the mass transfer rates of the species in the melt. When these observations are taken in combination with conclusion 2 it is also clear the bubble, having reacted, does not go through a transition toward true equilibrium.
-
5.
Manganese loss can be explained by considering multiple mechanisms in parallel: oxide formation and vapor formation, and evaporation-condensation.
References
B.C. De Cooman, O. Kwon, and K.G. Chin: Mater. Sci. Technol., 2012, vol. 28, pp. 513–27.
O. Bouaziz, H. Zurob, B. Chehab, J.D. Embury, S. Allain, and M. Huang: Mater. Sci. Technol., 2011, vol. 27, pp. 707–9.
L. Chen, Y. Zhao, and X. Qin: ACTA Metall. Sin., 2013, vol. 26, pp. 1–15.
K. Chin, W. Cho, S. Kim, Y. Kim, T. Song, and T. Kim: in The METEC and 2nd European Steel Technology and Application Days (ESTAD) conference, Düsseldorf, 2015, pp. 65–68.
R. Elliott, K. Coley, S. Mostaghel, and M. Barati: JOM, 2018, vol. 70, pp. 691–9.
O. Bouaziz, S. Allain, C.P. Scott, P. Cugy, and D. Barbier: Curr. Opin. Solid State Mater. Sci., 2011, vol. 15, pp. 141–68.
O. Grässel, L. Krüger, G. Frommeyer, and L.W. Meyer: Int. J. Plast., 2000, vol. 16, pp. 1391–1409.
R. Elliott, K. Coley, S. Mostaghel, and M. Barati: JOM, 2018, vol. 70, pp. 680–90.
L. Mujica, S. Weber, G. Hunold, and W. Theisen: Steel Res. Int., 2011, vol. 82, pp. 26–31.
T. Furukawa, H. Huang, and O. Matsumura: Mater. Sci. Technol. (United Kingdom), 1994, vol. 10, pp. 964–70.
D.S. Kozak and L.R. Matricardi.: Iron Steelmak., 1981, vol. 8, pp. 28–31.
S.E. Olsen, M. Tangstad, and T. Lindstad: Production of Manganese Ferroalloys, 2007, pp. 56-57.
M. Tangstad: Manganese Ferroalloys Technology, Elsevier, Trondheim, 2013, p.257.
C.F. Redeker, H.-J. Renner, and J. Schöttler: Cost Efficient Metallurgy for the Production of Novel Ultra-High Strength Deep Drawable Steel Gades with High Mn Contents from 10 to 25 Wt.-%, Luxembourg:, 2008.
R.J. Fruehan: Ironmak. Steelmak., 1976, vol. 3, pp. 153–8.
T. Ohno and T. Nishida: Tetsu-to-Hagane, 1977, vol. 63, pp. 2094–9.
J. Reichel and J. Szekely: Iron Steelmak., 1995, vol. 22, pp. 41–8.
J. Wei and D. Zhu: Metall. Mater. Trans. B, 2002, vol. 33, pp. 111–9.
J.H. Wei: J. Shanghai Univ., 2002, vol. 6, pp. 1–23.
V.-V. Visuri, M. Järvinen, A. Kärnä, P. Sulasalmi, E.-P. Heikkinen, P. Kupari, and T. Fabritius: Metall. Mater. Trans. B, 2017, vol. 48, pp. 1850–67.
B.-D. You, B.-W. Lee, and J.-J. Pak: Met. Mater., 1999, vol. 5, pp. 497–502.
B.-D. You, J. Han, and J. Pak: Steel Res., 2000, vol. 71, pp. 22–6.
B.-D. You: J. Korean Institue Met. Mater., 1995, vol. 33, pp. 1508–13.
B.-D. You, K.-Y. Park, J.-J. Pak, and J.-W. Han: Met. Mater., 1999, vol. 5, pp. 395–9.
K. Yamamoto, T. Mimura, S. Ito, and T. Onoye: Tetsu-to-Hagane, 1986, vol. 72, pp. 1034–46.
Y.E. Lee and L. Kolbeinsen: ISIJ Int., 2005, vol. 45, pp. 1282–90.
E.T. Turkdogan, P. Grieveson, and L.S. Darken: J. Am. Chem. Soc., 1963, vol. 67, pp. 1647–54.
H. Liu, J. Liu, J. Schenk, F.M. Penz, L. Sun, R. Zhang, and Z. An: Metall. Mater. Trans. B, 2020, vol. 51, pp. 756–62.
A. Rafiei, G.A. Irons, and K.S. Coley: Steel Res. Int., https://doi.org/10.1002/srin.202000480.
A. Rafiei, G.A. Irons, and K.S. Coley: Metall. Trans. B, 2021. https://doi.org/10.1007/s11663-021-02180-z.
J. Chu, Y. Bao, X. Li, F. Gao, and M. Wang: Steel Res. Int., 2020, vol. 2000333, pp. 1–9.
D. R.Gaskell and D.E. Laughlin: Introduction to the Thermodynamics of Materials, Sixth Edit., CRC Press, 2018, p.709.
Y.E. Lee: Metall. Mater. Trans. B, 1998, vol. 29, pp. 397–403.
G.A. Irons and R.I.L. Guthrie: Metall. Trans. B, 1978, vol. 9, pp. 101–10.
Acknowledgments
The authors acknowledge financial support from the Natural Sciences and Engineering Research Council of Canada. Additional thanks goes to ArcelorMittal Dofasco, Stelco, Praxair, and Hatch Ltd. for in-kind support and technical expertise.
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Manuscript submitted December 28, 2020; accepted April 18, 2021.
Appendices
APPENDIX A: detailed calculations of manganese vapor pressure in Section IV–D–1
Each gas bubble is composed of CO, Ar, and Mn (g), and the total pressure inside each bubble, Pt, is approximately 1 atm. Hence, the vapor pressure of manganese can be calculated from Equation A1 with known amount of CO and MnO removed per bubble during each stage.
The number of moles of each species removed by end of stages 1 and 3, and the partial pressure of CO and Mn are listed in Tables AI and AII. In these experiments, the total gas flow rate and gas composition were 5 × 10−6 Nm3/s and Ar-6.7 pct O2. The calculated bubble diameter according to the work of Irons et al.[34] was 0.0148 m at 1823 K and 0.0149 m at 1893 and 1913 K. The frequency of bubble formation was 18, 18.30, and 18.38 s−1 at 1823, 1893, and 1913 K, respectively. For this reason, the number of moles of argon was slightly different depending on the bubble size. The required temperature to achieve these manganese vapor pressures are calculated from Equation A2 where \( {\text{a}}_{\text{Mn}} \) is the activity of manganese in the melt, and Tm is the melt temperature.
It should be noted that the \( \Delta T \) values tabulated in Tables AI and AII are the difference between the required temperature and the initial temperature of the melt. As indicated, the required temperature for evaporation decreases with increasing manganese concentration in the alloy. This is because the vapor pressure and activity of manganese increase with its content in the melt.
APPENDIX B: Detailed Calculations of Manganese in the Heated Layer in Section IV–D–2
To determine the flux of manganese in the gas phase by the proposed evaporation-condensation mechanism, Equation B1 can be used where; kMn is the mass transfer coefficient of manganese in unit of m/s. R is the gas constant in the unit of \( {\text{J}}/{\text{K}}\;{\text{mol}} \). Tb and Ts are the temperatures inside the bubble and at its surface, in units of K. The surface area of bubble A is 6.9 × 10−4 m2 at 1823 K and 7 × 10−4 m2 at 1893 and 1913 K. \( {\text{dMn}}_{\text{evap}} /{\text{d}}t \) is the rate of evaporation of Mn in unit of mol/s.
The requirement for this mechanism to operate is that \( p_{\text{Mn}}^{\text{s}} /T_{\text{s}} \) being higher than \( p_{\text{Mn}}^{\text{mist}} /T_{\text{b}} \). The activity of manganese in the mist inside the bubble \( a_{\text{Mn}}^{\text{mist}} \)is equal to 1 and \( a_{\text{Mn}}^{\text{s}} \) is the activity of manganese in equilibrium with the liquid. Therefore, the temperature at the surface of bubble Ts can be calculated from Equation B3.
As mentioned earlier, in these experiments the reactions take place very quickly, and the actual manganese evaporation rate is unknown. For the purpose of these calculations, the authors have chosen to use the minimum value possible by assuming the reaction occurs over the total residence of the bubble (0.11 seconds). For instance, for Fe–15Mn–0.42C at 1893 K, \( {\text{dMn}}_{\text{evap}} /A \cdot {\text{d}}t \) is equal to 9.6×10−11 mol/m2 s. The temperature at the surface of the bubble (Ts) is 2211 K. kMn is calculated to be 2.62 m/s. Because of the assumptions behind the calculations, this value is conservative. But is consistent with the values quoted in the literature based on similar assumptions.[26]
In the case of Fe–15Mn–0.42C alloy at 1893 K in stage 1, 0.27J heat is generated from MnO and CO formation. The temperature at the surface of the bubble (Ts) is 2211 K, constant pressure molar heat capacity (Cp) of liquid steel is 46 J/mol.K. Hence, 0.27J can heat 1.8 × 10−5 mole steel (1.5 × 10−10 m3) from 1893 to 2211 K. As the activity of manganese in this alloy is 0.15, the heated layer contains 2.8 × 10−6 moles of Mn. In stage 1 of this experiment, total manganese loss is 2.5 × 10−6 moles. Therefore, there is sufficient manganese in the heated layer to account for the excess manganese loss by the evaporation-condensation mechanism. The ratio of the volume of metal in that layer (1.5 × 10−10 m3) to the surface area of the bubble (7 × 10−4 m2) gives the thickness of the heated layer which is 2.1×10−7 m (0.21 μm).
Rights and permissions
About this article

Cite this article
Rafiei, A., Irons, G.A. & Coley, K.S. Argon-Oxygen Decarburization of High Manganese Steels: Effect of Temperature, Alloy Composition, and Submergence Depth. Metall Mater Trans B 52, 2509–2525 (2021). https://doi.org/10.1007/s11663-021-02196-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-021-02196-5