
Abstract
Direct reduction of iron ore pellets using hydrogen gas has the potential to significantly reduce CO2 emissions from the ironmaking process. In this work, green pellets of titanomagnetite ironsand from New Zealand were oxidatively sintered to form titanohematite. These sintered pellets were then reduced by H2 gas at temperatures ≥ 1043 K, and a maximum reduction degree of ~ 97 pct was achieved. Fully reduced pellets contained metallic Fe as the main product phase, but several different (Fe, Ti) oxides were also present as minor inclusions. The phase distribution of these oxides depended on the reduction temperature. With increasing temperature, the relative proportion of pseudobrookite in the final product increased, while the proportion of residual ilmenite and rutile decreased. The reduction kinetics were found to be well described by a pellet-scale single-interface shrinking core model, for reduction degrees up to 90 pct. At temperatures above 1143 K, the rate-limiting step was found to be solely an interfacial chemical reaction process, with a calculated apparent activation energy of 31.3 kJ/mol. For pellet sizes from 5.5 to 8.5 mm, the reaction rate was observed to increase linearly with decreasing pellet diameter, and this linear correlation extrapolated to intercept the axis at a pellet diameter of 2.5 mm. This is interpreted as the minimum length required for a shrinking core interface to develop within the pellet.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
There is increasing interest in the use of titanomagnetite (TTM) iron ore as a potential cheap source of iron,[1] as well as the possible co-production of other valuable minerals such as titania (TiO2) and vanadium pentoxide (V2O5). In New Zealand (NZ), large deposits of TTM ironsand are found along > 400 km of the west coast of the North Island.[2] The NZ ironsands typically contain ~ 8 wt pct TiO2. This is a significantly lower TiO2 content compared to other TTM deposits from around the world (e.g., 13 wt pct in China,[3] 10 wt pct in Indonesia,[4] and 14 wt pct in South Africa[5]). However, the presence of TiO2 means that the conventional blast furnace process is not suitable for reduction of these ironsands.[6] Instead, TTM ironsand is currently processed in NZ using a two-stage process. The TTM is initially partially reduced in a rotary kiln via a solid-state carbothermic reduction process, and then fully reduced and smelted in an electric furnace. This process is both energy and CO2 intensive. Therefore, developing new alternative processes for the reduction of TTM ironsand reduction is vital in order to commercially utilize this cheap source of iron into the future.
Gas-based direct reduction (DR) is widely regarded as a promising alternative process for TTM reduction. In particular, hydrogen reduction is attracting attention as a potential zero-CO2-emitting process.[7,8,9,10,11] NZ ironsands have a typical particle size distribution of 100 to 300 µm which means that it must be consolidated into pellets prior to processing in typical a vertical shaft furnace (e.g., MIDREX) or a rotary hearth process. Pelletization is generally carried out by sintering either in air (oxidative sintering) or in an inert atmosphere. Previous studies have found that pre-oxidation of TTM into titanohematite (TTH) increased the reduction rate.[12,13] Hematite ores were observed to reduce faster than magnetite ores,[12] and this was ascribed to an increase in surface area and gas permeability of the magnetite grains arising from microcracks formed during the conversion of the crystal structure from rhombohedral to cubic.[14] These microcracks form due to the large internal stresses imposed by the volume increase associated with this transformation. Park and Ostrovski[15] concluded that the increased reducibility of Ti-containing ironsand by pre-oxidation could be attributed to the same effect.
While there have been a number of studies on the gaseous reduction of TTM ironsand,[12,16,17,18,19] there are very few reports of the gas reduction of TTH, especially in the form of pellets. Wang et al.[20] found that the reduction rate of pre-oxidized Indonesian (East Java) TTH ironsand (11.41 wt pct TiO2) increased with the increasing content of H2 in a H2-Ar gas mixture. Similarly, McAdam et al.[21] found that H2 gas was much faster than CO in achieving 90 pct reduction of sintered NZ ironsand pellets, and that this reduction rate increased with both increasing gas flow rate and temperature. In several works, the morphologies of reduced TTM ironsand have been presented,[7,8,9,10,11] but the internal morphologies of reduced TTH ironsand pellets have not been comprehensively studied.
The gaseous reduction of TTH is complex and often considered to proceed via several intermediate products,[15,22] summarized as
Therefore, in the reduction of TTH pellets (or even just hematite (Fe2O3) pellets), multiple reaction stages are expected, each exhibiting different activation energies and rate constants.[23,24,25] Despite this, several previous works have successfully applied a simple single-interface reaction model to the kinetic analysis of the gaseous reduction of hematite/TTH pellets.[4,26,27,28,29] This has been demonstrated for both the widely applied shrinking core model (SCM) and finite element simulation methods.
The lower TiO2 content in the NZ ironsand distinguishes it from other internationally reported ores.[3,4,5] At present, the reaction mechanism and behavior of NZ TTH pellets with H2 at high temperatures is not fully understood. Therefore, the aim of this study is to develop an analytical kinetic description of the reduction mechanism of NZ TTH pellets in H2 and to assess the composition and morphology of the fully reduced pellets.
Experimental
Production of Sintered Pellets from Ironsand Concentrate
NZ TTM ironsand concentrate was obtained from Waikato North Head, New Zealand. Its elemental composition was determined by X-ray fluorescence (XRF), as shown in Table I. The ratio of Fe2+/Fe3+ within the ironsand was determined to be 0.56 by titration based on the ISO9035 method.
To make pellets, the ironsand was first ball milled to an average particle size of ~ 30 µm, as measured by a laser-scattering particle size analyzer (see Figure 1 for the particle size distribution of the milled ironsand).

Particle size distribution of wet milled NZ ironsand with an average particle size of ~ 30 µm
Spherical green pellets of approximately 7 mm diameter were then produced in a Lurgi-type disc pelletizer by mixing the milled ironsand with 1 wt pct bentonite and water. Table II shows the composition of the bentonite after roasting in air, as determined by XRF). During pelletization, water was manually sprayed onto the ironsand particles until spherical ‘green’ pellets began to form. At this point, no further water was added and the green pellets were removed and dried. The green pellets were then sintered at 1473 K for 2 hours in air to generate TTH pellets.
A range of TTH pellet diameters were obtained from the pelletization process. In this study, the TTH pellets from 5.5 to 8.5 mm were studied, which represents the median range obtained from the disc pelletizer. The compressive strength of twelve individual sintered TTH pellets was measured by crushing between parallel plates in a Tinius Olsen (UK) H10KT Universal Testing Machine. The average crushing strength found to be 1100 ± 200 N.
Experimental Set Up
The reduction behavior of pre-oxidized TTH pellets in hydrogen was measured through a series of TGA experiments, conducted at temperatures from 1043 K to 1443 K (at 100 K intervals). A schematic of the apparatus used to conduct these experiments is given in Figure 2.

Schematic of the TGA experimental arrangement used in this work, which employed a modified Mettler TA1 Thermobalance
In each experiment, a single TTH pellet was placed in the middle of a ceramic crucible located in the hot zone of the furnace. A gas inlet located close to the crucible ensured continuous gas flow around the pellet. After heating to the target temperature under nitrogen gas, the system was allowed to thermally stabilize for 30 minutes. Then, the gas flow was switched to hydrogen, marking the beginning of the reduction experiment. The mass of the sample was recorded every 10 seconds. Measurements proceeded until there was no further measurable weight loss of the pellet, at which point the gas flow was switched back to nitrogen and the system cooled to room temperature.
The degree of reduction, X, was calculated from this TGA output, and based on the weight of oxygen removed from the pellets using Eq. [1]:
where \( w_{0} \) is the initial weight of the pellet, \( w_{t} \) is the pellet weight after reduction time t, and 0.30 is the theoretical weight loss ratio for pure hematite reduction to metallic iron. The factor of 0.85 is the equivalent mass fraction of hematite in the oxidized sintered pellets, as established from the XRF data given in Table I. At the start of each TGA run, a delay of ~ 0.4 minutes was observed to occur between switching the flow of H2 gas into the chamber and the onset of weight loss. This delay was attributed to the time required for hydrogen gas to flow through the pipe and purge nitrogen from the chamber. Therefore, in all following analysis, the onset delay time was removed and the time t = 0 second considered to be the final data point collected before an initial weight loss was measured. At the end of each experimental run, the fully reduced pellet was characterized using SEM and XRD. Crystalline phase composition of the fully reduced pellets was obtained by quantitative-XRD analysis performed using Topas 4.2 (Bruker) software.
Prior to undertaking the experiments reported here, the critical flow rate of the instrument was investigated through monitoring the reduction rate as a function of hydrogen flow rate.[30] It was found that at flow rates above 380 ml/min, the reaction rate was not dependent on gas flow rate at any temperature investigated in this study. This eliminates gas transport effects in the experiments studied here. In order to confirm the reproducibility of the reduction measurements reported here, five separate runs were performed on different individual pellets under identical experimental settings (1343 K, 250 ml/min, ~ 7-mm-diameter pellets). The results from each of these runs are shown in Figure 3. Very close agreement is observed between the data obtained in each different run, providing a high degree of confidence in the reproducibility of the experimental methods employed.

Plots showing data obtained during reproducibility tests of the H2 reduction experiment performed in this work. Five sets of reduction degree data are shown, each obtained from the H2 reduction of similar TTH pellets under the same condition (1343 K, 250 ml/min, ~ 7-mm-diameter pellets)
The reduction swelling index (RSI) after hydrogen reduction was also measured for several sintered pellets (diameter 7.5 ± 0.5 mm) and found to be small, equating to a measured volume change of only 5 pct after reduction at 1343 K for 25 minutes.
Results and Discussion
SEM Characterization of Un-sintered and Oxidized TTM Ironsand
The as-received raw ironsand concentrate was characterized by SEM-EDS analysis. Figure 4(a) shows an optical microscope image illustrating the two types of particles present in the raw ironsand. One type of particle appears uniform throughout, whereas the other is non-uniform in character and contains lamellar structures. Under light microscopy, the uniform TTM particles are a similar color to the regions surrounding the lamellar structures in the non-uniform particles. Figure 4(b) shows back-scatter SEM microscopy of the raw ironsand. Here the lamellae structures appear darker than the surrounding regions, indicating a lower electron density and indicative of higher levels of oxygen. These naturally occurring lamellae have been identified in previous studies as exsolved TTH.[12] The elevated oxygen content of the lamellae structure was confirmed by EDS point analysis given in Table III of the points marked in Figure 4(b). The matrix compositions in both the uniform particles and non-uniform lamellar particles (points 1 and 2, respectively) are both determined to contain ~ 24 wt pct O, 66 wt pct Fe, and 4.4 wt pct Ti. This composition is consistent with a TTM phase. By contrast, the lamellar structures had significantly higher Ti content (~ 16 wt pct) and lower Fe content (~ 54 wt pct) (see points 3 and 4 in Table III). Other elements such as Al and Mg were also observed in the matrix but were not concentrated in the lamellae.

Two types of particles are observed in the raw as-received NZ TTM ironsand. (a) Light microscopy; (b) back-scatter SEM image
After oxidative sintering, XRD was used to determine the major phases present in the pellets. Figure 5 shows and compares the XRD patterns obtained from both the green and sintered pellets. From this figure it can be seen that the TTM within the green pellets was fully oxidized to TTH on sintering. Note that the small TTH peaks in the green pellet originate from the lamellae found within the non-uniform particles in the as-received ironsand.

XRD patterns obtained from green pellets and oxidized pellets following 2 h oxidative sintering at 1473 K in air (the radiation source is Co-α of 1.79026 Angstroms)
In Figure 6, SEM images of cross sections of oxidized ironsand are shown. Table IV gives the EDS spot analysis data for the points indicated in Figure 6(a), and the dark and light phases observed in Figures 6(b) through (d).

SEM pictures of particles in the oxidized ironsand: (a) uniform particles; (b), (c), and (d) non-uniform particles
Similar to the un-sintered TTM particles, it was found that most of the oxidized particles were uniform in appearance. Typical examples of these uniform particles are noted as 1 and 2 in Figure 6(a). As expected, the oxidized uniform particles had a higher O/(Fe + Ti) weight percentage ratio (~ 0.4) than the un-sintered uniform particles (~ 0.3). Interestingly, instead of only one type of non-uniform particle found in the un-sintered ironsand, the oxidized ironsand contained three different non-uniform morphologies. These non-uniform particles comprised ~ 15 pct of the total particle population. This estimated value was obtained from particle counting within a 5x optical image, taken at a randomly selected position on the cross section of a fully oxidized pellet. All incomplete particles at the edge of the image were excluded from particle counting, as were a small number of dark silica-like gangue particles (impure oxides). The remaining particles were all clearly identifiable as either homogeneous uniform particle or non-uniform particles. A total of 189 particles were counted, of which 29 particles were non-uniform, and 160 were uniform.
Typical SEM images of each type of non-uniform particle are shown in Figures 6(b) through (d). All of the non-uniform particles featured both darker and lighter phases when imaged back-scatter SEM mode. As summarized in Table IV, all of the lighter phases contained a similar O/(Fe + Ti) weight percentage ratio as that observed in the uniform particles. However, the darker phases showed differences.
In Figure 6(b), the dark areas were thick and long, and occupied most of the particle volume. These darker areas contained higher Ti content (19.2 wt pct) than the surrounding light phase. This type of non-uniform particles was the most common and accounted for ~ 12 pct of the total particle population. Approximately 3 pct of the total particle population had a morphology similar to that shown in Figure 6(c). These particles looked similar to the non-uniform TTH particles in the un-sintered ironsand, except that the darker areas were no longer continuous lamellae. The dark areas contained ~ 13.6 wt pct Ti.
Figure 6(d) shows a further type of non-uniform particle morphology, which accounted for ~ 1 pct of the total grain population. In this case, a regular and densely hatched lamellar structure is observed. The darker areas were not continuous, but instead formed individual “islands” bounded by the lighter lamellae. These dark areas contained higher Al and Mg contents (9.8 and 9.1 wt pct, respectively) and a lower Ti content (1.93 wt pct) than the lighter phase. This phase segregation of Al and Mg was not observed in any of the raw ironsand particles, which suggests that Al and Mg were redistributed during oxidation.
Influence of Reduction Temperature
Figure 7 presents the results of TGA experiments studying the reduction of oxidized pellets in flowing H2 gas at temperatures between 1043 K and 1443 K. From this figure it can be seen the reduction rate increased significantly with increasing temperature, and that at all temperatures, a similar maximum reduction degree of 97 pct ± 0.8 pct was achieved. At 1043 K, it took ~ 40 minutes to reach the maximum reduction degree, while it took less than 20 minutes to achieve maximum reduction at all other temperatures. Attempts to measure the reduction kinetics of the TTH pellets at a higher temperature of 1543 K were not successful, due to the onset of weight loss caused by partial thermal decomposition, which occurred before the temperature of the furnace had fully stabilized.

Dependence of reduction degree on reduction time for oxidized pellets in flowing H2 gas. Data shown for temperatures at 100 K intervals between 1043 K and 1443 K
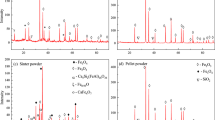
Post reduction, XRD analysis was performed on the fully reduced pellets. These results are shown in Figure 8, and clearly show the presence of residual iron-titanium oxides at the end of each experiment. The weight proportions of these minor phases (ilmenite (FeTiO3), rutile (TiO2), pseudobrookite (PSB, Fe2TiO5), and an undefined spinel) changes with reduction temperature, as is shown in Figure 8(b). The presence of these residual oxides is consistent with a final reduction degree of <100 pct. Further, from Figure 8(b) it can be seen that on increasing the reduction temperature, both FeTiO3 and TiO2 decreased while the PSB content increased. The spinel concentration did not change significantly with temperature. These observations are consistent with previous reports that PSB is more stable at higher temperatures.[32] A similar trend was also observed by Prabowo et al.,[10] where it was suggested that rutile combines with ilmenite to form PSB under high-temperature reducing conditions.

(a) Magnified XRD diffractograms obtained from fully reduced pellets following reduction at each temperature. (Note that the metallic iron peak (A) is truncated in order to enable minor peaks to be clearly shown.) (b) Weight proportions of minor phases in final fully reduced pellets at each reduction temperature. Values obtained from quantitative-XRD analysis using Topas 4.2 (Bruker) software
Morphology Analysis
Cross-section microscopy was carried out on the reduced TTH pellets to characterize the effects of temperature on the pellet morphology. Figures 9(a) through (e) show representative back-scatter SEM images of the pellet morphology at each experimental temperature. Lower temperature reduction (1043 K and 1143 K, Figures 9(a) and (b)) resulted in metallic Fe having an irregular dendritic structure. As the reduction temperature increased, this dendritic structure coarsened and became less defined, with Fe instead forming bulbous clusters. The higher the temperature, the stronger the coarsening effect. This is consistent with the expected increased diffusion mobility of Fe at higher temperatures.

Back-scatter SEM images of cross-sectioned fully reduced pellets showing the final morphologies of particles at each reducing temperature studied: (a) 1043 K; (b) 1143 K; (c) 1243 K; (d) 1343 K; and (e) 1443 K
Influence of Pellet Size
Pellets produced via industrial processes typically have a size distribution covering a range of diameters. It is therefore important to understand the effect of pellet size on reduction kinetics.
The effect of pellet diameter on the reduction rate at 1343 K is plotted in Figure 10. As the pellet size increases from 5.5 to 8.5 mm, the reduction rate decreases steadily. This is consistent with the findings in McAdam et al.’s work,[21] and indicates that pellet-scale processes must play a role in the rate-controlling step of the reduction process.

Dependence of reduction degree on reduction time of oxidized spherical pellets with different diameters. Data obtained at 1343 K in flowing H2 gas
Kinetic Analysis of the Reduction Process
The reduction of conventional hematite pellets by H2 proceeds via the following steps[31]:
-
1.
Diffusion of gas reactant from the bulk gas to the surface of the sample;
-
2.
Penetration and diffusion of the gas reactant through the generated solid product layer to the surface of reaction site;
-
3.
Reaction of gas reactant with solid at this reaction site;
-
4.
Diffusion of gas product through the same solid product layer to the exterior surface of solid;
-
5.
Diffusion of the gas product from the solid exterior surface to the gas flow.
Various kinetic models have been developed to describe reactions involving one or more of these steps. Among them, the shrinking core model (SCM) is perhaps the most widely applied. Expressions that can be used to determine the rate-limiting step(s) in this model were derived by Levenspiel,[31] and have been shown to represent the behavior of iron ore particles/pellets in many studies (as summarized in the introduction). Despite the inherently multi-step nature of hematite reduction, several works[4,26,27,28,29] have shown that a reasonable description of kinetics can often be achieved through assuming a simplified single reaction step, Fe2O3 → Fe or FeO → Fe.
Reduction of TTH pellets may be expected to follow the same sequence of steps as for conventional hematite. The strong influence of pellet size upon reduction rate observed in this study is suggestive of a pellet-scale shrinking core mechanism. As such, the application of the SCM to the TTH reduction kinetics has been attempted.
The single-interface SCM assumes that the overall pellet size remains unchanged during reduction and the overall reaction rate is determined by the slowest or rate-limiting step, in the sequence of steps 1 to 5. For this work, only the interfacial chemical reaction (step 3) or gas diffusion through product layer (steps 2 and 4) needs to be considered. Steps 1 and 5, mass transfer effects in the gas phase, were eliminated by choice of gas flow rate in the experimental setup.
The ‘rate-limiting’ equations for a reaction controlled by a combined mix of gas diffusion and interfacial chemical reaction steps are as follows[31]:
where
and
Here, \( I\left( {X_{\text{a}} } \right) \) denotes the interfacial chemical reaction dependence, \( D\left( {X_{\text{a}} } \right) \) is the diffusion dependence, t is time, \( \rho_{\text{B}} \) is the density of the pellet, \( R \) is the pellet diameter, \( b \) is the stoichiometric coefficient, \( C_{\text{A}} \) is the concentration of the reducing gas at the reaction interface, \( D_{\text{e}} \) is the effective gas diffusion coefficient through the product layer, \( k_{\text{g}} \) is the chemical reaction coefficient, \( X_{\text{a}} = {\raise0.7ex\hbox{$X$} \!\mathord{\left/ {\vphantom {X {100}}}\right.\kern-0pt} \!\lower0.7ex\hbox{${100}$}} \) is the reduction degree expressed as a decimal ratio, \( k^{\prime}_{\text{g}} \) is the apparent reaction rate constant, and \( D^{\prime}_{\text{e}} \) is the apparent diffusion rate constant.
Equation [2] can be linearized by dividing both sides either by \( I\left( {X_{\text{a}} } \right) \) or \( D\left( {X_{\text{a}} } \right) \) to generate Eqs. [7] and [8]. This enables the dependence on each term to be analyzed using the plots shown in Figures 11(a) and (b), respectively.

Plots enabling values for the apparent diffusion constant and apparent reaction rate constant to be determined from Eqs. [7] and [8]. Linear fits are shown between X = 10 pct and X = 90 pct for experimental data obtained for pellet diameter of ~ 7 mm at temperatures from 1043 K to 1443 K. These fits are extrapolated to the y-axis. (a) Plot relating to Eq. [7], where the gradient describes the contribution of the diffusion component to the rate-limiting step, while the y-axis intercept determines \( 1/k^{\prime}_{\text{g}} \). (b) Plot relating to Eq. [8], where the gradient determines the relative contribution of the interfacial reaction component to the rate-limiting step, while the y-axis intercept determines \( 1/D^{\prime}_{\text{e}} \)
Taken together, Figures 11(a) and (b) present strong evidence that for each of the temperatures experimentally studied, the single-interface SCM model provides a good description of the period during which most of the reaction occurs. Reasonable linear fits are obtained for all data sets between X = 10 pct and X = 90 pct, while the poorer agreement observed during the very early and very late stages of the reaction is to be expected. During the very early (initial) stages, the reaction interface will take time to develop (as it is not infinitesimally thick), while any experimental measurement errors are amplified by the ratio of a short time and a small mass loss. Similarly, once the reduction degree exceeds 90 pct (late stage), the morphologies shown in Figure 9 will have become established throughout the pellets with the remaining reactions sites interspersed within the dendritic iron such that they can no longer be considered as a single core.
In Figure 11(a), we see that the linear fits at all temperatures exhibit a non-zero y-axis intercept, but that only the data at 1043 K exhibit a linear gradient that is appreciably more than zero (the other lines are all close to horizontal). This implies that at T ≥ 1143 K the diffusion component of the rate-limiting step is zero (as \( 1/D^{\prime}_{\text{e}} \approx 0) \). This indicates that the reaction is controlled solely by the rate of the chemical reaction at the interface. However, at 1043 K we observed mixed control in which both diffusion and interfacial chemical reaction play a role.
This is confirmed by the linear fits shown in Figure 11(b). In this plot we see that all lines exhibit a positive non-zero gradient indicating that in each case the reaction rate is at least partly controlled by the I(X) component. This indicates that at no temperature in the range considered are the kinetics solely controlled by diffusion. However, at all temperatures ≥ 1143 K we also observe that the y-axis intercept is zero, confirming that the diffusion coefficient can be neglected at these temperatures, and we have chemical reaction control. As before, it is only at 1043 K that a non-zero contribution from the diffusion component is observed.
Figures 12(a) and (b) show a similar analysis of the applicability of the SCM model to the experiment data obtained from different size pellets at 1343 K (raw data shown in Figure 10). These plots show that both the gradient in Figure 12(a) and the intercept in Figure 12(b) are close to zero for all pellet sizes, indicating that diffusion did not contribute to the rate-controlling step in any of these experiments. Instead, interfacial chemical reaction is found to be the dominant rate-controlling step for all pellet sizes at 1343 K.

Plots enabling the relative contribution of diffusion component, \( D(X_{\text{a}} ) \), and interfacial chemical reaction component, \( I(X_{\text{a}} ) \), using Eqs. [7] and [8]. Linear fits are shown between X = 10 pct and X = 90 pct, for experimental data obtained for pellet diameters of different sizes (5.5, 6.5, 7.5, and 8.5 mm) at 1343 K. These linear fits are shown extrapolated to the y-axis. (a) Plot relating to Eq. [7], where the gradient describes the contribution of the diffusion component to the rate-limiting step, while the y-axis intercept determines \( 1/k^{\prime}_{\text{g}} \). (b) Plot relating to Eq. [8], where the gradient determines the relative contribution of the interfacial reaction component to the rate-limiting step, while the y-axis intercept determines \( 1/D^{\prime}_{\text{e}} \)
Kinetic Analysis Discussion
At temperatures from 1143 K to 1443 K, the interfacial chemical reaction rate controlled the reduction rate. In this case, Eq. [2] simplifies to
Figure 13 shows linear fits of Eq. [9] to the experimental data obtained from 1143 K to 1443 K. In this case, fitting was conducted using all data for X ≤ 90 pct. R2 values for each fit are listed in Table V, and values of 0.998 or higher were obtained in each case, confirming a close fit with experimental data.

Fitting of Eq. [9] to experimental data obtained at temperatures from 1143 K to 1443 K with pellets of ~ 7 mm diameter. These fits are conducted to X ≤ 90 pct. Data for X > 90 pct are highlighted by gray area
The activation energy of the interfacial chemical reaction is defined by the Arrhenius equation:
where \( k_{\text{g}} \) is the reaction rate constant, \( R \) is the universal gas constant, \( A \) is a frequency factor and \( T \) is the absolute temperature in Kelvin. However, the calculation of kg requires prior knowledge of \( C_{\text{A}} \) (the concentration of the reducing gas at the reaction interface). This is not easily determined. Instead, the apparent rate constant \( k^{\prime}_{\text{g}} \) was applied. If B is approximately constant with temperature, then the activation energy can be obtained from
where \( B = \rho_{\text{B}} R/bC_{\text{A}} \). The gradient of each fitted line in Figure 13 was determined and used to calculate \( k^{\prime}_{\text{g}} \). The values obtained are shown in Table V, while Figure 14 plots \( \ln k^{\prime}_{\text{g}} \) against 1/T. The apparent activation energy can be calculated from the gradient of this plot, and is found to be 31.3 ± 1.3 kJ/mol.

Arrhenius plot for determining apparent activation energy of TTH pellets of ~ 7 mm reduced by H2 from 1143 K to 1443 K
Minimum Pellet Size for the Shrinking Core Model
Equation [9] also applies to the experimental data obtained for different pellet sizes at 1343 K. Figure 15(a) plots linear fits to these data and show that the gradient increases with increasing pellet diameter. From Eqs. [5] and [9], the gradient is given by \( \frac{1}{{k^{\prime}_{\text{g}} }} = \frac{{\rho_{\text{B}} R}}{{bk_{\text{g}} C_{\text{A}} }} \). Since each of these reductions were conducted under identical conditions (except for pellet diameter), \( \rho_{\text{B}} \), \( b \), \( k_{\text{g}} \), and \( C_{\text{A}} \) should all be the same. Therefore, a simple linear relation between \( 1/k^{\prime}_{\text{g}} \) and \( R \) is expected. Table VI shows the fitted values obtained for the slope of each line in Figure 15(a) (which is equal to \( 1/k^{\prime}_{\text{g}} \)) for each pellet diameter. These values are plotted in Figure 15(b). While a linear correlation is observed, this line extrapolates to intercept the x-axis at 2.5 mm. This is not expected from a direct application Eq. [5]. This x-axis intercept is considered to represent the minimum critical pellet diameter at which a shrinking core interface will occur. If the pellet diameter is less than 2.5 mm, the pellet may be too small to accommodate the fully developed interface width. As such, the single-interface pellet-scale SCM can no longer be applied to represent the data, and an alternative grain scale model is required to describe the reduction behavior.

(a) Plot showing fits of interfacial chemical reaction control (Eq. [9]) to experimental data obtained at different pellet diameters, at a reduction temperature of 1343 K. These fits are conducted to X ≤ 90 pct. Data for X > 90 pct are indicated by the shaded gray area. (b) Plot showing the linear relationship between pellet diameter and the fitted slopes \( ( = 1/k^{\prime}_{\text{g}} ) \) obtained from (a)
Accuracy of the Shrinking Core Model
An accurate analytical description of the reduction rate of NZ TTH pellets in H2 has potential industrial importance, as this can inform the design and optimization of new prototype H2-DRI reactors. Figure 16(a) shows comparisons between experimental data for T ≥ 1143 K and reduction values calculated from Eqs. [9] and [11] using the fitted values for EA and B obtained before. Figure 16(b) shows a similar comparison between experimental data at 1043 K and calculated values for X, obtained using values for \( 1/D^{\prime}_{\text{e}} \) and \( 1/k^{\prime}_{\text{g}} \) obtained from the intercept and gradients shown in Figure 11. The close agreement with experimental data up to 90 pct reduction degree is observed at all temperatures, indicating the pellet reduction kinetics are broadly consistent with the SCM.

Comparison of reduction degrees obtained from experiment with those calculated from the kinetic models developed in this work. (a) Reduction temperature from 1143 K to 1443 K. (b) Reduction temperature = 1043 K
When the reducing temperature is above 1143 K, only parameters that affect the interfacial chemical reaction rate constant will influence the reduction rate (e.g., temperature, initial pellet diameter). However, at 1043 K other parameters which affect the diffusion rate can also have an influence on the reduction kinetics (e.g., pellet porosity). This has implications for future optimization of a pelletizing process. Optimizing pellet porosity is likely to be more important in the design of lower temperature H2-DRI processes for TTH pellets, while optimizing the production and handling of smaller pellets will play a more important role for higher temperature processes.
Conclusion
In this work, the reduction characteristics of oxidized NZ ironsand pellets in an H2 gas flow were studied using a TGA system. The pellets were reduced isothermally over the temperature range of 1043 K to 1443 K. An analytical single-interface shrinking core model (SCM) was investigated and applied to describe the reaction and compare with experiment results. Based on the analysis and observations, the key findings are as follows:
-
1.
After oxidation of the as-received un-sintered NZ TTM ironsand, the uniform TTM particles in the ironsand were completely transformed into TTH particles. These comprised the great majority of the particles within the ironsand, and XRD confirmed that TTH was the dominant crystalline phase present within the oxidized pellets. The raw TTM ironsand also included a minority of non-uniform particles containing lamellar structures, but these were not found in the oxidized ironsand. Instead, three different types of non-uniform particles were observed.
-
2.
The reduction rate of the oxidized TTH pellets in 100 pct H2 increased with increasing temperature, while the final reduction degree was approximately equal at all temperatures studied. The maximum reduction degree achieved in these experiments was ~ 97 pct ± 0.8 pct. 100 pct reduction was not achieved at any temperature. This was principally due to the formation of residual Fe-Ti-O phases that once formed remained present to the end of each experiment.
-
3.
The specific Ti-bearing phases present in the final reduced pellets depended on the reaction temperature. It was found that the content of ilmenite and rutile decreased with increasing temperature, while the content of PSB increased.
-
4.
The internal micro-morphology of the fully reduced pellets changed with increasing reduction temperature. Dendritic whiskers were observed to be the main type of micro-structure at the lowest temperature (1043 K), while more globular ‘coarsened’ clusters were observed to occur at higher temperatures.
-
5.
The reduction rate increased with decreasing pellet size at 1343 K, which indicated that pellet-scale mechanisms determined the reduction rate. A simple single-interface SCM was used to analyze and describe the reduction data. Generally, good agreement between experiment and the SCM was obtained for reduction degrees up to 90 pct. However, agreement was poor at higher reduction degrees. This may be due to the formation of Fe-Ti-O phases, found dispersed throughout the pellet, during the latter stages of reduction.
-
6.
Kinetic modeling using the shrinking core approach enabled the different rate-controlling process above and below 1143 K to be identified. At 1043 K, the reduction kinetics were determined by a mixed control mechanism including both diffusion and interfacial reaction components. However, at temperatures from 1143 K to 1443 K, reaction control depended only on an interfacial reaction process, which exhibited an activation energy of 31.3 ± 1.3 kJ/mol.
References
1 H. Sun, A.A. Adetoro, Z. Wang, F. Pan, and L. Li: ISIJ Int., 2016, vol. 56, pp. 936–43.
2 R.L. Brathwaite, M.F. Gazley, and A.B. Christie: J. Geochemical Explor., 2017, vol. 178, pp. 23–34.
3 D.S. Chen, B. Song, L.N. Wang, T. Qi, Y. Wang, and W.J. Wang: Miner. Eng., 2011, vol. 24, pp. 864–9.
4 Y. Li, H. Guo, and L. Li: Chinese J. Eng., 2011, vol. 40, pp. 154–57.
A.A. Adetoro, H. Sun, S. He, Q. Zhu, and H. Li: Metall. Mater. Trans. B, 2018, vol. 49B, pp. 846–57.
Iron and steel – Te Ara Encyclopedia of New Zealand, https://teara.govt.nz/en/iron-and-steel/page-2. Accessed 28 June 2017.
K.-H. Tacke and R. Steffen: 1st Chinese-German Seminar on Fundamentals of Iron and Steelmaking, Beijing, 2004.
8 V. Vogl, M. Åhman, and L.J. Nilsson: J. Clean. Prod., 2018, vol. 203, pp. 736–45.
9 A. Ranzani da Costa, D. Wagner, and F. Patisson: J. Clean. Prod., 2013, vol. 46(May), pp. 27–35.
S.W. Prabowo, R.J. Longbottom, B.J. Monaghan, D. del Puerto, M.J. Ryan, and C.W. Bumby: Metall. Mater. Trans. B, 2019, 50: 1729.
11 M.E. Choi and H.Y. Sohn: Ironmak. Steelmak., 2010, vol. 37, pp. 81–8.
12 E. Park and O. Ostrovski: ISIJ Int., 2003, vol. 43, pp. 1316–25.
R.J. Longbottom, O. Ostrovski, J. Zhang, and D. Young: Metall. Mater. Trans. B, 2007, vol. 38, pp. 175–84.
14 Y.L. Sui, Y.F. Guo, T. Jiang, X.L. Xie, S. Wang, and F.Q. Zheng: Int. J. Miner. Metall. Mater., 2017, vol. 24, pp. 10–7.
15 E. Park and O. Ostrovski: Isij Int., 2004, vol. 44, pp. 74–81.
16 E. Park and O. Ostrovski: ISIJ Int., 2004, vol. 44, pp. 999–1005.
17 J. Dang, G.H. Zhang, X.J. Hu, and K.C. Chou: Int. J. Miner. Metall. Mater., 2013, vol. 20, pp. 1134–40.
18 X.F. She, H.Y. Sun, X.J. Dong, Q.G. Xue, and J.S. Wang: J. Min. Metall. Sect. B Metall., 2013, vol. 49, pp. 263–70.
19 R.J. Longbottom, O. Ostrovski, and E. Park: ISIJ Int., 2006, vol. 46, pp. 641–6.
20 Z. Wang, J. Zhang, K. Jiao, Z. Liu, and M. Barati: J. Alloys Compd., 2017, vol. 729, pp. 874–83.
21 G.D McAdam, R.E.A Dall, T. Marshall: NEW Zeal. J. Sci., 1969, vol. 12, pp. 649–68.
J. Tang, M.S. Chu, Z.W. Ying, F. Li, C. Feng and Z.G. Liu: Metals (Basel)., 2017, vol. 7: 153.
23 Y.D. Wang, X.N. Hua, C.C. Zhao, T.T. Fu, W. Li, and W. Wang: Int. J. Hydrogen Energy, 2017, vol. 42, pp. 5667–75.
24 B. Hou, H. Zhang, H. Li, and Q. Zhu: Chinese J. Chem. Eng., 2015, vol. 23, pp. 974–80.
25 C. Li, H. Lin, Y. Chen: Thermochim. Acta, 2003, vol. 400, pp. 61–7.
26 A. Bonalde, A. Henriquez, and M. Manrique: ISIJ Int., 2005, vol. 45, pp. 1255–60.
M. Kazemi, M.S. Pour, and D. Sichen: Metall. Mater. Trans. B 48: 1114 (2017).
28 H. Zuo, C. Wang, J. Dong, K. Jiao, and R. Xu: Int. J. Miner. Metall. Mater., 2015, vol. 22, pp. 688–96.
B. Kennedy, N.H. Daniels, T. Marshall: N. Z. J. Sci., 1969, vol. 12, pp. 701–20.
A. Zhang, R.J. Longbottom, M. Nusheh, C.W. Bumby, and B.J. Monaghan: Proc. 8th International Congress on science and Technology of Ironmaking, Art. No. 282, Vienna, 2018.
Levenspiel O (1999) Chemical Reaction Engineering. Wiley, New York
32 P. den Hoed and A. Luckos: Oil Gas Sci. Technol., 2011, vol. 66, pp. 249–63
Acknowledgments
This research was supported by funding received from the Endeavour Fund of the New Zealand Ministry of Business Innovation and Employment (Grant No. RTVU1404)
Author information
Authors and Affiliations
Corresponding author
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Zhang, A., Monaghan, B.J., Longbottom, R.J. et al. Reduction Kinetics of Oxidized New Zealand Ironsand Pellets in H2 at Temperatures up to 1443 K. Metall Mater Trans B 51, 492–504 (2020). https://doi.org/10.1007/s11663-020-01790-3
Received:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-020-01790-3