
Abstract
Steel produced in an electric arc furnace (EAF) contains a high amount of Cu that causes a surface-cracking phenomenon called surface hot shortness. Ni reduces the risk for surface hot shortness, and this work focuses on investigating the following two phenomena caused by Ni during oxidation at 1150 °C for Fe–Cu–Ni alloys: (1) the decrease in oxidation rate and (2) the formation of a wavy liquid-Cu/oxide and of liquid-Cu/γ-iron (γFe) interfaces, which promote Cu occlusion into the scale. Thermogravimetry, scanning electron microscopy, and transmission electron microscopy-energy dispersive spectroscopy techniques were applied. A numerical model also was developed to explain the experimental results. High Ni contents cause higher liquid-Cu/γFe interface nickel concentrations and more potential for an interface breakdown. The decrease in oxidation rate by adding nickel can be explained qualitatively by the decrease in Fe cation transport through the wüstite layer.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
In 2008, about 42 pct of the steel in the United States (40 million tons) was produced by recycling scrap in electric arc furnaces (EAFs), and it is expected that this amount will continue to increase.[1] Although EAF-steelmaking saves energy, raw material, and cost compared with the blast furnace/basic oxygen furnace (BF/BOF) route,[2] an important problem exists called surface hot shortness that limits steel manufacturing through EAF-based techniques.[3,4] Because the feed materials of EAFs are predominantly steel scrap, it inevitably contains Cu, usually in the range between 0.25 wt pct and 0.45 wt pct,[5] which mostly originates from scraped cars and wirings. When the Fe in the steel oxidizes at temperatures above 1100 °C, Cu enriches at the oxide/steel interface, and when the content exceeds the Cu solubility in the γFe phase, a Cu-rich liquid layer forms. The liquid-Cu can penetrate into the γFe grain boundaries, and as a result, the steel surface can crack during rolling. This process is the origin of the surface hot shortness problem caused by Cu.
Because other residual elements in the steel scrap could affect Cu behavior in the γFe phase,[4,6] one remedy for hot shortness is to control these elements by mixing various scrap sources. Ni, which is usually present in steel scrap, is the key element to suppress hot shortness by increasing the solubility of Cu in the γFe phase,[7] decreasing the oxidation rate,[8] and promoting occlusion.[9] Occlusion is a phenomenon whereby the Cu-rich phase becomes incorporated into Fe oxides when the internal Fe oxides bridge with the external scale. It was suggested that internal oxidation and an uneven interface are necessary conditions for occlusion to occur.[10,11] Fukagawa found that an addition of 0.02 wt pct Ni was sufficient to cause a wavy oxide/steel interface, whereas 0.001 wt pct Ni content did not result in a significantly perturbed interface.[10] Akamatsu et al. suggested that the Ni concentration at the liquid-Cu/γFe interface increases along the ternary phase boundary as oxidation proceeds.[12] Based on this suggestion, Webler et al.[8] proposed a qualitative description of the liquid-Cu/γFe and liquid-Cu/oxide interface morphology evolution in terms of the concepts of constitutional super-saturation and diffusion path[13] and suggested that the decrease in the oxidation rate by adding Ni was attributed to the breakdown of the liquid-Cu layer into isolated liquid pockets.
This article focuses on the effects of Ni on the liquid-Cu/oxide and liquid-Cu/γFe interface morphologies and on the oxidation rate for Fe–Cu–Ni alloys. Thermogravimetry (TG), scanning electron microscopy (SEM) and transmission electron microscopy-energy dispersive spectroscopy (TEM-EDS) techniques were used to investigate oxidation behavior, interface morphology, and liquid-Cu/γFe interface concentrations, respectively. A numerical model is presented to support the experimental results, which predict the liquid-Cu/γFe interface concentrations and interface perturbation.
Methodology
Experiment Methodology
Materials
Fe–Cu–Ni alloys were chosen to investigate the effects of Ni; their compositions are listed in Table I. Fe, Fe-0.3 wt pct Cu and Fe-0.3 wt pct Cu-0.10 wt pct Ni alloys from a previous study conducted in similar conditions also are presented for comparison and are marked with an asterisk.[8] These alloys were produced by the Materials Preparation Center at the Ames Laboratory.[14] The ferrite grain size of these samples was approximately 500 μm.
Methods
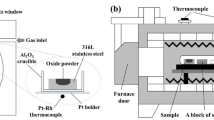
TG was used to measure the oxidation kinetics of alloys in the air at 1150 °C for 1 and 10 minutes. The temperature profile in the current experiment and the TG setup are shown in Figure 1. Samples were polished down to 320 SiC with abrasive paper before oxidation and had dimensions of 19 mm × 10 mm × 5 mm. Oxidation data were recorded every second and were reported as mass change per unit area (Δm/A). To achieve rapid heating, an infrared gold image furnace with high-energy-density halogen lamps was used. The constant temperature hot zone in the furnace was 14 cm length and 1 cm diameter, and its temperature range was ±1 °C. A K-type thermocouple was inserted into the bottom of the tube to measure the hot-zone temperature. Samples were attached to a balance by a platinum wire, and the bottom of the sample was placed as close as possible to the thermocouple to obtain an accurate temperature of the sample. The placement of the thermocouple was determined by the point at which molten Cu was found. Although the oxidation reaction was exothermic, and this could heat the sample and the surrounding area, this effect would be counteracted by adjusting the input power of the thermocouple automatically. As will be shown later, the fact that the oxidation rate of pure Fe in the current set up was consistent with the previous reported values for isothermal oxidation is an indirect validation for temperature control.

(a) Temperature profile in the current experiment and (b) TG setup
The air flow rate was maintained at 3789 standard cubic centimeters per minute (sccm), which corresponds to a linear velocity of 3.22 cm/s at 25 °C, to supply enough oxygen but not causing sample cooling.[15,16] The air used had a water-vapor partial pressure of 1.4 × 10−4 atm at 23 °C.
After oxidation samples were cold-mounted in epoxy to preserve oxide/metal interfaces and subsequently sectioned by a low-speed diamond saw. The microstructure of these samples was investigated in a Philips XL30 SEM (Amsterdam, The Netherlands) in backscatter electron (BSE) mode. The liquid-Cu/oxide (or oxide/γFe if no liquid was present) interface roughness, fraction of interface covered by the Cu-rich phase, and occluded Cu-rich phase were analyzed from ten SEM images for each chemistry by an open-source software.[17] Interface roughness was obtained by normalizing the tracking interface length to the length of the image to interpret the degree of perturbation, and the total area of the occluded Cu-rich phase in the scale was divided by the length of the image to characterize the amount of occluded material. Standard deviation of ten quantification results for each sample was used as the error bar.
An FEI NOVA 600 focused ion beam (FIB) (FEI, Hillsboro, OR) was used to prepare TEM samples containing liquid-Cu/γFe interfaces, as illustrated in Figure 2. A Tecnai F20 field emission TEM (Hillsboro, OR) installed with an EDS detector was used to investigate concentration profiles of Fe, Cu, and Ni near the interfaces. ESVision-Offline software was used to do quantification analysis of the EDS spectra.

Preparation procedure of TEM samples in FIB. (a) Isolation of thin material near the interface. (b) The thin material was lifted out by microprobe. (c) The final TEM sample after polishing in FIB
Modeling Methodology
A numerical model simulating the diffusion process of Ni and Cu in the γFe during oxidation at 1150 °C was developed to compare quantitatively with the experimental results. Oxygen was not considered as a component in the Fe–Cu–Ni ternary system. Liquid-Cu/γFe (or oxide/γFe if liquid is absent) interface concentrations and concentration gradients in the γFe were calculated as functions of time.
Figure 3(a) shows the arrangement of phases and the Ni concentration profiles. The quantities used in the model are summarized in Table II. During oxidation, the liquid-Cu/γFe and liquid-Cu/oxide interfaces would move toward the right in Figure 3(a) with consumption of the γFe phase. The equilibrium compositions on both sides of the liquid-Cu/γFe interface can change with time because of the additional degree of freedom in the two-phase region of the Fe–Cu–Ni ternary system. By virtue of the shape of the phase boundaries in the isothermal ternary Fe–Cu–Ni phase diagram at 1150 °C calculated from Thermo-Calc® software (Stockholm, Sweden)[18] (Figure 3(b)), the Cu rejected from the oxidized Fe was incorporated mostly in the liquid phase, whereas only a small portion of the rejected Ni incorporated into the liquid-Cu phase because of a limited solubility. Back diffusion of Ni in γFe was slow compared with the rate of Ni rejection because of the fast oxidation in the early oxidation stage, and consequentially, Ni built up in the γFe along the liquid-Cu/γFe phase boundary, i.e., the interface composition at the γFe side changed along the phase boundary following a, b, c, and d in Figure 3(b), as stated by Akamatsu.[12] However this Ni enrichment could only be sustained below a critical value C *Ni (the maximum Ni concentration at the phase boundary as shown in Figure 3(b)) beyond which, the growth of the liquid would necessitate lowering the interface Ni concentration. Thus, the liquid cannot be sustained, and the interface would be oxide/γFe with a composition on the alloy side, e.g., point e in Figure 3(b).

(a) Phases arrangement and Ni profiles. (b) Fe–Cu–Ni phase diagram at 1150 °C calculated from Thermo-Calc (Stockholm, Sweden)
The model was formulated based on the following assumptions:
-
(a)
Liquid-Cu formed immediately after the oxidation began, and therefore, the initial interface concentration had the same Cu/Ni ratio as the bulk Cu/Ni ratio. Calculations[19] showed that the time for the formation of liquid-Cu was about 10 milliseconds, which justified this assumption;
-
(b)
The composition of the liquid phase was uniform because diffusion in the liquid-Cu (~10−9 m2/s[20]) was rapid compared with diffusion in the oxide (~10−11 m2/s[21]) and diffusion in γFe (~10−15 m2/s[22]);
-
(c)
Nondiagonal terms of ternary interdiffusion coefficients were assumed to be zero and the on diagonal terms were assumed to be concentration independent;
-
(d)
Concentration profiles in γFe were not distorted by the moving interface when Fe was consumed (γFe shrinks) through oxidation;
-
(e)
The amount of occluded Cu-rich phase and the migration of the Cu-rich phase into the scale were negligible. Our previous[8] and current works show that Ni alone did not cause much occlusion within 10 minutes of oxidation, as will be shown later (Figure 7(c)). Although some reports[23] suggested that Cu could migrate into scales on industrial steel samples, our previous work[24] showed that this amount was negligible for Fe–Cu alloys.
A global mass-balance method was adopted in which a constant diffusion-affected zone was used to integrate the concentration profiles in γFe, and therefore, Fe was considered to be consumed continuously from the diffusion unaffected zone. The mass balance equations for Cu and Ni are as follows:
To solve Eqs. [1] and [2], the liquid-Cu/γFe interface Cu and Ni compositions on the phase boundaries of the Fe–Cu–Ni ternary phase diagram in both the γFe and liquid-Cu phases were collected by Thermo-Calc (Stockholm, Sweden). The transient diffusion model in Comsol Multiphysics[25] (Burlington, MA) was used to solve the diffusion process in the γFe phase using the finite element method. The following solution strategy was implemented by coupling Comsol Multiphysics and Matlab (Natick, MA)[26] to calculate interface compositions and interface concentration gradients as a function of time:
-
(a)
Obtain oxidation TG data, initial interface concentrations, and liquid thickness;
-
(b)
Obtain the interface Ni concentration that satisfies Eq. [2] (using L(t) of the last time step);
-
(c)
Obtain the solution of L(t) using Eq. [1];
-
(d)
Repeat step (b) and (c).
The input data of the model are listed in Table III. The length of the diffusion-affected zone was chosen to be longer than the diffusion depth of Fe, Cu, and Ni in the γFe phase.
The theory for interface breakdown was used to interpret the interface morphology. A nonplanar interface can develop in ternary systems, such as ternary diffusion couples,[27–29] the oxidation of binary alloys (using oxygen as the third component),[30–32] and the solidification of binary or multicomponent alloys (using thermal diffusion as the third field).[33,34] The interface in a binary diffusion couple is always flat because no concentration gradient is allowed in the two-phase region, and interface breakdown has no driving force.[35] However, in a ternary diffusion system, the additional degree of freedom in a two-phase region gives the possibility to develop nonplanar interfaces.
Kirkaldy and Brown[28] suggested that calculated diffusion paths assuming a flat interface (virtual path) could predict the breakdown of interfaces in ternary systems. Coates and Kirkaldy[13] then proposed a quantitative constitutional super-saturation criterion (elementary criterion) in analogy to the constitutional super-cooling criterion in solidification, which represents the same idea as a “diffusion path,” to explain interface morphology. The constitutional super-saturation criterion deals with two independent diffusion fields, whereas the constitutional super-cooling criterion involves a thermal field and a diffusion field. This theory is applicable to the current situation. Coates and Kirkaldy[36] also developed a more complicated criterion following the Mullins–Sekerka criterion.[33] But this standard assumed that alloy diffusion controls the moving boundary velocity, which is mostly not valid in our problem (wüstite diffusion control), as will be shown later. This theory, thus, was not adopted.
The constitutional super-saturation criterion is as follows: because a concentration gradient is permitted along the interface in a ternary system, a constitutional super-saturation layer could form, and an interface needs to break down to relax it. By calculating the interface concentration gradient, interface breakdown could be predicted. Because diffusion in the liquid phase and wüstite is fast, any concentration gradient at the liquid-Cu/oxide interface will vanish rapidly. Therefore, interface breakdown develops most likely at the liquid-Cu/γFe interface initially, and subsequently, the oxide/liquid-Cu interface creeps to become uneven. Although the motion of the liquid-Cu/γFe interface is a balance between two independent processes—the removal of Fe because of oxidation and the back-diffusion of Ni and Cu—it was assumed that the development of interface morphology results mainly from the back-diffusion process. Besides super-saturation, internal oxides[11] and changes in surface tension[37,38] caused by Ni also could affect the interface roughness. However, as will be shown later in Figure 6, the interface becomes uneven and few internal oxides are present. The change in surface tension caused by Ni could be a possibility, but the data available for the Fe–Cu–Ni system[38] are at significantly higher temperatures. More work is needed to explore this possibility.
At every time instance, the perturbation value was calculated through Eq. [3][13] to predict the liquid-Cu/γFe interface breakdown, and if the perturbation criterion value was shown to be negative, then the interface was assumed to break down to a nonplanar interface.
G γ/LNi and G γ/LCu are concentration gradients for Ni and Cu, respectively, on the γFe side of the liquid-Cu/γFe interface, assuming a flat interface. As shown in Figure 3(a), with a positive direction from the oxide to the γFe phase, both G γ/LNi and G γ/LCu would be negative. The slope of the liquid-Cu/γFe phase boundary mγ at the γFe side was obtained from Thermo-Calc® (Stockholm, Sweden). Curve fitting of this phase boundary in the low Ni concentration part of the ternary phase diagram gave the following:
The aforementioned model has two limitations:
-
(a)
The model is strictly valid until the interface is perturbed. Although the predictions would be reasonable for some time, ultimately, the perturbations will advance to a size whereby lateral diffusion cannot be ignored;
-
(b)
The model requires modification when Ni exceeds its maximum concentration on the phase boundary C *Ni , as shown in Figure 3(b). In this case, the liquid-Cu phase will be expected to solidify. A different model using the global mass-balance method was set up to handle this problem, and the equations are the same as Eqs. [1] and [2] without the terms for the liquid phase. It was assumed, however, that solidification of the liquid-Cu layer was instantaneous when C *Ni was reached.
Results and Discussion
TG Results
Figure 4 shows TG curves ((Δm/A) vs time) for all alloys. TG results for Fe, Fe-0.3 wt pct Cu and Fe-0.3 wt pct Cu-0.10 wt pct Ni alloys conducted in previous works (marked with an asterisk) also are presented for comparison.[8] The SEM characterization and analysis of these samples, and TEM characterization of the 0.1Ni alloy were carried out independently in the current work. All TG curves obeyed a linear rate law in the first 60–110 seconds and subsequently changed to a parabolic rate law after 150 seconds. Figure 4 illustrates that the transition from linear to parabolic oxidation regimes starts earlier for the Fe–Cu–Ni alloys (at about 60 seconds) when compared with the Fe and Fe–Cu alloys (at about 110 seconds). Measured linear and parabolic oxidation rate constants are summarized in Figure 5. Linear rate constants were obtained by linear fitting ((Δm/A) vs time) of the first 60-seconds or 110-seconds data, and parabolic rate constants k p were given by parabolic fitting ((Δm/A)2 vs time) of data after 150 seconds to eliminate the effect of differences in transition point. Calculated linear[16] and parabolic[39] rate constants for Fe in the air at 1150 °C are presented as straight lines in Figure 5. The equations and parameters to calculate the linear rate constant[16] and k p [39] for Fe are shown in Eqs. [5]–[7] and Table IV. These results show that the linear oxidation rates for all alloys were similar, which is reasonable because, in this regime, the oxidation rates would be expected to be controlled by a gas-phase transfer of oxygen and are chemistry independent.[16] By contrast, the parabolic rate was expected to be controlled by Fe cation diffusion through the wüstite layer.[40] The k p for Fe–Cu–Ni alloys were observed to be lower by a factor of three, compared with the Fe and Fe–Cu cases (Figure 5(b)), which will be discussed later. The change in k p and the time of transition did not vary dramatically with Ni content, so long as Ni was present. The surface-area reduction after oxidation was estimated to be less than 9 pct, and therefore, a constant surface area was assumed in the analysis.

TG-measured weight change for samples oxidized for 600 s (300 s for Fe).*[8]

(a) Linear oxidation rate constants. (b) Parabolic oxidation rate constants.*[8]
SEM Results
After oxidation, the interface morphology was observed under SEM for all alloys, and the results are shown in Figure 6. Some sample oxides separated from the metal after cooling in the furnace and, therefore, they are not visible in some images. Fe and Fe–Cu alloys had planar liquid-Cu/γFe and liquid-Cu/oxide interfaces, whereas Fe–Cu–Ni alloys had uneven interfaces. The 0.10Ni and 0.15Ni alloys had rougher interfaces than the 0.03Ni and 0.06Ni alloys whose interfaces were nearly planar. The measured interface roughness, liquid-Cu cover fraction, and the amount of occluded Cu-rich phases are shown in Figures 7(a–c), respectively, and summarized in Table V. In Figure 7(b), for Fe–Cu and all Fe–Cu–Ni alloys, most interfaces were covered by liquid-Cu, which was pertinent for later analysis because it indicates that more or less no contact occurred between the wüstite and the γFe. The amount of occluded Cu-rich phase was negligible, which justifies the fifth assumption in the model.

BSE-SEM images of oxide/γFe interface

(a) Interface length. (b) Fraction of interface covered by liquid-Cu. (c) Amount of the occluded Cu-rich phase
Figure 8 shows examples of SEM images at a lower magnification, compared with those in Figure 7. Many large pores were observed in the wüstite layer for Fe–Cu alloys but not for the Fe–Cu–Ni alloys. Most oxides have a wüstite/magnetite/hematite thickness ratio close to 95:4:1.

Typical oxide layers after 10 min. oxidation. (a) Fe-0.3Cu. (b) Fe-0.3Cu-0.06Ni
TEM Results
To characterize interface concentrations at the liquid-Cu/γFe interfaces, TEM images and TEM-EDS profiles were obtained for 0.03Ni, 0.10Ni, and 0.15Ni alloys oxidized 10 minutes, which are shown in Figure 9. The positions chosen for the FIB-cross sectioning are indicated on the SEM images in Figure 9, and images obtained during the lift-out procedure in the FIB are presented in Figure 2. Interface concentrations were collected by averaging data from three profiles for each alloy and are summarized in Table V. The accuracy of the TEM-EDS data was estimated to be 3 wt pct by comparing EDS results with the known alloy matrix concentrations.

Cross-sectioning positions in SEM, TEM images, and concentration profiles near the interface. (a–c) 0.03Ni alloy oxidized for 10 min. (d–f) 0.10Ni alloy oxidized for 10 min. (g–i) 0.15Ni alloy oxidized for 10 min
Modeling Results and Comparison with Experimental Results
The aforementioned model was set up to simulate the evolution of liquid-Cu/γFe interface concentrations and predict liquid-Cu/γFe interface morphology, to explain the SEM results of the perturbations (Figure 6) as well as the TEM results of liquid-Cu/γFe interface compositions (Figure 9). As shown in Figure 3(b), the interface Ni concentration was expected to change during the course of oxidation at the liquid-Cu/γFe interface. This change was confirmed by the modeling results, and the calculated evolution of the Ni interface concentration C γ/LNi is shown in Figure 10. Figure 10 also illustrates the calculated times at which the interface broke down and C *Ni was reached. The predicted concentrations were plotted as thin dashed lines after C *Ni was reached because, after that point, the liquid would have solidified; another similar model was set up to handle the evolution of interface concentrations in the solid state until interface concentrations returned to the phase boundary. All alloys followed a reasonable trend, i.e., C γ/LNi first increased quickly to a peak value at about 60 seconds because of the fast linear oxidation rate and then decreased slowly in the parabolic oxidation regime as the rate of back diffusion became significant in comparison with the decreasing rate of accumulation. Modeling results predicted that, except for the 0.03Ni alloy, all liquid-Cu/γFe interfaces would break down within the linear oxidation regime with a breakdown occurring faster with increasing bulk Ni contents (Figure 10). It also was predicted that the 0.10Ni and 0.15Ni alloys could reach C *Ni within the linear oxidation but after the interfaces broke down, as shown in Figure 10. The calculated evolution of interface concentrations also is shown in Figure 3(b). For 0.03Ni and 0.06Ni alloys, interface concentration changes along the phase boundary; following case (1) in Figure 3(b): C γ/LNi increased from point a to c and decreased to b. The evolution of 0.10Ni and 0.15Ni alloys is shown in case (2) in Figure 3(b): interface concentrations increased along the phase boundary until C *Ni was reached and liquid-Cu solidified. C γ/LNi increased to point e until oxidation slowed down and subsequently decreased as a result of the lowered oxidation rate. Eventually interface concentrations returned to the phase boundary (point d), liquid Cu reappeared, and C γ/LNi decreased along the phase boundary.

Evolution of C γ/LNi and time points when interface perturbs and reaches C *Ni . Thin dashed lines are the evolution after reaching C *Ni
If liquid-Cu was present, then Fe at the liquid-Cu/γFe interface would diffuse quickly through the liquid-Cu layer, and the oxidation rate would be controlled by Fe cation transport in wüstite. When liquid-Cu disappeared, the oxidation process would be controlled by Fe diffusion through the alloy if the liquid-Cu/oxide interface Fe concentration decreased to zero because of the lack of Fe supply at the interface. Otherwise, Fe at the interface would diffuse directly through the wüstite layer, and the oxidation rate would be controlled by Fe cation diffusion through the wüstite. Calculations showed that zero Fe interface concentration exists only for a short time during the transition stage from linear to parabolic oxidation (long after interface breakdown) for 0.10Ni and 0.15Ni alloys. This finding suggests that after the linear oxidation regime, oxidation always is controlled by diffusion through the wüstite layer rather than diffusion in the alloy, which would be much slower (0.145 mg2/cm4/s based on calculations in Comsol Multiphysics® (Burlington, MA), assuming zero Fe interface concentration). This result is consistent with the fact that no sudden decrease in oxidation rate was observed.
Figure 11(a) illustrates the calculated maximum values of C γ/LNi and C γ/LNi after 10 minutes of oxidation, and the solid line indicates C *Ni . Concentrations above C *Ni are plotted as open symbols. As given in Figure 11(a), a higher-bulk Ni content resulted in a higher Ni rejection rate and, therefore, a higher-interface Ni concentration. Figure 11(b) shows that higher Ni contents have faster interface breakdown, which is reasonable according to Eq. [3]. The greater the C γ/LNi , the greater the Ni concentration gradient at the interface (more negative) and the greater the slope of the phase boundary (Eq. [4]). Therefore, a higher C γ/LNi has more chance of making the value of m γ G γ/LNi −G γ/LCu negative, which suggests an interface breakdown. As shown in Figure 11(a), alloys with higher-bulk Ni contents have higher C γ/LNi and, therefore, higher-bulk Ni contents have more potential for interface breakdown.

(a) Predicted maximum interface Ni concentration and interface Ni concentration after 10-min oxidation (open squares are concentrations greater than C *Ni ). (b) Time points when interface perturbs
Table V summarizes the results of the SEM and TEM characterizations and model predictions for 10 minutes of oxidation.
In Table V, the predicted interface concentrations are reasonably close to the experimental EDS results when considering the uncertainties in the available experimental data. Calculated results show the same trend as the experimental results—namely that the alloys with higher-bulk Ni contents have higher Ni interface concentrations, which also is shown in Figure 12.

Comparison of interface concentrations obtained from modeling with TEM-EDS data plotting on the ternary phase diagram
Table V (column 4 and column 9) also shows that the prediction of interface breakdown is consistent with the SEM results, except for the 0.03Ni alloy which does show a slight perturbation (Figure 6(d) and (e)) despite the model prediction that the interface should remain planar (Figure 10). This result could possibly be a consequence of the choice for the interdiffusion coefficients of Ni.
\( \widetilde{\text{D}}_{\text{NiNi}}^{\text{Fe}} \) was assumed to be the same as the chemical and impurity diffusion coefficients of Ni in the Fe-rich phase (10−16 to 10−15 m2/s at 1150 °C).[22,41,42] Effects of \( \widetilde{\text{D}}_{\text{NiNi}}^{\text{Fe}} \) for the 0.03Ni alloy on C γ/LNi are given in Figure 13(a). A lower \( \widetilde{\text{D}}_{\text{NiNi}}^{\text{Fe}} \) lead to a higher C γ/LNi because lower diffusivities resulted in slower back diffusion, and consequently, more Ni could stay at the interface to increase C γ/LNi as well as the potential for interface breakdown. As shown in Figure 13(b), a lower \( \widetilde{\text{D}}_{\text{NiNi}}^{\text{Fe}} \) could decrease the perturbation criterion value to an extent that it becomes negative.

Effects of \( \widetilde{\text{D}}_{\text{NiNi}}^{\text{Fe}} \) on the Ni interface concentration and perturbation criterion values for Fe-0.3Cu-0.03Ni
Possible Explanations for the Observed Decrease in Oxidation Rate for Fe–Cu–Ni Alloys
As shown in the TG results section in Figure 5(b), k p of Fe–Cu–Ni alloys decrease by a factor of three compared with Fe and Fe–Cu alloys. Several possible causes are as follows:
-
(a)
It has been noted in the literature[43] that the creation of pores near the interface should result in lower oxidation rate because pores could block the outward diffusion of Fe cation. However, this finding is not the case because large pores were observed for the Fe–Cu alloy but not the Fe–Cu–Ni alloys (Figure 8).
-
(b)
In many cases, separation between oxides and metal was observed in the SEM after sample preparation. Oxide scales formed on Fe in the air between 700 and 1250 °C consisted of wüstite, magnetite, and hematite, with the relative thickness ratio of 95:4:1.[44] If separation occurred between the oxides and the metal during oxidation, then the wüstite thickness ratio would deviate distinctly from 95 pct because of the interruption of the Fe cation diffusion through the wüstite layer. Therefore, the wüstite thickness ratio is an alternative method to check whether oxides separate from the metal during oxidation. Although the wüstite thickness ratio in some area deviated from 95 pct in some samples, the total thickness of the oxide layer in these regions was not far away from the oxides in which the wüstite thickness ratio was 95 pct, which suggests that oxides were separated from the metal only for a short time. Moreover, alloys without separations also had parabolic rates that decreased by a factor of about two. Therefore, some regional separations could not account for such a decrease in oxidation rate.
-
(c)
The breakdown of the liquid-Cu layer into isolated liquid pockets because of interface waviness could block Fe cation diffusion where no liquid-Cu coverage occurred, as stated by Webler et al.[8] However, this finding does not seem likely because most interfaces were covered by liquid-Cu with different thicknesses as shown in Figures 6(b) and 7. Moreover, the reduction in the oxidation rate of the 0.03Ni alloy was close to that of the others (Figure 5(b)), although the interface perturbation was minimal in this alloy, and the interface clearly was covered by a liquid-Cu layer (Figure 6(e) or Figure 9(a)). Even if some regions do not have liquid-Cu coverage, Fe cation diffusion was not likely blocked unless zero Fe liquid-Cu/oxide interface concentration was observed. TEM-EDS investigation on regions without clear liquid-Cu coverage did not show high enrichment of Cu or Ni, which shows that Fe diffusion was not stopped in these regions.
-
(d)
The formation of Ni oxides. It is known that the Ni oxide could suppress wüstite formation and decrease parabolic rates.[45] However, no Ni oxides were found.
-
(e)
A remaining explanation could be that Ni changes the transportation behavior of Fe cation through the wüstite layer by changing the Fe activity in the liquid-Cu phase (most γFe interfaces were covered by liquid-Cu especially for the 0.03Ni alloy, as shown in Figure 7(b)) such that a decrease occurred in the concentration difference of Fe vacancies across the wüstite layer, which implies a lowered growth rate of the wüstite layer. Although parabolic rates of Fe–Cu–Ni alloys decreased by a factor of three, most oxides had a wüstite thickness ratio close to 95 pct, as shown in Figure 8, and at a first consideration, this result would seem inconsistent with a change in the wüstite growth rate. Yurek, et al. have developed a theory that quantitatively describes the relative thickness of the growth of double-layer oxides on pure metals.[46] Garnaud, et al. applied this theory to the formation of Fe oxides at 1100 °C, and it agreed with the experimental oxides thickness ratio.[47] A calculation based on Yurek’s expression using molar volume, diffusivity, and oxide composition of wüstite and magnetite at 1100 °C showed that if the exclusive parabolic rate constant of wüstite k w p deceased by a factor of two or three, it would only result in a small deviation from the ideal oxide ratio of 95:4:1. This result is because the diffusivity of Fe in wüstite is much faster than that in magnetite, and therefore, the oxide thickness ratio is not sensitive to the decrease in k w p . Therefore, it is indeed reasonable for Fe–Cu–Ni alloys to have an oxide thickness ratio close to the ideal ratio, whereas the total parabolic rate constant kp (includes all oxide layers and is proportional to k w p ) decreases by a factor of three.
At the wüstite/liquid-Cu interface, wüstite/Fe equilibrium was maintained as follows:
The activity of wüstite was assumed to be unity, and the partial pressure of oxygen \( p_{{{\rm O}_{2} }}^{\prime} \) (1 atm O2 at 1150 °C as the standard state) at the γFe/wüstite interface was inversely proportional to the square of the Fe activity (with pure Fe as the standard state) in the liquid-Cu phase. The decrease in Fe activity, therefore, would increase \( p_{{{\rm O}_{2} }}^{\prime } \). According to Myers and Eugster,[48] the equilibrium \( p_{{{\rm O}_{2} }}^{\prime } \) at the γFe/wüstite interface at 1150 °C is 4.12 × 10−13 atm when the Fe activity equals unity.
At the other end of the wüstite layer, equilibrium was maintained at the wüstite/magnetite interface. If the activities of wüstite and magnetite were assumed to be unity, then \( p_{{{\text{O}}_{ 2} }}^{\prime \prime } \) at the wüstite/magnetite interface would have been kept constant and equaled 1.34 × 10−10 atm at 1150 °C.[48]
Wüstite is known to have a metal deficient defect structure, and the Fe vacancy concentration [\( V^{\prime\prime}_{{{\text{Fe}}}} \)] would be affected by the partial pressure of oxygen. The experimental dependence of [\( V^{\prime\prime}_{{{\text{Fe}}}} \)] on \( P_{{{\text{O}}_{ 2} }} \) at 1150 °C for FeOx was converted from Swaroop and Wagner[49] by using the following equilibrium constant for \( {\text{CO}} + {\frac{1}{2}}{\text{O}}_{2} = {\text{CO}}_{2} \) from Gaskell[50]:
The k p is proportional to the Fe vacancy concentration difference across the wüstite layer \(([V_{\text{Fe}}^{\prime\prime}]^{\prime\prime}-[V_{\text{Fe}}^{\prime\prime}]^\prime)\), assuming a constant diffusivity of the Fe vacancy \( D_{{V_{\text{Fe}}^{\prime \prime } }}^{w} \) in the wüstite layer.[43] Therefore, k p could be calculated as a ratio of k Fe through Eqs. [9] and [10] for different a Fe.
To evaluate the change of Fe activity in the liquid phase, the activity coefficient of Fe in the Cu-rich liquid was needed. Choudary[51] gave quadratic equations for the activity coefficients of Fe and Cu at 1600 °C for the Fe–Cu system, which matched the quadratic equations given by Timberg,[52] and its extrapolated data to 1550 °C agreed with the results of Morris.[53] Therefore, the quadratic equation for the Fe activity coefficient at 1150 °C was obtained by extrapolating Choudary’s results to 1150 °C, following Choudary’s temperature dependent relation.[51] The resulting activity coefficient of Fe γ Fe in the liquid-Cu at 1150 °C (with face-centered-cubic Fe at 1150 °C as the standard state) then was given in Eq. [11] and plotted in Figure 14. XCu is the molar fraction of Cu in the liquid-Cu phase. The activity coefficient calculated by Thermo-Calc® (Stockholm, Sweden) was not adopted because the calculated values at 1600 °C did not reasonably agree with the experimental data from Choudary[51] in the Cu-rich region as illustrated in Figure 14.

Activity coefficient of Fe at 1600 °C from Choudary,[51] calculated by Thermo-Calc (Stockholm, Sweden), and the extrapolated activity coefficient of Fe at 1150 °C
By using the average calculated Fe and Cu liquid-Cu/γFe interface concentrations in the liquid-Cu phase (the concentration was assumed to be uniform in the liquid phase because of the fast diffusivity ~10−9 m2/s[20]), estimates of k p is given in Table VI.
As shown in columns 6 and 7 in Table V, the calculated k p has the same trend as the experimental k p , whereby Fe and Fe–Cu alloys have higher k p values than all Fe–Cu–Ni alloys, and k p for Fe–Cu–Ni alloys are similar. Because Fe concentrations in the both Fe–Cu and Fe-Cu-Ni liquid phases are similar, the difference in activity could be achieved by changing the activity coefficients. The presence of Ni in the liquid-Cu phase was predicted to cause a decrease of Cu in the liquid phase. Although Cu concentrations were found to decrease only a small amount, it could cause a sharp change in the activity coefficient of Fe in the Cu-rich regime, as shown in Figure 14. Thus, a big change in Fe activity could have occurred, which was consistent with the trend of the 0.03Ni alloy activity being smaller than in the Fe and Fe–Cu alloys. If the Cu concentration in the liquid phase decreased to a certain amount, then γFe would not be so sensitive to XCu. Therefore, the activity of Fe would not change so much, which explains the similar activity among Fe–Cu–Ni alloys. It should be noted, however, that although the calculated k p show a trend that agrees with the experimental trend, a significant difference exist between the actual predicted values and the measured values. This discrepancy might be because of the sensitivity to the activity coefficient curve in the Cu-rich region that was based on the extrapolation of experimental measurements. Future work is needed to confirm this proposed mechanism.
In Figure 4, it is shown that the transition points from the linear rate to the parabolic rate occur earlier for the Fe–Cu–Ni alloys compared with the Fe and Fe–Cu alloys. This finding is reasonable becuase linear oxidation will change to parabolic oxidation when diffusion through the wüstite layer proceeds at a rate lower than that of the gas-phase transfer of oxygen. Therefore, Fe–Cu–Ni alloys with lower Fe cation transport in the wüstite layer would have earlier transition points.
Summary of the Likely Effects Caused by Ni Relevant to Hot Shortness
The presence of Ni would result in (1) the enrichment of Ni in the γFe phase and (2) a decrease of the Cu content in the liquid-Cu phase.
The first effect would cause the liquid-Cu/γFe interface concentration to increase, and eventually, the interface would break down. Higher Ni contents have a higher potential for interface breakdown, and it is expected that a minimum Ni content for interface breakdown is required, which is estimated to be around 0.03 wt pct. This result is consistent with findings from Fukagawa that 0.02 wt% pct Ni caused a wavy interface, whereas 0.001 wt% pct Ni did not.[10] However, the presence of Ni alone does not promote appreciable occlusion for such short oxidation times (occlusion could be significant for a long oxidation time[54]), which could be caused by an insufficient amount of internal oxides needed to bridge the enriched regions between protrusions to form occlusions.
The second effect would decrease the oxidation rate because of the decrease in Fe activity in the liquid-Cu phase, which would lower the difference of defects across the wüstite scale. The oxidation rates are similar among all the Fe–Cu–Ni alloys, and therefore, the presence of a little Ni (0.03 wt pct) is sufficient to decrease the oxidation rate by a factor of three, which is effective in suppressing hot shortness because the decrease in oxidation rate would decrease the amount of liquid-Cu phase. This fact is consistent with the finding that a low amount of Ni is sufficient to decrease the sensitivity to hot shortness.[10]
Conclusions
The effects of Ni on interface morphology and oxidation rate were investigated for Fe–Cu–Ni alloys, and the following conclusions were reached:
-
1.
According to modeling results, liquid-Cu/γFe interface Ni concentrations first increase to a peak value and then decrease slowly for all the Fe–Cu–Ni alloys during the course of oxidation.
-
2.
TEM-EDS results show that higher bulk Ni contents have higher liquid-Cu/γFe interface Ni concentrations, and this trend also was confirmed by the modeling results. According to constitutional super-saturation criterion, higher bulk Ni contents have more potential for interface breakdown.
-
3.
Predictions of interface morphology for Fe–Cu–Ni alloys showed that the interface breaks down within the linear oxidation regime except for the 0.03Ni alloy. These results are consistent with SEM results except for the 0.03Ni alloy, and the discrepancy could result from the uncertainty of \( \widetilde{\text{D}}_{\text{NiNi}}^{\text{Fe}} \).
-
4.
The decrease in the parabolic oxidation rate is observed when Ni is added, which can be explained qualitatively by the decrease in Fe activity in the liquid-Cu phase because of the enrichment of Ni at the liquid-Cu/γFe interface. Quantitatively this explanation needs further work before it can be confirmed.
References
U.S. Geological Survey: Mineral Commodity Studies 2008, p. 86, Department of Interior, Washington, DC, 2008
Energetics Inc.: Energy and Environmental Profile of the U.S. Iron and Steel Industry, p. 10–26, U.S. Department of Energy, Office of Industrial Technologies, Washington, DC, 2000.
A. Nicholson and J.D. Murray: J. Iron Steel Inst., 1965, vol. 203, pp. 1007-18.
D.A. Melford: Philos. Trans. R. Soc. Lond., 1980, vol. 295, no.1413, pp. 89-103.
J.K.S. Tee and D.J. Fray: Ironmaking Steelmaking, 2006, vol. 33, no. 1, pp. 19-23.
D.A. Melford: J. Iron Steel Inst., 1962, vol. 200, pp. 290-99.
W.J.M. Salter: J. Iron Steel Inst., 1966, vol. 204, pp. 478-88.
B.A. Webler, L. Yin, and S. Seetharaman: Metall. Mater. Trans. B, 2008, vol. 39, no. 5, pp. 725-37.
G.L. Fisher: J. Iron Steel Inst., 1969, vol. 207, pp. 1010-16.
T. Fukagawa and H. Fujikawa: Oxid. Met., 1999, vol. 52, nos. 3–4, pp. 177-94.
R.Y. Chen and W.Y.D. Yuen: ISIJ Int., 2005, vol. 45, no. 6, pp. 807-16.
S. Akamatsu, T. Senuma, Y. Takada, and M. Hasebe: Mater. Sci. Technol., 1999, vol. 15, no. 11, pp. 1301-07.
D.E. Coates and J.S. Kirkaldy: J. Cryst. Growth, 1968, vol. 3, no. 4, pp. 549-54.
Materials Preparation Center: Ames Laboratory, US DOE Basic Energy Sciences, www.mpc.ameslab.gov.
K. Sachs and C.W. Tuck: Proc. Conf. Reheating for Hot Working, Iron and Steel Institute, 1968, pp. 1–17.
H. Abuluwefa, R.I.L. Guthrie, and F. Ajersch: Oxid. Met., 1996, vol. 46, no. 5–6, pp. 423-40.
W.S. Rasband: ImageJ, U.S. National Institutes of Health, http://rsb.info.nih.gov/ij/, 1997.
Thermo-Calc Software AB: Version Q on WinNT and the TCFe3 Database, Stockholm, Sweden, 2004.
S. Pötschke and A.R. Buchner: Steel Res. Int., 2006, vol. 77, no. 6, pp. 416-22.
M.M.G. Alemany, L.J. Gallego, L.E. González, and D.J. González: J. Chem. Phys., 2000, vol. 113, no. 22, p. 10410-11.
W.K. Chen and N.L. Peterson: J. Phys. Chem. Solids, 1975, vol. 36, no. 10, pp. 1097-103.
C.J. Smithells: Smithells Metals Reference Book, 8th ed., W.F. Gale and T.C. Totemeier, eds., Elsevier, New York, 2004, pp. 13-12–13-22.
Y. Kondo: ISIJ Int., 2004, vol. 44, no. 9, pp. 1576-80.
B.A. Webler and S. Sridhar: ISIJ Int., 2008, vol. 48, no. 10, pp. 1345-53.
COMSOL AB: FEMLAB Version 3.2, Reference manual, 2005.
MATLAB: MATLAB 7.0.4 Release 14. The Mathworks Inc., Natick, MA, 2005.
J.B. Clark and F.N. Rhines: Trans. ASM, 1959, vol. 51, pp. 199-221.
J.S. Kirkaldy and L.C. Brown: Can. Metall. Q., 1963, vol. 2, no. 1, pp. 89-115.
L.E. Wirtz and M.A. Dayananda: Metall. Trans. A, 1977, vol. 8A, pp. 567-75.
C. Wagner: J. Electrochem. Soc., 1956, vol. 103, no. 10, pp. 571-80.
D.P. Whittle, D.J. Young, and W.W. Smeltzer: J. Electrochem. Soc., 1976, vol. 123, no. 7, pp. 1073-79.
A.D. Dalvi and D.E. Coates: Oxid. Met., 1972, vol. 5, no. 2. pp. 113-35.
W.W. Mullins and R.F. Sekerka: J. Appl. Phys., 1964, vol. 35, no. 2, pp. 444-51.
R.F. Sekerka: J. Appl. Phys., 1965, vol. 36, no. 1, pp. 264-67.
J.S. Kirkaldy and D.G. Fedak: Trans. Metall. Soc. AIME, 1962, vol. 224, no. 3, pp. 490-94.
D.E. Coates and J.S. Kirkaldy: Trans. ASM Q., 1969, vol. 62, no. 2, pp. 426-36.
J. Brillo and I. Egry: J. Mater. Sci., 2005, vol. 40, nos. 9–10, p. 2213-16.
J. Brillo, I. Egry, and T. Matsushita: Int. J. Thermophys., 2006, vol. 27, no. 6, pp. 1778-91.
R.Y. Chen and W.Y.D. Yuen: Oxid. Met., 2003, vol. 59, nos. 5–6, pp. 433-68.
L. Himmel, R.F. Mehl, and C.E. Birchenall: J. Met., 1953, vol. 5, no. 6, pp. 827-43.
H. Bakker, H.P. Bonzel, C.M. Bruff, M.A. Dayananda, W. Gust, J. Horvath, I. Kaur, G.V. Kidson, A.D. LeClaire, H. Mehrer, G.E. Murch, G. Neumann, N. Stolica, and N.A. Stolwijk: Diffusion in Solid Metals and Alloys, Springer-Verlag, Berlin, Germany, 1990.
Y. Hanatate, K. Majima, and H. Mitani, Trans. Jpn. Inst. Met., 1978, vol. 19, pp. 669-73.
N. Birks, G.H. Meier, and F.S. Pettit: High-Temperature Oxidation of Metals, 2nd ed., pp. 49–62, 75–78, Cambridge University Press, Cambridge, UK, 2006.
J. Païassi: Acta Metall., 1958, vol. 6, no. 3, pp. 184-94.
R.T. Foley: J. Electrochem. Soc., 1962, vol. 109, no. 12, pp. 1202-06.
G.J. Yurek, J.P. Hirth, and R.A. Rapp: Oxid. Met., 1974, vol. 8, no. 5, pp. 265-81.
G. Garnaud and R.A. Rapp: Oxid. Met., 1977, vol. 11, no. 4, pp. 193-98.
J. Myers and H.P. Eugster: Contrib. Mineral. Petrol., 1983, vol. 82, no. 1, pp. 75-90.
B. Swaroop and J.B. Wagner: Trans. Metall. Soc. AIME, 1967, vol. 239, no. 8, pp. 1215-18.
D.R. Gaskell: Introduction to the Thermodynamics of Materials, 4th ed. p. 582, Taloy & Francis, Oxford, UK, 2003.
U. Choudary, J. Serkin, and G. Belton: Metall. Trans. B, 1975, vol. 6, no. 3, pp. 399-403.
L. Timberg, J.M. Toguri, and T. Azakami: Metall. Trans. B, 1981, vol. 12, no. 2, pp. 275-79.
J.P. Morris and G.R. Zellars: Trans. AIME, 1956, vol. 206, no. 8, pp. 1086-90.
N. Imai, N. Komatsubara, and K. Kunishige: ISIJ Int., 1997, vol. 37, no. 3, pp. 224-31.
Acknowledgments
Financial support from the Center for Iron and Steelmaking Research (CISR) is gratefully acknowledged. Valuable discussions with Professor Brian Gleeson at the University of Pittsburgh are appreciated. The author also is indebted to colleagues Jingxi Zhu and Adam Wise at Carnegie Mellon University for help on FIB and TEM techniques.
Author information
Authors and Affiliations
Corresponding author
Additional information
Manuscript submitted October 3, 2009.
An erratum to this article can be found at http://dx.doi.org/10.1007/s11663-010-9382-4
Rights and permissions
About this article
Cite this article
Yin, L., Balaji, S. & Sridhar, S. Effects of Nickel on the Oxide/Metal Interface Morphology and Oxidation Rate During High-Temperature Oxidation of Fe–Cu–Ni Alloys. Metall Mater Trans B 41, 598–611 (2010). https://doi.org/10.1007/s11663-009-9334-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11663-009-9334-z