
Abstract
The cohesive properties of Ni-Sn intermetallics (stable, metastable, and virtual), hitherto unexplored by density-functional theory (DFT) methods, are reported. Specifically, the total energies and cohesive properties of Ni, Sn, and 27 Ni-Sn intermetallics are calculated from first-principles, using ultrasoft pseudopotentials (USPP) and both local-density approximation (LDA) and generalized-gradient approximation (GGA) for the exchange-correlation functional. Among the intermetallics considered, the ground-state structures are consistent with experimental observations; however, not all of them are registered in the equilibrium-phase diagram. An important result of this systematic study, using both USPP-LDA and USPP-GGA, is that oC20-NiSn4 is predicted to be the ground-state structure. Only recently, this phase has been observed as a product of the interfacial reaction in Ni/Sn diffusion couples. In addition, we find that the thermodynamic stability of a tetragonal phase, tP10-NiSn4, is very similar to that of oC20-NiSn4. The elastic stability of both tP10-NiSn4 and oC20-NiSn4 is confirmed by calculating their single-crystal elastic constants. The calorimetric data for the enthalpy of formation of stable intermetallics show an agreement that is better for those calculated with USPP-LDA than those calculated with USPP-GGA. In general, the experimental lattice parameters of stable and metastable phases are found to lie between those calculated using USPP-LDA and those calculated using USPP-GGA; however, in several cases, the values calculated using USPP-GGA agree to within 1 pct of the experimental data. The Ni3Sn2(ht) ⇌ Ni3Sn2(lt) transformation is discussed in terms of supergroup-subgroup relations. The bonding between the Ni and the Sn is discussed based on the analyses of the density of states (DOS) and the charge densities.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The factors controlling the reliability of modern solder interconnects in electronic packages constitute a very active area of current research. This is primarily driven by the increasingly dense arrays of interconnects, the miniaturization of electronic packages, the requirement of new packaging technology, and the use of lead-free solders. The critical issues in microelectronic solder interconnects that affect reliability are the dynamics of the wetting behavior, the interfacial reaction with the underlying base metal governing the microstructure evolution during processing and in the service of devices, and electromigration.
Due to the decreasing size of modern solder joints, the intermetallic layer continues to be proportionately thicker. In addition to the intrinsic properties of solder, the strength and reliability of solder joints are determined by the interfacial intermetallic(s), the interfacial properties (between solder and intermetallic), and the microstructural coarsening of solder. The phases that form in electronic and optoelectronic interconnects during fabrication and under normal service conditions may affect the reliability of the devices through the following intrinsic properties: the coefficient of thermal expansion, the ductile/brittle transition (due to a change in composition and temperature), the elastic constants, the inelastic behavior, the heat capacity, and the thermal conductivity. While the formation of a thin intermetallic layer is desirable for achieving a good metallurgical bond, excessive intermetallic growth may have a deleterious effect on the mechanical reliability of the solder joints. The importance of some of these issues has been discussed in several recent reviews.[1–5] The intermetallics in solder interconnects may become the “weakest link,” if they are inherently brittle.
Under-bump metallurgy (UBM) ranging from pure Ni to Ni-Cu, Ni-Pd, Ni-P, Ni-Ti, and Ni-V alloys is either used or is currently being developed for use in microelectronics packaging.[6–12] Even though Cu is the most commonly used solderable surface finish in a printed circuit board, Ni or a suitable Ni-base alloy is considered a better candidate, because the kinetics of the interfacial reaction between Ni and solder is much slower than that between Cu and solder. When used as UBM, the Ni layer should be thick enough to prevent its complete consumption to form Ni-Sn and other Ni-containing intermetallic(s) during reflow and during the lifetime of a device that may experience a range of temperatures. The evolving microelectronic soldering technology requires a quantitative understanding of processing-microstructure-property-performance links; hence, it has become necessary to generate new knowledge about thermodynamics, kinetics, and the thermophysical properties of relevant phases.
In addition to their importance in soldering technology for electronic packaging, Ni-Sn intermetallics are attractive alternatives to graphite as an anode material in rechargeable Li-ion batteries.[13–17] Some of the key properties of interest include their charge-storage capacity, cyclic charge-discharge performance, and electronic conductivity. In a recent development, a nanostructured Ni3Sn4 electrode has achieved a capacity of 500 mA h g−1, with no decay over 200 cycles.[17]
The results we present here are only a part of our larger study on the phase stability and elastic and inelastic properties of intermetallics relevant to microelectronic packaging. Specific to this article, as a part of our effort to create a reliable multicomponent thermodynamic database to facilitate the calculation of stable and metastable phase diagrams and a diffusion-path analysis of modern solder interconnects, we present the results of a comprehensive study of the energetics and equilibrium cohesive properties of Ni-Sn intermetallics using ab-initio computational techniques. Understanding the thermodynamic stability of Ni-Sn intermetallics is very important in understanding phase stability and phase equilibria in ternary and multicomponent systems containing Ni and Sn.
The plan for the remainder of the article is as follows. In Section II, we briefly review experimental data for Ni-Sn intermetallics. In Section III, we present the computational methodology. In Section IV, we present the calculated equilibrium structural and cohesive properties of the intermetallic compounds; these include the formation energies, bulk moduli, lattice parameters, and degrees of freedom (when applicable) for atomic positions. The calculated cohesive properties of selected intermetallics are compared with the corresponding experimental data. In Section V, we discuss the results of ab-initio total energy and electronic structure calculations. The conclusions are summarized in Section VI.
2 Literature Review
2.1 Crystal Chemistry
Earlier experimental information on Ni-Sn phases has been assessed from time to time.[18–21] More recently, several reports on the crystal chemistry of intermetallics have appeared in the literature.[22–31] The crystallographic data for Ni-Sn intermetallics considered in this study are summarized in Table I.
The intermetallics in Table I are conveniently classified as stable, metastable, and virtual. We consider the stable phases as those present in the assessed equilibrium-phase diagram,[21] or the temperature or compositional ranges of stability.
Three intermetallics, Ni3Sn, Ni3Sn2, and Ni3Sn4, have been accepted in the assessed Ni-Sn phase diagram.[21] However, earlier studies of bulk alloys reported other phases such as Ni4Sn and NiSn that were not confirmed in subsequent investigations.
In the equilibrium-phase diagram, there are two forms of Ni3Sn: the high-temperature form (DO 3-BiF3 type), Ni3Sn(ht), stable between 1123 and 1447 K,[21] and the low-temperature form (DO 19-type), Ni3Sn(lt), stable below 1250 K.[21,32] It has been argued that the transformation of Ni3Sn(ht) to Ni3Sn(lt) is an order-disorder type. However, two other structures of Ni3Sn have also been reported: a cubic (L12-AuCu3 type) at high pressure[33] and an orthorhombic (DO a -Cu3Ti type).[34–36] Pak et al.[36] suggested that DO a -Ni3Sn is the result of a martensitic transformation of Ni3Sn(ht) upon rapid quenching.
In the equilibrium-phase diagram, Ni3Sn2 is shown is to have two forms: the high-temperature form (InNi2/NiAs, B82/B81-type Ni3Sn2(ht), stable between 873 and 1540 K),[21,37] and the low-temperature form, Ni3Sn2(lt), stable below 873 K.[21,37] However, in recent years, the solid-state phase equilibria around 40 at. pct Sn have been reinvestigated in great detail. References 24 through 27 and 29 through 31 identify three different forms of Ni3Sn2, one commensurate and two incommensurate phases. The commensurate phase, stable between 39.3 and 40.8 at. pct Sn, has an orthorhombic structure (Pearson symbol: oP20; space group: Pnma), and it is a lock-in phase between the two incommensurate phases, Ni3Sn2-LT′ and Ni3Sn2-LT′′. The former has an orthorhombic symmetry and is stable between 40.8 and 42.5 at. pct Sn; the latter is stable at a Sn content less than 39.2 at. pct Sn, but the crystallographic details are not known. These incommensurate phases are not considered in our first-principles calculations.
Even though the existence of equilibrium-phase Ni3Sn4 has been confirmed many times,[21,38] the reported structural details differ. For example, Furuseth and Fjellvag[39] confirmed the monoclinic symmetry of Ni3Sn4, but claimed that it is isotypic with CoGe; thus, the ideal stoichiometry should correspond to NiSn rather than to Ni3Sn4.
Metastable phases are usually transient in nature; during annealing/heat treatment they eventually undergo transformation to stable phases, as governed by the equilibrium-phase diagram. The metastable phases may form during nonequilibrium processing such as mechanical alloying, rapid solidification, or vapor deposition; they also sometimes form during solid-solid phase transformations, solid-solid (liquid) interfacial reactions in diffusion couples, thin-film reactions, and the crystallization of amorphous phases. Metastable phases may also be stabilized by extraneous effects such as the impurity elements in an otherwise pure alloy. Sometimes, the intermetallics that are thermodynamically metastable in a binary system may become stable in ternary and higher-order systems. This may have a significant influence on the phase diagrams, diffusion paths, and interfacial microstructure in diffusion couples of multicomponent systems.
The formation of metastable crystalline phases has been reported in at least two distinct experiments: (1) during the interfacial reaction in Ni/Sn diffusion couples[22,40] and (2) in electrodeposited thin films.[41] Watanabe et al.[41] observed two metastable phases, M1 and M2, in electrodeposited Ni-Sn thin films: single-phase M1 in the range of 29 to 56 at. pct Sn and M2 forms in the composition range of 75.5 to 90 at. pct Sn. The latter was reported to have a tetragonal structure with a = 0.623 nm and c = 0.577 nm. Haimovich[40] reported the formation of NiSn3 during the solid-state aging of Ni/Sn diffusion couples at 160 °C or below, but its structure was not determined. Very recently, Boettinger et al.[22] reported the formation of NiSn4 in solder joints involving electroplated layers of Ni and Sn that were subjected to thermal cycling between −40 °C and +130 °C; the NiSn4 is reported to have orthorhombic symmetry (isotypic with AuSn4), as determined by the electron backscattered diffraction method in a scanning electron microscope.
Based on the extrapolated liquidus and solidus boundaries of the equilibrium-phase diagram, Liu et al.[42] ruled out the possibility of amorphous-phase formation in Ni-Sn system. However, amorphous phases have been prepared by mechanical alloying[43] and vapor quenching methods.[44,45] Tianen and Schwarz[43] prepared single-phase amorphous alloy at 25 at. pct Sn, through the mechanical alloying of Ni and Sn powders at 240 K. Geny et al.[44] prepared amorphous Ni1−x Sn x 0.57 ≤ x ≤ 0.73 thin films through the codeposition of elemental vapors on substrates at 77 K. Amorphous phases at Ni3Sn0:89 and Ni3Sn1:94 compositions have also been prepared through the electron-beam evaporation of pure elements on silicon substrate with a spincoated film of polymethylmethacralate.[45] Of course, the noncrystalline structures are not considered in our first-principles calculations.
The concept of a virtual phase is a mathematical one in the context of the calculation of phase diagrams (CALPHAD) modeling of intermetallics with a finite homogeneity range, using a sublattice model.[46] For example, Ni3Sn2 (oP20) and Ni3Sn4 (mC14) are stable phases, while Ni2Sn3 (oP20) and Ni4Sn3 (mC14) are virtual phases in the Ni-Sn system. The energetics of virtual phases is important in understanding the site occupancy of third elements and defect energetics in binary intermetallics. We have considered several virtual phases that are ordered structures based on simple lattices, such as (1) bcc-based ordered structures (B2 and DO 3), (2) fcc-based ordered structures (L12, DO 22, DO 23, and L10), and (3) hcp-based ordered structures (B19, DO 19, and DO a ). A tetragonal phase tP10-NiSn4, which is prototypical of the tP10-AuSn4 observed in the Au/Sn thin-film reaction,[47] is also considered as virtual.
2.2 Calorimetry Data
The heats of formation (Δ f H) of Ni-Sn intermetallics have been measured by both solution calorimetry[48–52] and direct-reaction calorimetry.[53,54] Hultgren et al.[55] assessed the data of Korber and Oelsen,[53] who carried out the direct-reaction calorimetry by pouring liquid Ni and liquid Sn near 293 K. Hultgren et al. noted that both the attainment of equilibrium and the final state of the alloys were very uncertain; therefore, their assessed data for Ni3Sn(lt) and Ni3Sn2(lt) should be regarded as tentative. Subsequent investigations carried out solution calorimetry using liquid Sn as a solvent, to determine the heats of formation of the following intermetallics: Ni3Sn(ht),[50] Ni3Sn2(ht),[48–50] and Ni3Sn4.[49–51] Very recently, two sets of calorimetric data have been reported. Vassilev et al.[54] carried out direct-reaction calorimetry to determine the enthalpies of the following intermetallics: Ni3Sn(ht), Ni3Sn(lt), Ni3Sn2(ht), Ni3Sn2(lt), and Ni3Sn4. On the other hand, Flandorfer et al.[52] carried out solution calorimetry using liquid Sn in a Calvet-type microcalorimeter and reported the standard enthalpy of formation of Ni3Sn(lt), Ni3Sn2(ht), and Ni3Sn4.
2.3 CALPHAD Modeling of Phase Stability and Phase Diagram
The CALPHAD modeling of the phase stability[56,57] and phase equilibria of the Ni-Sn system has been reported by four different groups.[42,58–60] Nash et al.[58] treated all intermetallics as line compounds and did not make a distinction between high- and low-temperature forms of Ni3Sn2. The first comprehensive modeling of phase stability and phase equilibria was carried out by Ghosh.[59] Subsequently, Miettinnen[60] presented another thermodynamic assessment of the Ni-Sn system, using simplified models of Ni3Sn and Ni3Sn2 phases. For example, Miettinnen treated cF16-Ni3Sn(ht) as a solid-solution phase and did not distinguish between the high- and low-temperature forms of Ni3Sn2. Later, Liu et al.[42] reassessed the phase stability and phase equilibria of the Ni-Sn system.
3 Computational Methodology
3.1 Ab-initio Total Energy Calculations
The ab-initio calculations presented here are based on the electronic density-functional theory (DFT) and have been carried out using the ab-initio total-energy program known as the Vienna Ab-initio Simulation Package (VASP).[61–63] The current calculations make use of fully nonlocal Vanderbilt-type ultrasoft pseudopotentials (USPP),[64] as implemented in VASP,[65] to describe the electron-ion interaction and an expansion of the electronic-wave functions in plane waves with a kinetic-energy cutoff of 314 eV, which is at least 30 pct more than the recommended default value. The USPPs used in this work explicitly treat ten valence electrons for Ni (3d 10) and 14 valence electrons for Sn (4d 105s 2 p 2). All calculated results were derived using spin polarized Hamiltonian, using both local-density approximation (LDA), and generalized-gradient approximation (GGA) for the exchange-correlation functional. As implemented in VASP, the LDA calculations use the Ceperly–Alder[66] form of exchange-correlation (xc) energy, as parametrized by Perdew and Zunger.[67] For the GGA calculations, the exchange-correlation functional used is due to Perdew and Wang,[68] with the Vosko–Wilk–Nussair[69] interpolation of the correlation energy.
Brillouin zone integrations were performed using Monkhorst–Pack[70] k -point meshes, and the Methfessel–Paxton[71] technique, with the smearing parameter of 0.1 eV. Tests were carried out using different k-point meshes, to ensure the absolute convergence of the total energy to a precision of greater than 1.0 meV/atom (≈0.1 kJ/mol-atom). For example, the k-point meshes for Ni3Sn (cF16), Ni3Sn (hP8), Ni3Sn2(oP20), and Ni3Sn4 (mC14) were 10 × 10 × 10, 8 × 8 × 10, 8 × 11 × 7, and 3 × 8 × 6, respectively. Depending on the structure of the phases listed in Table I, up to 324 k points were used in the irreducible Brillouin zone. All calculations are performed using the “high” setting of precision within the VASP code. The total energy was converged numerically to less than 5 × 10−6 eV/atom, with respect to the electronic, ionic, and unit-cell degrees of freedom; the latter two were relaxed, using the Hellman–Feynman forces with a preconditioned conjugate gradient algorithm. After structural optimization, the Hellman–Feynman forces (when permitted by the space group symmetry) were less than 4 meV/A in magnitude. With the chosen plane-wave cutoff and k-point sampling, the reported formation energies are estimated to be converged to a precision of greater than 2 meV/atom (≈0.2 kJ/mol-atom).
The cohesive properties of pure elements and selected intermetallics are also calculated using pseudopotentials constructed by the projector-augmented wave (PAW) method,[72] as implemented in VASP.[73] In the PAW method, which retains the all-electron character, a plane-wave basis set is used to express the auxiliary description of the electron density, which is transformed into full density by projectors that add the effect of the local atomic density. In practice, this loosely corresponds to a frozen-core approximation, with a plane-wave expansion of the valence electrons. The frozen-core approximation is not strictly necessary; in fact, very recently, a relaxed-core PAW method has been proposed[74] that yields results with an accuracy comparable to the FLAPW (full-potential linearized augmented plane-wave) method.[75] The PAW method includes a nonlinear core correction, accounts for core-core overlap, and is free of any shape approximation for both the charge density and the electronic potential. Therefore, PAW pseudopotentials are an improvement over Vanderbilt-type USPP. The valence configurations of PAW pseudopotentials and the exchange-correlation functional were the same as in USPP; however, a kinetic-energy cutoff of 350 eV was used for the expansion of the electronic-wave functions in plane waves.
3.2 Equation of State and Formation Energy
As described in a previous publication,[76] we take the formation energy (Δ f E) at zero temperature and zero pressure, and without the zero-point motion of an intermetallic Ni p Sn q , in which p and q are integers, as a key measure of the relative stability of competing structures (ϕ 1, ϕ 2, ϕ 3...). The formation energy per atom of Ni-Sn intermetallics is evaluated relative to the composition-averaged energies of fcc Ni and A5-Sn (or β-Sn).
For most of the intermetallics, we calculated the total energy (E) as a function of the volume (V). Then we used the zero-temperature equation of state (EOS) due to Vinet et al.[77] to define the equilibrium volume (V o ) and the corresponding total energy (E o ), and the bulk modulus (B o ) and its derivative (B o ′), with respect to the hydrostatic pressure (P).
3.3 Single-Crystal Elastic Constants of Pure Elements
To check the accuracy of the computational procedure, we have calculated the single-crystal elastic constants of the pure elements for which reliable and low-temperature experimental data are available. In particular, we are interested in comparing the calculated values with the experimental data, to assess the role of the exchange-correlation functional and the type of pseudopotential. Generally, the single-crystal elastic constants can be obtained by ab-initio electronic-structure methods, by calculating the total energy as a function of the appropriate lattice deformations. Depending on the crystal system and the type of imposed lattice deformation, the curvature of the total energy vs strain curves defines either a particular elastic constant or a combination of elastic constants.
For a phase with cubic lattice symmetry, there are three single-crystal elastic constants, C 11, C 12, and C 44. For a tetragonal phase, there are six single-crystal elastic constants, C 11, C 12, C 13, C 33, C 44, and C 66. Mehl et al.[78] summarized the lattice deformations needed to derive the C ij in both cubic and tetragonal crystal systems. Accordingly, we have calculated total energies by imposing appropriate strains up to ±3 pct at a 0.5 pct interval. The total energies vs strain were fit with the functional forms provided by Mehl et al.,[78] to extract the elastic constants.
4 Results
4.1 Lattice Stability and Cohesive Properties of Ni and Sn
The lattice stability of pure elements is very important in the CALPHAD method of modeling phase diagrams,[56,57] which begins with the Gibbs energy description of pure elements with different structures. We have calculated the total energies of Ni and Sn as a function of volume for the following structures: fcc (A1 or cF4), bcc (A2 or cI2), hcp (A3 or hP2), diamond cubic (A4 or cF8), and tetragonal (A5 or tI4 and A6 or tI2). The resulting zero-temperature cohesive properties of Ni are compared with the available experimental data in Table II. A detailed discussion of the lattice stability and cohesive properties of Sn may be found in an earlier publication.[79]
The lattice parameter of fcc Ni at 0 K is obtained by the extrapolation of the experimental data[80,81] at higher temperatures. The experimental bulk moduli of fcc Ni[82] are based on the extrapolated values of C 11 and C 12 to 0 K, with B o = (C 11 + 2C 12)/3. The extrapolated lattice parameter of fcc Ni and its B o agree very well with those calculated using USPP-GGA; they differ by only 0.4 and 5 pct, respectively. The calculated magnetic moment of fcc Ni also shows good agreement with the experimental values at 0 K.[83,84] On the other hand, the USPP-LDA values manifest the effect of overbinding, causing a smaller lattice parameter and a higher bulk modulus by approximately 2.3 and 25 pct, respectively.
The experimental B o values of fcc Ni, listed in Table II, are at 298 K.[85] It has been pointed out[85] that, depending on the measurement technique (ultrasonic resonance) vs the initial slope of the locus of the Hugoniot states in shock-velocity particle-velocity coordinates, the values of B o may differ, even though, ideally, they should be the same.
An important issue in the theoretical study of phase stability is the elastic stability of crystals, which can be assessed from knowledge of the single-crystal elastic constants. The calculated single-crystal elastic constants are compared with the experimental data in Table III. The requirements of elastic stability in a cubic crystal are[86] C 11 > 0, C 12 > 0, (C 11 – C 12) > 0, and C 44 > 0. The calculated values of C ij satisfy these criteria, for both fcc Ni and A4-Sn.
In addition, as shown in Table III, the calculated values of C ij of fcc Ni made using USPP-GGA and PAW-GGA agree within 5 pct of the experimental values,[82] while those made using USPP-LDA are overestimated by as much as 35 pct of the experimental values. Furthermore, our calculated values for the C ij of fcc Ni made using USPP-GGA and PAW-GGA show a very good agreement with those made using FLAPW-GGA.[87]
We have also calculated the values C ij of A5-Sn; the results are listed in Table III. The requirements of elastic stability in a tetragonal crystal are[86] C11 > 0, C12 > 0, C13 > 0, C33 > 0, C 44 > 0, C 66 > 0, (C 11 – C 12) > 0, (C 11 + C 33 – 2C 13) > 0, and (2C 11 + C 33 + 2C 12 – 4C 13) > 0. Our calculated values demonstrate that they satisfy the criteria of the elastic stability of A5-Sn. The calculated values of C ij made using USPP-LDA agree within 6 pct of the experimental values,[88,89] while those made using USPP-GGA and PAW-GGA are underestimated by as much as 55 pct. Adkim et al.[90] reported the calculated values of C ij of both A4- and A5-Sn using the tight-binding method; their results are summarized in Table III. As seen, while their results for A4-Sn agree reasonably well with our calculated values made using USPP-LDA, the agreement for A5-Sn is very poor, with both the experimental data and our calculated values.
The calculated lattice stabilities of Ni at 0 K are compared with those from the Scientific Group Thermodata Europe (SGTE) database,[91] as provided in the Thermo-Calc software package[92] in Table IV. The corresponding properties of Sn may be found in an earlier publication.[79] Our calculated lattice stabilities of Ni involving bcc, fcc, and hcp structures based on USPP-GGA, listed in Table IV, are in very good agreement (within 0.5 kJ/mol-atom) with recently reported values.[93] For simple structures, such as bcc, fcc, and hcp, the quantitative differences between our calculated values and the SGTE database are approximately 1 kJ/mol-atom; thus, the agreement should be considered as good.
Dinsdale[91] (also the SGTE database) and Wang et al.[93] did not report the lattice stabilities of Ni and Sn in A4, A5, and A6 structures. These lattice stabilities are important in the thermodynamic modeling of Sn-based binary and ternary phase diagrams.
4.2 Phase Stability and Cohesive Properties of Ni-Sn Intermetallics
There are many important aspects of an investigation of the phase stability and cohesive properties of solids using DFT methods. These include an understanding of the electronic structure and bonding mechanisms; a search for the ground-state structures; calculation of the thermodynamic properties of stable phases, which may be compared with the experimental data (when available) and with the properties of the metastable and virtual phases that cannot be accessed experimentally; calculation of the electronic, magnetic, structural, physical, and thermophysical properties (when possible); an exploration of the roles of LDA and GGA for the exchange-correlation functional on the calculated properties, etc. In the case of alloy systems hitherto unexplored using electronic DFT methods, it is difficult to predict whether the calculated cohesive properties using LDA or GGA will yield better agreement with the experimental data, and also to what extent the properties calculated using LDA and GGA will differ with each other.
In the preceding Section A, we showed that the cohesive properties of fcc Ni (the lattice parameter, bulk modulus, and C ij ) calculated using USPP-GGA agree very well with the corresponding experimental data, while those of A5-Sn (or β-Sn) calculated using USPP-LDA agree very well with the experimental data (Table III). These findings warrant calculation of the cohesive properties of Ni-Sn intermetallics using both USPP-LDA and USPP-GGA. Consequently, these results are presented here.
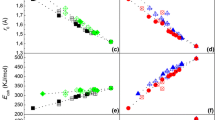
The results of ab-initio calculations for Ni-Sn intermetallics are summarized in Tables V through VIII and are plotted in Figures 1 and 2. The calculated Δ f E using USPP-LDA and USPP-GGA are plotted in Figures 1(a) and 2(a), respectively, as a function of Sn content. In both cases, the ground-state convex hull is asymmetric and skewed toward the Ni side, with a minimum in Δ f E at Ni3Sn2. Among all the phases considered (Table I), our calculations using USPP-LDA yield a ground-state convex hull defined by the following phases: Ni3Sn (hP8), Ni3Sn2 (oP20), NiSn (hP4), Ni3Sn4 (mC14), and NiSn4 (oC20). In other words, these are the structures (of the 27 intermetallics considered) that are predicted to be stable alloy phases at zero temperature. When USPP-GGA is used, the same phases, with the exception of Ni3Sn (cP4) instead of Ni3Sn (hP8), also define the ground-state convex hull. Consistent with the expected behavior, the Δ f E values computed using USPP-LDA are consistently more negative than those obtained using USPP-GGA. Similarly, the equilibrium volumes computed using USPP-LDA are consistently smaller than those computed using USPP-GGA (Figures 1(b) and 2(b)), while the reverse is the case for the computed bulk moduli (Figures 1(c) and 2(c)).

Calculated cohesive properties, using LDA exchange correlation, of Ni-Sn intermetallics: (a) the formation energy (Δ f E), (b) the equilibrium volume (V o ), and (c) the bulk modulus at equilibrium volume (B o ). In (a), the solid line defines the ground state convex hull, and in (b) and (c), the solid line is drawn through the same structures that form the ground state convex hull in (a)

Calculated cohesive properties, using GGA exchange correlation, of Ni-Sn intermetallics: (a) the formation energy (Δ f E), (b) the equilibrium volume (V o ), and (c) the bulk modulus at equilibrium volume (B o ). In (a), the solid line defines the ground state convex hull, and in (b) and (c), the solid line is drawn through the same structures that form the ground state convex hull in (a)
It is important to note that the ground-state structures predicted by our first-principles calculations (both LDA and GGA) have been experimentally observed, but not all of them are registered in the assessed phase diagram.[21] Four different structures of Ni3Sn have been reported. While the high-temperature form is believed to be the DO 3 type,[21] the reported structures of low-temperature Ni3Sn are the DO 19, DO a , and L1 2 types.[21] As presented in Table VI, the structural energy difference between the latter three structures is very small. In particular, the structure energy difference between L1 2-Ni3Sn and DO 19-Ni3Sn is predicted to be less than 0.2 kJ/mol-atom. Experimentally, these three structures have been observed at room temperature, and it is not clear whether impurities have played any role in their stability. The Ni3Sn2(ht) phase, which is prototypical of NiSn (hP4), has been observed at high temperature. While the phases Ni3Sn2 (oP20)[21,37] and Ni3Sn4 (mC14)[21,38] were established long ago, only recently has NiSn4 (oC20) been identified as a product of the interfacial reaction in Ni/Sn diffusion couples;[22] its equilibrium relations with other phases in the Ni-Sn phase diagram, however, are yet to be established.
Both the USPP-LDA and the USPP-GGA calculations show that the formation energy of the metastable phases Ni3Sn (oP8),[34–36] NiSn (oP32),[94] and Ni3Sn4 (mC16)[39] lie approximately 1 kJ/mol-atom above the ground-state convex hull (Figures 1(a) and 2(a) and Table VI). On the other hand, the formation energies of the virtual phases considered in Table I lie several kJ/mol-atom above the ground-state convex hull (Figures 1(a) and 2(a)). The only exception is tP10-NiSn4. Both the USPP-LDA and USPP-GGA Δ f E values show that tP10-NiSn4 is only slightly less stable than oC20-NiSn4; their formation energies differ by less than 1 kJ/mol-atom.
The available calorimetric data for the enthalpies of formation show a better agreement with those calculated using USPP-LDA than with those calculated using USPP-GGA. In particular, the Δ f E calculated using USPP-LDA for Ni3Sn(ht) (cF16) and Ni3Sn(lt) (hP8) agree well with the corresponding calorimetric Δ f H 298.15[53,54] values. The enthalpy of formation of Ni3Sn2(lt) (oP20) reported by Predel and Ruge[47] could not be reproduced in a later investigation.[54] In the direct-reaction calorimetry experiment, Vassilev et al.[54] noted an incomplete reaction; thus, their Δ f H 298:15 value should be considered as approximate. In the case of Ni3Sn4 (mC14), both the solution calorimetry[49,51] and direct-reaction calorimetry[54] values of Δ f H 298.15 show an agreement with the Δ f E calculated that is better when using USPP-LDA than when using USPP-GGA.
All the results discussed here were obtained using the USPPs described in Section III–A. In addition, several calculations at the LDA and GGA levels for a few stable, metastable, and virtual phases were also performed with alternative settings, to gage the overall accuracy of the our results. Specifically, additional calculations were performed using PAW-GGA pseudopotentials in the case of Ni3Sn4 (mC14) and Ni3Sn (cP4); an alternative USPP was used for Sn, in which four electrons, 5s 2 p 2, were explicitly treated as valence (i.e., they did not include semicore 4d states as valence) in the case of Ni3Sn (cP4), NiSn (hP4), and NiSn3 (cP4). The effects of the alternative pseudopotentials on Δ f E are compared in Table VI. It is seen that the calculations for the Δ f E of Ni3Sn4 (mC14) and Ni3Sn (cP4) done using USPP-GGA and PAW-GGA agree within approximately 1 kJ/mol-atom. Similarly, the calculations for the Δ f E of Ni3Sn (cP4), NiSn (hP4), and NiSn3 (cP4) done using two different USPPs of Sn (valence configuration: 4d 105s 2 p 2 vs 5s 2 p 2) also agree within approximately 1.5 kJ/mol-atom, at both LDA and GGA levels.
Tables VII and VIII present detailed comparisons of the calculations and measurements for lattice parameters and atomic positions, respectively. As shown in Table VII, the lattice parameters of both the stable and metastable phases measured at ambient temperature lie between the lattice parameters calculated using USPP-LDA and USPP-GGA. However, we note that, in many cases, the experimental lattice parameters are much closer to those calculated using USPP-GGA than to those calculated using USPP-LDA. Specifically, the lattice parameters of Ni3Sn (cP4), Ni3Sn (cF16), Ni3Sn (hP8), Ni3Sn2 (oP20), and Ni3Sn4 (mC14) calculated using USPP-GGA agree to within 1 pct of the experimental data at ambient temperature.
5 Discussion
5.1 Phase Stability of Ni-Sn Intermetallics
This is the first study of the cohesive properties of Ni-Sn intermetallics that uses DFT methods. Based on the results presented in Table VI, we find that the formation energies of stable and metastable phases obtained using USPP-LDA are more negative by up to 5.5 kJ/mol-atom than those obtained by USPP-GGA. The effect of LDA and GGA on the formation energy of many metallic systems is well established in the literature; however, usually the difference tends to be a few kJ/mol-atoms, with LDA giving more negative values. In this study, we find that the quantitative difference is somewhat larger than expected.
To establish a trend, we have carried out several test cases of intermetallics involving Sn and another transition element; we also carry out test cases involving Sn and a filled d-shell element. Specifically, several test cases are carried out for Pd-Sn (because Pd is in the same group as Ni, but is nonmagnetic), Ag-Sn, Au-Sn, and Cu-Sn intermetallics in the composition range of 25 to 50 at. pct Sn, using both USPP-LDA and USPP-GGA. For this purpose, relatively simple structures among the stable and metastable phases listed in Table I are selected. In the case of Pd-Sn intermetallics, we find that the LDA- and GGA-based formation energies differ by approximately 4 kJ/mol-atom. On the other hand, when similar calculations are carried out for Ag-Sn, Au-Sn, and Cu-Sn intermetallics, we find that the LDA- and GGA-based formation energies differ by 2 kJ/mol-atom or less. Therefore, these calculations suggest that the somewhat large differences between the LDA- and GGA-based formation energies reflect a limitation in the LDA/GGA methods in DFT while dealing with transitional metal-Sn (TM-Sn) systems, in which the TM is a transition element.
As seen in Table I, we have considered two phases at 80 at. pct Sn, tP10-NiSn, and oC20-NiSn4; their structural models are shown in Figure 3. It is interesting to note the following computational results: (1) the thermodynamic stability of tP10-NiSn4 and oC20-NiSn4 is very similar; the latter is only slightly more stable (Table VI) and (2) the lattice parameters of tP10-NiSn4 calculated using USPP-LDA, a = 0.61559 nm and c = 0.57265 nm, are in reasonably good agreement with those of “a tetragonal phase” having a = 0.623 nm and c = 0.577 nm observed in the electrodeposited Ni-Sn alloy.[41]

Structural models of (a) tP10-NiSn4 and (b) oC20-NiSn4
In discussing the thermodynamic stability of tP10-NiSn4, it is relevant to examine the phase stability in the Au-Sn system. An in-situ study of the Au/Sn thin-film reaction has shown that the formation of tP10-AuSn4 precedes that of oC20-AuSn4.[47] However, such a formation sequence is yet to be established in the Ni-Sn system. Nevertheless, our computational results warrant the additional structural characterization of solid phases at Ni20Sn80. Midgley et al.[47] have proposed a possible mechanism of transformation from a tP10 to a oC20 structure. It involves a shift of one Sn layer by a 1/2-unit cell, with respect to the other Sn layer in the tP10 structure, leading to a small orthorhombic distortion and a concurrent doubling of the c-axis lattice parameter. This would be a first-order reconstructive phase transformation, because these two structures are not related by a maximal supergroup/subgroup.
An additional important result of our systematic study is the identification of energetically low-lying, but slightly above the ground-state convex hulls, structures that may be both metastable and virtual types. In the present case, the low-lying structures include oP8-Ni3Sn, oP32-NiSn, mC16-Ni3Sn4, and tP10-NiSn4. One or more of these may become stable phase(s) in ternary and higher-order systems containing Ni and Sn. For the case at hand, we note that the metastable oP8-Ni3Sn becomes stable in the Cu-Ni-Sn system. Its homogeneity range is described as Cu x Ni6–x Sn2 with 0.8 ≤ x ≤ 1.41;[95,96] it is present in both 240 °C[97,98] and 200 °C[99] isothermal sections. We also find that the experimental lattice parameters of CuNi5Sn2, a = 0.538, b = 0.4285, and c = 0.4495 nm,[96] are very close to those of oP8-Ni3Sn calculated using USPP-GGA (Table VII).
In comparing the heat of formation from first-principles calculations, calorimetry, and CALPHAD model parameters, we see a spread of several kJ/mol-atom (Table VI). The spread in the experimental data may originate from incomplete reactions in the calorimetry and may also be due to intrinsic factors associated with the solution and direct-reaction calorimetry techniques. The accuracy of the CALPHAD-based model parameters relies heavily on the accuracy of the experimental thermodynamic parameters; it also relies on the judgments made by the assessors in optimizing the thermodynamic model parameters.
Negative heats of formation imply strong Ni-Sn bonds. Normally, this is expected to cause a decrease in the equilibrium atomic volume and an increase in the bulk modulus relative to a concentration-weighted average of the pure-element values. Indeed, as shown in Figures 1(b) and 2(b), there is a decrease in the equilibrium atomic volume relative to the concentration-averaged value. However, there is a “decrease” in the bulk modulus, in general, relative to a concentration-weighted average of the pure-element values, as shown in Figures 1(c) and 2(c). Our observation is true irrespective of whether the comparison is made based on LDA- or GGA-based calculated values. In a previous study of the Cu-Sn system, we observed a similar trend.[79]
Our observations of an increase in the equilibrium atomic volume and a concomitant decrease in the bulk modulus are consistent with the earlier first-principles results of bcc Fe1−x Ge x and Fe1−x Sn x alloys, using coherent potential approximation.[100] Both Ge and Sn increase the atomic volume of the bcc solid solution of Fe; they also decrease the bulk modulus. This was attributed to the increasing number of p-p and p-d bonds with the addition of Ge and Sn in Fe.[100] Taking L12-Ni3Sn as a prototypical case, in Section B, we show that the bonding mechanism is dominated by p-d hybridization.
Finally, it is worth noting that our calculated values of Δ f E using USPP-GGA and those predicted by Miedema’s model[101] agree fairly well. This is merely a coincidence, as there are many limitations inherent in Miedema’s model for deriving accurate values for heats of formation.[102] This issue has been discussed in detail elsewhere.[76,103]
5.2 The Ni3Sn2(ht) ⇌ Ni3Sn2(lt) Phase Transformation
In recent years, there have been several studies on the stability of Ni3Sn2(ht) and Ni3Sn2(lt) forms,[24–28] including the kinetics and mechanism of Ni3Sn2(ht) ⇌ Ni3Sn2(lt) transformation,[23,28–30] and the diffusion mechanism in Ni3Sn2(ht).[30,104,105] In the following, we examine the stability of Ni3Sn2(ht) and Ni3Sn2(lt) forms, based on our calculated results.
Due to its composition, the structure of Ni3Sn2(ht) falls between the prototypical structures of B81-NiAs and B82-InNi2. In an ideal B81-NiSn, the Sn atoms from packed layers in hexagonal-stacking BC and Ni atoms on site A are located at the center of all octahedral interstices in the Sn layers. The Ni has six Sn neighbors and the Sn is surrounded by six Ni in a right-trigonal prism. The Sn octahedra share faces normal to the c axis, so that the Ni atoms are direct neighbors along the [001] direction; the ratio of Ni:Sn atoms is then 1:1. There are tetrahedral interstices, two of which are connected to a double tetrahedral interstice, due to a low c/a compared to an ideal hcp lattice. A significant departure from the ideal composition may be caused by a high degree of filling of double tetrahedral interstices by Ni atoms that may occupy Sn sites as antistructure defects.
The B82 structure is an important type, because of the many defects and filled-up structures derived from it. In an ideal B82-Ni2Sn, the Sn atoms also have a hcp BC stacking sequence; Ni(1) atoms are located at the centers of octahedral holes in this array, so they form trigonal prisms surrounding the Sn atoms. The Ni(2) atoms occupy the centers of the other Ni(1) trigonal prisms (or tetrahedral positions). The Ni3Sn2(lt) is considered to be the superstructure derived from the high-temperature “disordered” B82-Ni2Sn. The Ni(2) atoms have ordered sites; the other atoms are displaced slightly from their sites of the B82-Ni2Sn.
The formation energies of the Ni3Sn2(ht) and related structures are plotted in Figure 4. Both the LDA- and GGA-level calculations show that the driving energy for the Ni3Sn2(ht) → Ni3Sn2(lt) transformation is approximately −3 to −3.5 kJ/mol-atom.

Calculated formation energy (Δ f E) of selected Ni-Sn intermetallics to demonstrate the driving energy (Δ(Δ f E)) for Ni2Sn/NiSn-type (hP6/hP4) to Ni3Sn2 (oP20) transformation
The Ni3Sn2(ht) ⇌ Ni3Sn2(lt) transformation has been treated both as a second-order[59] and a first-order[42] type in the CALPHAD modeling of the Ni-Sn phase diagram. In the former treatment, Ghosh[59] assumed the validity of the second-order transformation, as implied in the assessed Ni-Sn phase diagram.[21,106] However, Liu et al.[42] treated Ni3Sn2(ht) ⇌ Ni3Sn2(lt) as a first-order phase transformation. Because the second-order nature of the Ni3Sn2(ht) ⇌ Ni3Sn2(lt) transformation was never justified in the assessed Ni-Sn phase diagram,[21,106] we further consider the nature of this transformation in light of the supergroup/subgroup analysis.
The Landau theory of phase transitions[107] is based on the assumptions that the symmetry of the product phase is a subgroup of the parent phase and that the structures of these phases can be related by a set of order parameters. For a given space group, the maximal group/subgroup relations and the chains of maximal subgroups via an intermediate structure the space group of which is either a supergroup of both structures or a subgroup of both structures[108] can be obtained from a suitable crystallographic database.[109] Figure 5 shows the chain of maximal subgroups, which involves the space groups P63/mmc (194) and Pnma (62); this is relevant to the Ni3Sn2(ht) ⇌ Ni3Sn2(lt) transformation. Also shown in Figure 5 is the index, [n], defined by the ratio of the number of symmetry elements in the supergroup and subgroup. In the Landau theory, an odd index leads to a first-order transition, while an even index is compatible with either a first- or a second-order character.

Group-subgroup symmetry relations involving P63/mmc (194) and Pnma (62) relevant to Ni3Sn2 (ht) ⇌ Ni3Sn2 (lt) phase transformation.[109] The index (n) defines the ratio of the number of symmetry elements in the supergroup and subgroup
In Figure 5, we note that the space group of the Ni3Sn2(lt) (Pnma, 62) is not a maximal subgroup of Ni3Sn2(ht) (P63/mmc, 194). Therefore, from a symmetry perspective (i.e., group-subgroup criteria) and at the mean field level, the Ni3Sn2(ht) ⇌ Ni3Sn2(lt) is a first-order phase transition. Theoretically, if fluctuations are considered at a renormalization level, this can be a continuous, or second-order, phase transition. However, consistent with our group-subgroup argument, we have also observed the latent heat associated with the Ni3Sn2(ht) ⇌ Ni3Sn2(lt) transformation,[110] further confirming its first-order nature.
5.3 Elastic Stability of tP10-NiSn4 and oC20-NiSn4
As discussed in Section IV–B, and as shown in Figures 1(a) and 2(a), an important result of our systematic study is the prediction that NiSn4 (oC20) is the ground-state structure. Only recently has this phase been identified as the product of the interfacial reaction in Ni/Sn diffusion couples thermally cycled between −40 °C and +130 °C.[22] In addition, we also find that the thermodynamic stability of tP10-NiSn4 is very similar to that of oC20-NiSn4.
In considering the relative thermodynamic stability of competing stable and metastable phases, it is instructive to verify the elastic stability in their bulk form. This issue of elastic stability can be assessed by calculating the single-crystal elastic constants. In addition, the elastic constants of solids are closely related to many fundamental solid-state properties, such as the EOS, specific heat, thermal expansion, Debye temperature, Grüneisen parameter, melting point, etc. Also, from the elastic constants, one can obtain valuable information about the binding characteristic between adjacent atomic planes and about the anisotropic character of the bonding and structural stability.
We have calculated the C ij of tP10-NiSn4 and oC20-NiSn4 by the stress-tensor method. For this purpose, we have used the generalized Hooke’s law (due to Cauchy), σ i = C ij ε j , at small deformations, to account for the multiaxial loading conditions and the elastic anisotropy. Starting with the fully optimized geometry of the oC20-NiSn4 unit cell at zero pressure, we have applied the following four deformations: (1) ε 1 = δ, (2) ε 2 = δ, (3) ε 3 = δ, and (4) γ 4 = γ 5 = γ 6 = δ. In the case of tP10-NiSn4, we have used the first, third, and fourth deformations. For each type of deformation, we have used strains (δ) of ±0.25 and ±0.5 pct; the unlisted strains are set to zero.
Subsequently, the stresses are calculated after optimizing the atomic positions; the calculations for the C ij are then extracted by a least-square method.[111,112] For geometry optimization and the calculation of stresses, we have used a k mesh of 14 × 14 × 15 and 13 × 13 × 7 for tP10-NiSn4 and oC20-NiSn4, respectively. When the crystal symmetry is conserved, these k meshes correspond to 224 and 196 k points in the respective irreducible Brillouin zone. All calculations are performed using the high setting of precision within the VASP code. The calculations for the C ij using USPP-LDA and USPP-GGA are listed in Table IX.
The requirement that a tetragonal phase in its bulk form be stable against any homogeneous lattice deformation places restrictions on the elastic constants. The necessary conditions for elastic stability in an tetragonal crystal are[86]
and
Similarly, the necessary conditions for elastic stability in an orthorhombic crystal are[86]
The calculated zero-temperature elastic constants in Table IX satisfy the criteria for the elastic stability of both tP10-NiSn4 and oC20-NiSn4, suggesting that their bulk form is experimentally accessible. However, so far, oC20-NiSn4 has been observed only as an interfacial phase in Ni/Sn diffusion couples.
5.4 Electronic Structure and Bonding Mechanism in L12-Ni3Sn
Negative formation energies of the stable phases in Table VI are indicative of strong bonding tendencies between the constituent atoms. To obtain additional insight into the nature of the bonding, we present the electronic density of states (DOS) and the charge density in L12-Ni3Sn that have been experimentally observed in its bulk form.[33] In the following, we present both total and partial DOS; the latter quantities were computed using projections into site-centered atomic spheres, each with radii equal to half the nearest-neighbor distance. We also present the total and bonding-charge densities. The latter, also called the deformation-charge density, is defined as the difference (point-by-point, using an identical fast Fourier transform grid) between the self-consistent charge density of the intermetallic and a reference charge density constructed from the superposition of a noninteracting atomic-charge density at the crystal sites. Both the DOS and the charge densities are calculated using PAW pseudopotentials.
Total and partial spin-polarized DOS are shown in Figure 6. An important feature of the DOS in Figure 6 is the presence of a dip approximately 0.25 eV above the Fermi level. Such pseudogap features have been extensively discussed in the context of the bonding in TM trialuminides, where they are associated with the hybridization between TM-d states and Al-p states (references cited by Ghosh et al.[103]). The total DOS at the Fermi level is rather low: 0.27 states/eV/atom. The Fermi level lies at the left edge of the pseudogap minimum in the DOS, i.e., the bonding states are almost completely occupied. When comparing the up and down DOS, both total and partials, we find that they are symmetric in the entire energy range. Both this and the very low DOS at the Fermi level imply that L12-Ni3Sn is nonmagnetic. Divis[113] reported the band structure and electronic DOS of cF16-Ni3Sn (DO 3), using a tight-binding Hamiltonian, while the DOS at the Fermi level was observed to be very small. Consequently, cF16-Ni3Sn was predicted to be paramagnetic.

Angular momentum and site decomposed electronic density of states, n(E), for L12-Ni3Sn calculated (spin polarized) using all-electron PAW pseudopotentials
The partial DOS in Figure 6 provide direct evidence for hybridization processes. From the Fermi level to about 2.5 eV below, the s and p states of Sn exhibit hybridization with Ni-d, but their contributions are relatively small. The contributions due to the Ni-s and Ni-p states are very small. However, it is seen that at approximately 3.1 eV below the Fermi level, pronounced peaks of Sn-p exhibit strong hybridization with Ni-d. The pseudogap can thus be interpreted as separating bonding and antibonding states associated with such hybridizations. The antibonding states (or the unoccupied conduction region) beyond the Fermi level are the result of delocalized Ni-d states, with only a small contribution from Sn-p states. The Ni-s, Ni-p, Sn-s, and Sn-d contributions to the conduction band are negligibly small.
Figure 7(a) shows a total-charge density and Figure 7(b) shows a bonding-charge density (also referred to as the deformation-charge density) in the (001) plane in which Ni and Sn are the nearest neighbors. In Figure 7(b), while there is the delocalization of the bonding charge in the interstitial region that resembles metallic bonding, there is also evidence of the directionality of the electron density along the [110] direction, which may contribute to covalent character. The distribution of the bonding-charge density is clearly asymmetric; the maximum is located along the Ni-Sn bond but toward the Ni site. The presence of the covalent character is further supported by the fact that the energy interval for bonding is much larger than for antibonding, as seen in Figure 6. In Figure 7(b), it is interesting to note that the bonding-charge-density (δρ) isocontour lobes are oriented perpendicular to the Ni-Sn bonding direction. Combining the angular-momentum-resolved DOS and the bonding-charge-density plot, it may be concluded that the mechanism of cohesion is dominated by the short-range band mixing between the Ni-d and Sn-p states, along with the long-range charge-transfer (electrostatic) effect. A somewhat similar distribution of the bonding-charge density in hP8-Ni3Sn (DO 19) has been reported to be the primary bonding mechanism.[32]

Electronic structure of L12-Ni3Sn calculated (spin polarized) using all-electron PAW pseudopotentials showing the distribution of (a) total and (b) bonding (or deformation) charge density in the (001) plane. The total charge density ranges from 0.149 (ρ min) to 13.983 (ρ max) e/Å3, while the bonding charge density ranges from −0.367 (depleted region: δρ(−)) to 0.032 (enhanced region: δρ(+)) e/Å3. In each case, the contour lines are drawn at a constant interval
6 Conclusions
A systematic and comprehensive study of the phase stability of the intermetallic phases in the Ni-Sn system is carried out using DFT methods. Specifically, the phase stability and cohesive properties of Ni, Sn, and 27 Ni-Sn intermetallic phases are calculated using a Vanderbilt-type USPP in combination with both LDA and GGA for the exchange correlation functional. In addition, the fidelity of our results is demonstrated by calculating the cohesive properties of Ni, Sn and selected intermetallics, using pseudopotentials constructed by the PAW method. The following conclusions are drawn.
-
1.
We have calculated the lattice stabilities and cohesive properties of Ni and Sn with six structures, A1 (cF4), A2 (cI2), A3 (hP2), A4 (cF8), A5 (tI4), and A6 (tI2), using both USPP-LDA and USPP-GGA. Whenever possible, our calculated results are compared with the previously reported ab-initio results and with the experimental data.
-
2.
Single-crystal elastic constants, C ij , of Ni and Sn are calculated using USPP-LDA, USPP-GGA, and PAW-GGA. In the case of fcc Ni (A1, cF4), the experimental data agree very well with those calculated using both USPP-GGA and PAW-GGA, while in the case of tetragonal-Sn (A5, tI4), the experimental data agree very well with those calculated using USPP-LDA.
-
3.
The formation energies (Δ f E) of Ni-Sn intermetallics are calculated using both USPP-LDA and USPP-GGA. The calorimetric data for the enthalpies of formation of Ni-Sn intermetallics show a better agreement with those calculated using USPP-LDA than with those calculated using USPP-GGA.
-
4.
In general, we find that the experimental lattice parameters of both stable and metastable phases measured at ambient temperature lie between those calculated using USPP-LDA and those calculated using USPP-GGA; however, in several cases, the values calculated using USPP-GGA agree to within 1 pct of the experimental data.
-
5.
The ground-state structures based on USPP-LDA and USPP-GGA calculations are essentially the same. Even though the ground-state structures have been experimentally observed, not all of them are registered in the assessed Ni-Sn phase diagram.
-
6.
An important result of our systematic study is the prediction that oC20-NiSn4 is the ground-state structure (using both USPP-LDA and USPP-GGA). A related phase, tP10-NiSn4, is found to have a very similar thermodynamic stability. Only recently, oC20-NiSn4 has been identified as the product of the interfacial reaction in Ni/Sn diffusion couples. The calculation of the C ij confirmed the elastic stability of both the tP10-NiSn4 and the oC20-NiSn4. Additional experiments are needed for the structural characterization of these two intermetallics and for their presence in bulk alloys.
-
7.
Contrary to what is shown in the assessed Ni-Sn phase diagram,[21,106] it is argued that Ni3Sn2(ht) ⇌ Ni3Sn2(lt) is a first-order phase transformation.
-
8.
The DOS calculations of L12-Ni3Sn show the presence of a pseudogap. The site and angular-momentum decomposed DOS show that the bonding mechanism between Ni and Sn is dominated by p-d hybridization.
-
9.
The oP8-Ni3Sn is a metastable phase in the binary Ni-Sn system, but it is a stable phase in the ternary Cu-Ni-Sn system. In the ternary regime, the homogeneity range of this phase is represented as Cu x Ni6−x Sn2 with 0.8 ≤ x ≤ 1.41, i.e., the composition range of stability is very close to the Ni-Sn binary edge. The present study clearly demonstrates the importance of a systematic study of binary intermetallics using first-principles methods in understanding the origin of the stability of a ternary phase. Additional low-lying, but metastable, structures are also identified; these may become stable in ternary and higher-order systems containing Ni and Sn.
-
10.
The lattice stability of Ni and Sn and the Δ f E values of the Ni-Sn intermetallics presented here may be integrated within CALPHAD formalism[56,57] to create a reliable thermodynamic database to facilitate the calculation of the stable and metastable phase diagrams of binary and multicomponent systems containing Ni and Sn.
References
M. Abtew, and G. Selvaduray: Mater. Sci. Eng., 2000, vol. R27, pp. 95–141
K.N. Tu, and K. Zeng: Mater. Sci. Eng., 2001, vol. R34, pp. 1–58
K. Zeng, and K.N. Tu: Mater. Sci. Eng., 2002, vol. R38, pp. 55–105
K.N. Tu, A.M. Gusak, and M. Li: J. Appl. Phys., 2003, vol. 93, pp. 1335–53
T. Laurila, V. Vuorinen, and J.K. Kivilahti: Mater. Sci. Eng., 2005, vol. R49, pp. 1–60
D.C. Abbott, R.M. Brook, N. McLelland, and J.S. Wiley: IEEE Trans. Compon. Hybrids Manuf. Technol., 1991, vol. 14, pp. 567–72
S.K. Kang, R.S. Rai, and S. Purusothaman: J. Electron. Mater., 1996, vol. 25, pp. 1113–20
G. Ghosh: J. Appl. Phys., 2000, vol. 88, pp. 6887–96
G. Ghosh: Acta Mater., 2001, vol. 49, pp. 2609–24
G. Ghosh: J. Electron. Mater., 2004, vol. 33, pp. 229–40
G. Ghosh: J. Electron. Mater., 2004, vol. 33, pp. 1080–91
R. Labie, W. Ruythooren, and J. Van Humbeeck: Intermetallics, 2007, vol. 15, pp. 396–403
H.Y. Lee, S.W. Jang, S.M. Lee, S.J. Lee, and H.K. Baik: J. Power Sources, 2002, vol. 112, pp. 8–12
H. Mukaibo, T. Sumi, T. Yokoshima, T. Momma, and T. Osaka: Electrochem. Solid-State Lett., 2003, vol. 6, pp. A218–A220
Q.F. Dong, C.Z. Wu, M.G. Jin, Z.C. Huang, M.S. Zheng, J.K. You, and Z.G. Lin: Solid State Ionics, 2004, vol. 167, pp. 49–54
I. Amadei, S. Panero, B. Scrosati, G. Cocco, and L. Schifini: J. Power Sources, 2005, vol. 143, pp. 227–30
J. Hassoun, S. Panero, P. Simon, P.L. Taberna, and B. Scrosati: Adv. Mater., 2007, vol. 19, pp. 1632–35
M. Hansen: Constitution of Binary Alloys, McGraw-Hill Book Company, Inc., New York, NY, 1958, pp. 1042–45
R.P. Elliott: Constitution of Binary Alloys, First Supplement, McGraw-Hill Book Company, Inc., New York, NY, 1965, p. 674
F. Shunk: Constitution of Binary Alloys, Second Supplement, McGraw-Hill Book Company Inc., New York, NY, 1969, pp. 555–56
P. Nash, and A. Nash: Bull. Alloy Phase Diagrams, 1985, vol. 6, pp. 350–59
W.J. Boettinger, M.D. Vaudin, M.E. Williams, L.A. Bendersky, and W.R. Wagner: J. Electron. Mater., 2003, vol. 32, pp. 511–15
A.K. Larsson, R.L. Withers, and L. Stenberg: J. Solid State Chem., 1996, vol. 127, pp. 222–30
A. Leineweber, M. Ellner, and E.J. Mittermeijer: J. Solid State Chem., 2001, vol. 159, pp. 191–97
A. Leineweber, O. Oeckler, and U. Zachwieja: J. Solid State Chem., 2004, vol. 177, pp. 936–45
A. Leineweber: J. Solid State Chem., 2004, vol. 177, pp. 1197–1212
A. Leineweber, E.J. Mittermeijer, M. Knapp, and C. Baehtz: Mater. Sci. Forum, 2004, vols. 443–444, pp. 247–50
A. Leineweber, and E.J. Mittermeijer: Z. Krist. Suppl., 2006, vol. 23, pp. 351–56
A. Leineweber, and E.J. Mittermeijer: Z. Kristallogr., 2007, vol. 222, pp. 150–59
A. Leineweber, E.J. Mittemeijer, M. Knapp, and C. Baehtz: Philos. Mag., 2007, vol. 87, pp. 1821–44
C. Schmetterer, H. Flandorfer, K.W. Richtera, U. Saeeda, M. Kaufman, P. Roussel, and H. Ipser: Intermetallics, 2007, vol. 15, pp. 862–84
A.L. Lyubimtsev, A.I. Baranov, A. Fischer, L. Kloo, and B.A. Popovkin: J. Alloys Compd., 2002, vol. 340, pp. 167–72
J.F. Cannon: MRS Symp. Proc., 1984, vol. 22, pp. 113–16
S.K. Shadangi, M. Singh, S.C. Panda, and S. Bhan: Cryst. Res. Technol., 1986, vol. 21, pp. 867–71
S.K. Shadangi, M. Singh, S.C. Panda, and S. Bhan: Indian J. Technol., 1986, vol. 24, pp. 105–07
H.R. Pak, T. Saburi, and S. Nenno: Bull. Jpn. Inst. Met., 1973, vol. 37, pp. 1128–34
H. Fjellvag, and A. Kejkshus: Acta Chem. Scand., 1986, vol. 40A, pp. 23–30
W. Jeitschko, and B. Jaberg: Acta Crystallogr., 1982, vol. 38B, pp. 598–600
S. Furuseth, and H. Fjellvag: Acta Chem. Scand., 1986, vol. 40A, pp. 695–700
J. Haimovich: Weld. J. Res. Suppl., 1989, vol. 8, pp. S102–S111
T. Watanabe, K. Arai, T. Hirose, and M. Chikazawa: J. Jpn. Inst. Met., 1999, vol. 63, pp. 489–95
H.S. Liu, J. Wang, and Z.P. Jin: Calphad, 2004, vol. 28, pp. 363–70
T.J. Tiainen, and R.B. Schwarz: J. Less-Common Met., 1988, vol. 140, pp. 99–112
J.F. Geny, G. Marchal, Ph. Mangin, Chr. Janot, and M. Piecuch: Phys. Rev. B, 1982, vol. 25, pp. 7449–66
S. Kim, and D.C. Johnson: J. Alloys Compd., 2005, vol. 392, pp. 105–111
B. Sundman, and J. Agren: J. Phys. Chem. Solids, 1981, vol. 2, pp. 297–301
P.A. Midgley, M.E. Sleight, and R. Vincent: J. Solid State Chem., 1996, vol. 124, pp. 132–42
B. Predel, and H. Ruge: Thermochim. Acta, 1972, vol. 3, pp. 411–19
H.D. Dannohl, and H.L. Lukas: Z. Metallkd., 1974, vol. 65, pp. 642–49
B. Predel, and W. Vogelbein: Thermochimica Acta, 1979, vol. 30, pp. 201–15
A.N. Torgersen, H. Bros, R. Castanet, and A. Kjekhus: J. Alloys Compd., 2000, vol. 307, pp. 167–73
H. Flandorfer, U. Saeed, C. Luef, A. Sabbar, and H. Ipser: Thermochim. Acta, 2007, vol. 459, pp. 34–39
F. Korber, and W. Oelson: Mitt. Kaiser Wilhelm Inst. Eisenforsch., 1937, vol. 19, pp. 209–19
G.P. Vassilev, K.I. Lilova, and J.C. Gachon: J. Alloys Compd., 2006, vol. 447, pp. 106–08
R. Hultgren, P. Desai, P. Hawkins, M. Gleiser, and K.K. Kelley: Selected Values of the Thermodynamic Properties of Binary Alloys, ASM, Metals Park, OH, 1973, pp. 1237–39
L. Kaufman, and H. Bernstein: Computer Calculation of Phase Diagram, Academic Press, Inc., New York, NY, 1970.
H.L. Lukas, S.G. Fries, and B. Sundman: Computational Thermodynamics: The Calphad Method, Cambridge University Press, Cambridge, United Kingdom, 2007.
P. Nash, H. Choo, and R.B. Schwarz: J. Mater. Sci., 1998, vol. 33, pp. 4929–36
G. Ghosh: Metall. Mater. Trans. A, 1999, vol. 30A, pp. 1481–94
J. Miettinen: Calphad, 2003, vol. 27, pp. 309–18
G. Kresse, and J. Hafner: Phys. Rev. B, 1994, vol. 49, pp. 14251–69
G. Kresse, and J. Furthmüller: Phys. Rev. B, 1996, vol. 54, pp. 11169–86
G. Kresse, and J. Furthmüller: Comput. Mater. Sci., 1996, vol. 6, pp. 15–50
D. Vanderbilt: Phys. Rev. B, 1990, vol. 41, pp. 7892–95
G. Kresse, and J. Hafner: J. Phys.: Condens. Matter, 1994, vol. 6, pp. 8245–57
D.M. Ceperley, and B.J. Alder: Phys. Rev. Lett., 1980, vol. 45, pp. 566–69
J.P. Perdew, and A. Zunger: Phys. Rev. B, 1981, vol. 23, pp. 5048–79
J.P. Perdew: in Electronic Structure of Solids '91, P. Ziesche H. Eschrig, eds., Akademie Verlag, Berlin, 1991, p. 11–20
S.H. Vosko, L. Wilk, and M. Nussair: Can. J. Phys., 1980; vol. 58, pp. 1200–11
M.J. Monkhorst, and J.D. Pack: Phys. Rev. B, 1976, vol. 13, pp. 5188–92
M. Methfessel, and A.T. Paxton: Phys. Rev. B, 1989, vol. 40, pp. 3616–21
P.E. Blöchl: Phys. Rev. B, 1994, vol. 50, pp. 17953–17979
G. Kresse, and D. Joubert: Phys. Rev. B, 1999, vol. 59, pp. 1758–75
M. Marsman and G. Kresse: J. Chem. Phys., 2006, vol. 125, pp. 104101-1–104101-12.
E. Wimmer, H. Krakauer, M. Weinert, and A.J. Freeman: Phys. Rev. B, 1981, vol. 24, pp. 864–75
G. Ghosh: Acta Mater., 2007, vol. 55, pp. 3347–74
P. Vinet, J.H. Rose, J. Ferrante, and J.R. Smith: J. Phys.: Condens. Matter, 1989, vol. 1, pp. 1941–63
M.J. Mehl, B.M. Klein, and K. Papaconstatopolous: in Intermetallic Compounds: Principles, J.H. Westbrook and R.L. Fleischer, eds., John Wiley & Sons, New York, NY, 1994, vol. 1, pp. 195–210
G. Ghosh, and M. Asta: J. Mater. Res., 2005, vol. 20, pp. 3102–17
R. Kohlhaas, P. Dünner, and N. Schmitz-Pranghe: Z. Angew. Phys., 1967, vol. 23, pp. 245–47
J. Bandopadhay, and K.P. Gupta: Cryogenics (London), 1977, vol. 17, pp. 345–47
G.A. Alers, and J.R. Neighbours: J. Phys. Chem. Solids, 1960, vol. 13, pp. 40–55
D.H. Martin: Magnetism in Solids, The MIT Press, Cambridge, MA, 1967, p. 10.
I. Nakai, F. Ono, and O. Yamada: J. Phys. Soc. Jpn., 1983, vol. 52, pp. 1791–98
J. Steinberg: J. Phys. Chem. Solids, 1982, vol. 43, pp. 1173–75
D.C. Wallace: Thermodynamics of Crystals, Wiley, New York, NY, 1972.
G.Y. Guo, and H.H. Wang: Chin. J. Phys. 2000, vol. 38, pp. 949–61
G. Simmons, and H. Wang: Single Crystal Elastic Constants and Calculated Aggregate Properties: A Handbook, 2nd ed., The MIT Press, Cambridge, MA, 1971, p. 115.
J.A. Rayne, and B.S. Chandrasekhar: Phys. Rev., 1960, vol. 120, pp. 1658–63
B. Akdim, D.A. Papaconstantopoulos, and M.J. Mehl: Philos. Mag. B, 2002, vol. 82, pp. 47–61
A. Dinsdale: Calphad, 1991, vol. 15, pp. 317–425
J.O. Andersson, T. Helander, L. Höglund, P.F. Shi, and B. Sundman: Calphad, 2002, vol. 26, pp. 273–312
Y. Wang, S. Curtarolo, C. Jiang, R. Arroyave, T. Wang, G. Ceder, L.Q. Chen, and Z.-K. Liu: Calphad, 2004, vol. 28, pp. 79–90
M.K. Bhargava, and K. Schubert: J. Less-Common Met., 1973, vol. 33, pp. 181–89
Y. Watanabe, Y. Murakami, and S. Kachi: J. Jpn. Inst. Met., 1981, vol. 45, pp. 551–58
J.S.L. Pak, K. Mukherjee, O.T. Inal, and H.-R. Pak: Mater. Sci. Eng., 1989, vol. 117A, pp. 167–73
C.-H. Lin, S.-W. Chen, and C.-H. Wang: J. Electron. Mater., 2002, vol. 31, pp. 907–15
C.-Y. Li, and J.-G. Duh: J. Electron. Mater., 2005, vol. 34, pp. 3118–24
S.-W. Chen, S.-H. Wu, and S.-W. Lee: J. Electron. Mater., 2003, vol. 32, pp. 1188–94
T. Khmelevska, S. Khmelevskyi, A.V. Ruban, and P. Mohn: J. Phys.: Condens. Matter, 2006, vol. 18, pp. 6677–89
F.R. de Boer, R. Boom, W.C.M. Mattens, and A.R. Miedema: Cohesion in Metals: Transition Metal Alloys, Elsevier, Amsterdam, 1998.
D.G. Pettifor: Solid State Phys., 1987, vol. 40, pp. 43–92
G. Ghosh, S. Delsante, G. Borzone, M. Asta, and R. Ferro: Acta Mater., 2006, vol. 54, pp. 4977–97
H. Schmidt, G. Frohberg, and H. Wever: Acta Metall. Mater., 1992, vol. 40, pp. 3105–11
H. Schmidt, G. Frohberg, W. Miekeley, and H. Wever: Phys. Status Solidi B, 1992, vol. 171, pp. 29–37
A. Nash and P. Nash: in Binary Alloy Phase Diagrams, T.B. Massalski, ed., ASM, Metals Park, OH, 1990, vol. 3, pp. 2863–64
L.D. Landau, and E.M. Lifschitz: Statistical Physics, Pergamon Press, Oxford, United Kingdom, 1976.
Y.A. Izyumov, and V.N. Syromyatnikov: Phase Transitions and Crystal Symmetry, Kluwer, Dordrecht, Holland, 1990.
G. Ghosh: Northwestern University, Evanston, IL, unpublished research, 2008
Y. Le Page and P. Saxe: Phys. Rev. B, 2001, vol. 63, pp. 174103-1–174103-8.
Y. Le Page and P. Saxe: Phys. Rev. B, 2002, vol. 65, pp. 104104-1–104104-14.
M. Divis: Phys. Status Solidi B, 1992, vol. 173, pp. K13–K17
Acknowledgments
This research was supported by the Semiconductor Research Corporation (Contract No. 2006-KJ-1393). In addition, this material is based upon work supported by the National Science Foundation (NSF) under the following programs: Partnerships for Advanced Computational Infrastructure, Distributed Terascale Facility, and Terascale Extensions: Enhancements to the Extensible Terascale Facility. Specific to NSF programs, this study has made use of IBM IA-64 Linux clusters at the National Center for Supercomputing Applications at the University of Illinois at Urbana–Champaign (Urbana, IL), and the San Diego Supercomputer Center, University of California at San Diego (La Jolla, CA) as a part of the TeraGrid sites, through Grant No. DMR070017N. The author thanks Dr. Avadh Saxena, Los Alamos National Laboratory (Los Alamos, NM), for helpful discussion on group/subgroup relations.
Author information
Authors and Affiliations
Corresponding author
Additional information
Manuscript submitted April 19, 2008.
Rights and permissions
About this article
Cite this article
Ghosh, G. First-Principles Calculation of Phase Stability and Cohesive Properties of Ni-Sn Intermetallics. Metall Mater Trans A 40, 4–23 (2009). https://doi.org/10.1007/s11661-008-9682-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11661-008-9682-3