
Abstract
This work has its roots in a long-term theoretical research line aimed at developing a complete database with structural, thermodynamic, cohesive and elastic properties of the intermetallic compounds (ICs) of the type MeaXb where Me = Cu, Ni and X = In, Sn and Sb. The paper reports the results of an ab initio study of various phases occurring in the Ni-Sb phase diagram, viz., the low-temperature Ni3Sb (orthorhombic oP8), the high-temperature Ni3Sb (cubic cF16), Ni5Sb2 (monoclinic mC28), NiSb (hexagonal hP4) and the NiSb2 (orthorhombic oP6) compounds. The molar volume, bulk modulus and its pressure derivative, the electronic density of states (DOS) and the energy of formation from the elements of these compounds are calculated ab initio using the relativistic projected augmented wave (PAW) method implemented in the VASP code. The Local Density Approximation of Ceperley and Alder and the Generalized Gradient Approximation due to Perdew and Wang are adopted to treat the exchange and correlation energies. Detailed comparisons between the current and previously reported theoretical and experimental values are reported.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The current contribution has practical as well as theoretical motivations. On the practical side, the work has its roots on a research line aimed at searching for candidates to be adopted in low-melting Pb-free soldering technology. To this aim various Me-X systems (with Me = Cu, Ni and X = In, Sn) have recently been explored theoretically, in particular, by the present authors.[1-3] The goal of those works was to develop a complete database with structural, thermodynamic, cohesive and elastic properties of the intermetallic compounds (ICs) occurring at the joints between the common contact materials Cu and Ni with the low-melting elements In or Sn. Specifically, Ramos et al.[3] performed a systematic comparative study of the electronic and cohesive properties of the Cu-In, Cu-Sn, Ni-In and Ni-Sn systems. On these bases, the thermophysical effects of replacing Cu by Ni were established.[3]
On the theoretical side, the current authors have also been interested in establishing and interpreting microscopically trends in the structural, thermodynamic and cohesive properties of a large class of CuaXb and NiaXb so-called “p-d bonded compounds”. The aim of this complementary research line was to provide information of interest for interpolations and extrapolations, as well as the assessment of the mentioned thermophysical properties.
In addition to the ICs formed with X = In, Sn, the effects of incorporating Sb were also investigated. In particular, the phase stability and thermophysical properties of a new Cu5+xIn2+xSb2−x B8-related structure were determined ab initio.[4] In addition, the physical properties of a specific compound of the Ni-Sb system, viz., the high-temperature Ni3Sb phase were also studied.[5] By going one step forward in this research line, the current paper focuses on the properties of various ICs occurring in the Ni-Sb system.
The phase diagram of the binary Ni-Sb system has been reviewed by Okamoto.[6] The following ICs have been detected at low temperature: Ni3Sb (orthorhombic oP8), Ni7Sb3 (unknown structure), NiSb (hexagonal hP4) and NiSb2 (orthorhombic oP8). At high temperatures, a non-stoichiometric stable phase usually represented as Ni5Sb2 has been reported.[7-9] In fact, the structure of this phase is still under discussion. This phase was classified by Okamoto[6] as monoclinic (mC28), whereas in the thermodynamic assessment of the phase diagram by Cao et al.[10] it was treated as a cubic, cF16 phase, and named as “high temperature (HT) Ni3Sb”. Luo et al.[11] performed low temperature resistivity and magnetization measurements on polycrystalline samples of four of these compounds: Ni3Sb, Ni5Sb2, NiSb, and NiSb2. In addition, ab initio band-structure calculations for the same ICs were performed with results which are consistent with those deduced from electrical and magnetic measurements.[11]
In the present work, the 0 K values of thermophysical and equation-of-state (EOS) parameters such as molar volume, bulk modulus and its pressure derivative, the electronic density of states (DOS), and the energy of formation from the elements of the stable Ni-Sb ICs: Ni3Sb, Ni5Sb2, NiSb, and NiSb2, are established theoretically. The calculations are performed in the framework of the density functional theory (DFT), using the VASP (Vienna Ab Initio Simulation Package) and the Projected Augmented Waves (PAW) method, with the generalized gradient approximation (GGA) and the Local Density Approximation (LDA) to evaluate the exchange and correlation energies. The results of the work are discussed in light of microscopic picture of the bonding trends previously developed by us for the closely related p-d bonded compounds of the Ni-In and Ni-Sn systems.[2,3]
2 Theoretical Method, Phases and Structures
Systematic ab initio DFT spin polarized calculations using the projected augmented wave (PAW) method[12,13] as implemented in the VASP[14] were performed. Both the LDA of Ceperley and Alder[15] and the GGA of Perdew and Wang[16] were adopted to evaluate the exchange-correlation contributions to the energy. We consider ten valence electrons for Ni (3d 84s 2) and 5 for Sb (5s 25p 3). The kinetic energy cut-off for the plane wave expansion of the electronic wave function was 330 eV. We use k-points grids defined according to the Monkhorst-Pack method[17]; the convergency of the k-point grids were checked until getting a precision in the energy higher than 1 meV/atom. We adopt the Methfessel-Paxton technique[18] with a smearing factor of 0.1 eV to occupy the electronic levels.
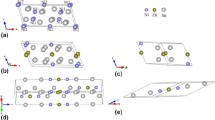
The following equilibrium ICs of the Ni-Sb phase diagram were treated: LT Ni3Sb (orthorhombic oP8), HT Ni3Sb (cubic cF16), Ni5Sb2 (monoclinic mC28), NiSb (hexagonal hP4) and NiSb2 (orthorhombic oP6). These compounds are listed in Table 1 and shown in Fig. 1.

Structures of the Ni-Sb compounds studied in the present work: (a) Ni3Sb (oP8), (b) Ni3Sb (cF16), (c) Ni5Sb2 (mC28), (d) NiSb (hP4) y and (e) NiSb2 (oP6). Ni atoms and Sb atoms are indicated
For these phases, the following k-point grids were considered: 7 × 9 × 9 for Ni3Sb (oP8), 11 × 11 × 11 for Ni3Sb (cF16), 5 × 11 × 5 for Ni5Sb2 (mC28), 11 × 11 × 9 for NiSb (hP4) and 7 × 5 × 11 for NiSb2 (oP6). These k-point meshes mean up to 140 k points in the irreducible Brillouin zone (IBZ), depending on the structure. The convergence criterion for the self-consistent total-energy calculations was 0.1 meV. The lattice parameters as well as the internal coordinates of the unit cell were fully optimized keeping the symmetry space group of the crystalline structure, until the forces were lower than 30 meV/Å, and the energy changes due to the ionic relaxation lower than 1 meV/atom.
To determine the EOS parameters, the total energy (E) and external pressure (P) were calculated for different volumes (V) varying around the equilibrium volume (V 0) by up to ± 5%, letting the relaxation of the internal coordinates. The bulk modulus (B 0) and its pressure derivative (\(B_{0}^{{\prime }}\)) were obtained by fitting the P vs V curves to the EOS by Vinet et al.[19]
The energy of formation of the ICs was calculated by computing the difference between the total energy of the compound and the weighted average of the energies of the elements Ni and Sb in their equilibrium structures, viz., the fcc and hR2 phases, respectively.
3 Results and Discussion
We first present and discuss the calculated structural and EOS parameters of fcc Ni and hR2 Sb. In Table 2, we report the currently calculated lattice parameters (LPs), equilibrium volume (V 0), bulk modulus (B 0) and its pressure derivative (\(B_{0}^{{\prime }}\)). Experimental data and previously reported theoretical results are also included for comparison.
In accord with our previously reported results,[1-4] the present PAW results show that the LPs are underestimated by less than 3% with LDA, and overestimated by less than 0.3% with GGA, with respect to the experimental results. On the other hand, these LPs agree well with those predicted by the FP-LAPW method[20] and other ab initio results based on ultra-soft pseudo potentials (US-PP).[21] As stated in previous works,[1,2,21] the calculated B 0 show larger discrepancies with experiments, and better agreement is found when the GGA is adopted.
The calculated LPs and cohesive properties for the Ni-Sb ICs studied in the present work are presented in Table 3, together with the experimental data and other theoretical results reported in the literature.
The calculated LPs deviates positively respect to the experimental results by less than 1.2% in the GGA approximation, whereas those given by the LDA are lower than the experimental ones by less than 3%. The calculated internal coordinates (Table 4) agree well with the experimental ones.
In Fig. 2 the GGA and LDA values of V 0 and B 0 of the Ni-Sb ICs of various Sb contents are compared with the experimental data available. The GGA approximation systematically overestimates V 0 while LDA underestimates it, as already found in other related systems.[1,2]

(a) Equilibrium volume per atom (V 0) and (b) Bulk modulus (B 0) of Ni-Sb compounds as functions of Sb atomic concentration. Filled symbols correspond to values calculated in this work (PAW method) by LDA (triangles) and GGA (circles). The crosses correspond to experimental values indicated in Table 1
The ab initio calculated V 0 and B 0 of the Ni-Sb ICs (Fig. 2) indicate, in general, a smooth variation with composition. The V 0 values tend to deviate negatively from the linear interpolation between the values corresponding to the elements. The B 0 values present positive as well as negative deviations with respect to the corresponding linear interpolation.
In Fig. 3 the ab initio calculated composition dependence of the energy of formation from the elements (ΔE f) is compared with experimental data and with the results of CALPHAD (i.e., Calculation of Phase Diagrams) assessments. All the phases studied in the present work are found to be thermodynamically stable with respect to the pure elements. Moreover, a very good qualitative agreement is found for the composition dependence of ΔE f. In particular, the most stable compound is correctly placed by the calculation at 50 at.%Sb. However, systematic quantitative discrepancies amounting at about 6 kJ/mol-atom between the GGA calculated ΔE f and the experimental data are found for the Ni3Sb oP8 phase. It is to be remarked that comparable discrepancies have already been reported for related systems (Ni-X, X = In, Sn).[2] A better agreement is found for the ΔE f obtained in the LDA approximation. In this case, the discrepancies between calculations and the available data amount at about 2 kJ/mol-atom. The systematically better prediction of the ΔE f values by LDA calculations for Ni-X (X = In, Sn) has already been reported.[2] Also, the present results reproduce a well established trend, with LDA calculated ΔE f being more negative than the GGA ones.[21]

Energy of formation of the Ni-Sb compounds as a function of Sb concentration. Filled circles and triangles correspond to the GGA and LDA values for the LT ground state phases, respectively, whereas symbols in grey correspond to those for HT phases. Squares and stars represent CALPHAD results extrapolated at 0 K[9,10] Crosses correspond to experimental results at 873 K[28]
We close the comparisons for ΔE f by focusing on the results for the NiSb compound. The current values are compared in Table 5 with two sets of CALPHAD estimates, which correspond to end-members of the three sublattice model (Ni,Va)1/3(Sb)1/3(Va,Ni)1/3 yielding the “NiSb” stoichiometry, viz., the (Ni)1/3(Sb)1/3(Va)1/3 and (Va)1/3(Sb)1/3(Ni)1/3 ICs.[9,10] The present calculations come closer to the CALPHAD estimates for the first compound.[9,10]
In Fig. 4 we plot the electronic DOS for the Ni3Sb-oP8, Ni3Sb-cF18, Ni5Sb2-mC28, NiSb-hP4 and NiSb2-oP6 ICs of the Ni-Sb system. We include the total DOS, and Ni-d, Sb-s and Sb-p partial DOS contributions.

Electronic DOS for the Ni-Sb compounds: (a) Ni3Sb-oP8, (b) Ni3Sb-cF16, (c) Ni5Sb2-mC28, (d) NiSb-hP4 and (e) NiSb2-oP6. The partial site contributions to the DOS of Ni 3d electrons, and Sb 5s and 5p electrons are also included. The origin of the energy scale corresponds to the Fermi level
The ICs treated in this work have a finite, relatively low value of the DOS at the Fermi level, indicating metallic character. The DOS for the current Ni-Sb ICs suggest an electronic behavior similar to that found in the related systems Ni-X[2] and Cu-X, (X = In, Sn).[1] Specifically, the main band in the DOS of this class of systems is determined by the high contribution of Ni-d electrons, whereas the contributions of the s- and p-electrons of Sb are relatively small. However, as found in related Me-X systems, the hybridization occurring between the Ni-d and Sb-p electrons is expected to contribute significantly to cohesion in this type of compounds.[1-3,21]
4 Summary and Concluding Remarks
The present study is part of a long-term research line focusing on the thermophysical properties of the compounds formed by Cu or Ni with In, Sn or Sb. By means of the ab initio methodology used in previous studies, the structural and thermodynamic properties of various Ni-Sb compounds have been determined. The calculated values agree reasonably well with the available experimental data. More specifically, the LPs of the currently studied compounds are better predicted by the GGA PW91 than by the Ceperley-Alder LDA treatment of the exchange-correlation contributions to the energy. However, the reverse is true for the energy of formation from the elements, for which the LDA results are closer to the experimental values. In addition, the study of the electron density of states obtained for the Ni-Sb intermetallics indicate that they may also be classified as “p-d bonded compounds”.
Finally, we emphasize that the present results open up the possibility of investigating the effects of varying the X alloying element of the Ni-X compounds in the sequence X = In → Sn → Sb. This problem will be dealt with in a forthcoming paper.
References
S. Ramos de Debiaggi, C. Deluque Toro, G.F. Cabeza, and A. Fernández Guillermet, Ab Initio Comparative Study of the Cu-In and Cu-Sn Intermetallic Phases in Cu-In-Sn Alloys, J. Alloys Compd., 2012, 542, p 280-292
S. Ramos de Debiaggi, C. Deluque Toro, G.F. Cabeza, and A. Fernández Guillermet, Ab Initio Study of the Cohesive Properties, Electronic Structure and Thermodynamic Stability of the Ni-In and Ni-Sn Intermetallics, J. Alloys Compd., 2013, 576, p 302-316
S. Ramos de Debiaggi, N.V. González Lemus, G.F. Cabeza, and A. Fernández Guillermet, Cohesive Properties of (Cu, Ni)-(In, Sn) Intermetallics: Database, Electron-Density Correlations and Interpretation of Bonding Trends, J. Phys. Chem. Sol., 2016, 93, p 40-51
C.J. Müller, S. Lidin, S.R. de Debiaggi, C.E. Deluque Toro, and A. Fernández Guillermet, Synthesis, Structural Characterization and Ab Initio Study of Cu5 + δIn2 + xSb2-x—A New B8 Related Structure Type, Inorg. Chem., 2012, 51, p 10787-10792
C.E. Deluque, S.B. Ramos, and A.J. Fernández, Elastic Properties and Electronic Structure of the Ni3Sb (cF16) Intermetallic, MRS Proceedings, 2016, 1816, p 1–7.
H. Okamoto, Ni-Sb (Nickel-Antimony), J. Phase Equilib. Diff., 2009, 30(3), p 301-302
R.P. Elliott, Constitution of Binary Alloys, McGraw-Hill, New York, 1965, p 671-672
F.A. Shunk, Constitution of Binary Alloys, McGraw-Hill, New York, 1969, p 553-554
Y. Zhang, Ch Li, Z. Du, and C. Guo, A Thermodynamic Assessment of Ni-Sb system, CALPHAD, 2008, 32, p 378-388
Z. Cao, Y. Takaku, I. Ohnuma, R. Kainuma, H. Zhu, and K. Ishia, Thermodynamic Assessment of the Ni-Sb Binary System, Rare Met., 2008, 27, p 384-392
X.-N. Luo, C. Dong, S.-K. Liu, Z.-P. Zhang, A.-L. Li, L.-H. Yang, and L.-C. Li, Low-Temperature Physical Properties and Electronic Structures of Ni3Sb, Ni5Sb2, NiSb2, and NiSb, J. Chin. Phys. B, 2015, 24(6), p 67201-067201
P.E. Blöchl, Projector Augmented-Wave Method, Phys. Rev. B, 1994, 50, p 17953-17979
G. Kresse and J. Joubert, From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method, Phys. Rev. B, 1999, 59, p 1758-1775
G. Kresse and J. Furthmüller, Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set, Comput. Mater. Sci., 1996, 6, p 15-50
D.M. Ceperley and B.J. Alder, Ground State of the Electron Gas by a Stochastic, Method Phys. Rev. Lett., 1980, 45, p 566-569
J.P. Perdew and Y. Wang, Accurate and Simple Analytic Representation of the Electron-Gas Correlation Energy, Phys. Rev. B, 1992, 45, p 13244-13249
H.J. Monkhorst and J.D. Pack, Special Points for Brillouin-Zones Integrations, Phys. Rev. B, 1976, 13, p 5188-5192
M. Methfessel and A.T. Paxton, High-Precision Sampling for Brillouin-Zone Integration in Metals, Phys. Rev. B, 1986, 40, p 3616-3621
P. Vinet, J.H. Rose, J. Ferrante, and J.R. Smith, Universal Features of the Equation of the State of Solids, J. Phys.: Condensed Matter, 1989, 1, p 1941-1963
C. Deluque Toro, S.R. de Debiaggi, and A.M. Monti, Study of Cohesive, Electronic and Magnetic Properties of the Ni-In Intermetallic System, Phys. B, 2012, 407, p 3236-3239
G. Ghosh, First-Principles Calculation of Phase Stability and Cohesive Properties of Ni-Sn Intermetallics, Metall. Mater. Trans. A, 2009, 40, p 4-23
R. Kolhaas, P. Dünner, and N. Schmitz-Pranghe, Über die Temperaturabhängigkeit der Gitter Parameter von Eisen, Kobalt and Nickel im Bereich Hoher Temperature, Z. Angew. Phys., 1967, 23, p 245-247
G. Alers and J.R. Neighbours, Temperature Dependent Magnetic Contribution to the High Field Elastic Constants of Nickel and Fe-Ni Alloy, J. Phys. Chem. Solids, 1960, 13, p 40-55
D.J. Steinberg, Some Observations Regarding the Pressure Dependence of the Bulk Modulus, J. Phys. Chem. Solids, 1982, 43, p 1173-1175
D.H. Martin, Magnetism in Solids, The MIT Press, Cambridge, 1967, p 10
P. Villars, Pearson’s Handbook, Desk ed., ASM, Materials Park, Ohio, 1997, p 2531
C. Kittel, Introduction to Solid State Physics, 7th ed., Wiley, New York, 1996, p 57
F. Korber and W. Oelsen, The formation enthalpy of binary alloys Fe-Sb, Co-Sb, Ni-Sb, Co-Sn, Ni-Sn, Cu-Sn and Cu-Zn in the cast condition, Mitt. Kaiser-Wilhelm-Inst. Eisenforsch. Düsseldorf, 1937, 19, p 209-219
Acknowledgment
This work was supported by Project PIP 112-20110100814 from CONICET and Project I197 from Universidad Nacional del Comahue.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Deluque Toro, C.E., Ramos, S.B. & Fernández Guillermet, A. Density-Functional-Theory Study of Cohesive, Structural and Electronic Properties of Ni-Sb Intermetallic Phases. J. Phase Equilib. Diffus. 38, 223–230 (2017). https://doi.org/10.1007/s11669-017-0534-y
Received:
Revised:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11669-017-0534-y