
Abstract
Shoot tips from four accessions (IC249250, IC 426442, IC 375976, and IC468878) of Bacopa monnieri (L.) Wettst., a commercially valuable memory revitalizing medicinal plant, were cryopreserved using a vitrification technique. Depending on the genotype, 0 to 20% plant regeneration without intermediary callus was achieved from cryopreserved shoot tips. Genetic stability of plants derived from cryopreserved shoot tips was assessed using biochemical and molecular markers. The regenerated plants from non-frozen controls and cryopreserved shoot tips exhibited morphological similarity to respective parental material when transferred to soil. On the basis of ten random amplified polymorphic DNA (RAPD) and bacoside A content using HPLC analysis, no significant reproducible variation was observed between the controls and in vitro-cryopreserved plants. Thus, after cryopreservation treatment, the regenerated plants exhibited molecular and biochemical genetic stability.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The Indian gene centre is immensely rich with a diversity of medicinal plants including 750 species with established folklore records and proven healthcare benefits in both traditional and modern pharmacopoeias. More than 80% of these bio-resources for industrial use still come through collection from wild stands (Chandel et al. 1996; Sharma and Pandey 2013). The threat of losing valuable gene pools of medicinal plant biodiversity is alarming and requires multiple approaches to prevent the loss. First, the available biodiverse and priority medicinal plants must be catalogued and characterized to promote their domestication and organized cultivation to meet industrial demands. Second, efficient methods must be developed for in-situ and ex-situ conservation of threatened gene pools. The last three decades have witnessed an increased use of in vitro cell and tissue culture techniques for biodiversity conservation of medicinal plants (Sarasan et al. 2006; Sharma and Pandey 2013).
Bacopa monnieri (L.) Wettst. is a popular ancient Indian Ayurvedic herb (‘Brahmi’) used as a nootropic, nervine tonic for neutralizing mental stress, epileptic disorders, and improving memory functions in humans (Anonymous 1988). It is also known to possess adaptogenic, anti-cancer, anti-oxidant, and immunomodulatory properties (Anonymous 1988; Sharma et al. 2007). The clinical efficacy of B. monnieri has been attributed to the presence of several triterpene saponins (bacosides) in its aerial tissues (Deepak et al. 2005). ‘Brahmighritam’, ‘Brahmirasayanam’, Memory Plus, and Mentat are some of the popular brand names of Bacopa-based drug preparations in the Indian herbal trade. As a predominantly vegetatively propagated species with a restricted habitat, B. monnieri has a narrow genetic base for herb and metabolite-contributing traits. Despite having abundant growth, large quantities of material are required to meet pharmaceutical demands due to a low drug content. Consequently, the intervention of biotechnological tools will be beneficial for conservation and genetic improvement. This herb is amenable to in vitro micropropagation and conservation techniques (Sharma et al. 2007, 2012, 2016). A single-step medium-term conservation protocol was successfully used to conserve over 20 accessions of B. monnieri from various ecogeographic regions of India (Sharma et al. 2007, 2016). For long-term conservation, cryogenic storage is the only method available. A basic protocol for cryopreservation of shoot tips using a vitrification technique has also been previously reported (Sharma et al. 2011). This protocol was developed using one accession. From the point of view of a conservationist, maintenance of genetic stability of plants regenerated after conservation is important to ensure sustainable use of conserved resources in the future.
The present study was, therefore, undertaken with the two objectives: (i) to assess the feasibility of cryopreserving shoot tips in three new accessions using a vitrification procedure and (ii) to monitor the genetic stability of plants regenerated from cryopreserved shoot tips using biochemical and molecular markers. This is the first report of genetic stability assessment of cryopreserved shoot tips in this important medicinal herb.
Material and Methods
Plant material
A total of four accessions of B. monnieri (IC249250, IC426442, IC375976, and IC468878; hereafter referred to as BC1, BCM, BCJ, and BC7, respectively) conserved in the In Vitro Genebank at ICAR-National Bureau of Plant Genetic Resources (ICAR-NBPGR), New Delhi, India, were used in the present study. The experimental material consisted of shoot tips (~1.0 mm in length) obtained from 24-wk-old proliferating shoot cultures. Stock cultures were multiplied on MS medium (Murashige and Skoog 1962) with 0.2 mg L−1 6-benzyladenine (BA) (Sigma-Aldrich®, St. Louis, MO) and 3% (w/v, 0.08 M) sucrose (Hi-Media®, Mumbai, India) at pH 5.8, henceforth, referred to as shoot multiplication (SM) medium. The medium was gelled using 0.8% (w/v) bacteriological agar (Hi-Media®) and autoclaved at 121°C and 1.06 kg cm−2 for 15 min. For multiplication of the cultures, single-node cuttings were subcultured onto SM medium every 4 to 8 wk under standard culture room conditions of 25 ± 2°C and a 16-h photoperiod (30 μmol m−2 s−1) provided by 30-W cool-white fluorescent tubes (Philips, Mumbai, India) (Sharma et al. 2007).
Cryopreservation
Cryopreservation of shoot tips was done following a slightly modified method described by Sharma et al. (2011) using BC1. Shoot tips (ca. 1.0 mm) dissected from 24-wk-old shoot cultures were precultured for 2 d on MS medium supplemented with 0.3 M sucrose, before dehydrating with plant vitrification solution 2 (PVS2) at 0°C. The PVS2 solution consisted of 30% (v/v, 3.26 M) glycerol (Sigma-Aldrich®), 15% (w/v, 2.42 M) ethylene glycol (EG) (Sigma-Aldrich®), 15% (v/v, 1.9 M) dimethylsulphoxide (DMSO) (Sigma-Aldrich®), and 0.4 M sucrose dissolved in pH 5.8 liquid MS medium (Sakai et al. 1990). In each experimental treatment, ten shoot tips were precultured on 10 ml of medium in a Petri dish (Hi-Media®) (60-mm diameter) at 25°C for 2 d. The sucrose-precultured shoot tips were placed in sterile 1.0ml Nunc™ cryovials (Thermo Fisher Scientific® Roskilde, Denmark) containing 1.0 ml PVS2 at 0°C for 30 min. After the 30 min in PVS2, cryovials containing shoot tips were quickly plunged into liquid nitrogen (LN) for at least 1 h. Thawing was done by stirring the cryovials in a water bath at 45°C for 2 min. After thawing, the PVS2 solution was removed using a Pasteur pipette and shoot tips were rinsed five times over a 20-min period with a deloading solution of 1.2 M sucrose dissolved in pH 5.8 liquid MS medium. Shoot tips were then transferred onto sterile filter paper strips. For regrowth, shoot tips were placed on 10 ml of SM medium in Petri dishes. The control shoot tips (precultured and PVS2-treated but not frozen) were recultured in a similar manner. After 1-wk incubation in the dark, these were transferred to the light.
All the cultures were incubated at 25 ± 2°C at a 16-h photoperiod and a light intensity of 30 μmol m−2 s−1, provided by 30-W cool white fluorescent tubes (Philips). After 8 wk, surviving shoot tips were transferred onto the same medium in culture tubes (Qualigens Fine Chemicals Pvt., Ltd., Mumbai, India) (150 mm × 25 mm) for multiplication and root formation. Ten shoot tips were tested for each of two replications in every experiment. The experiment was repeated at least three times. Survival of the cultures was assessed by visual examination and by the ability of the explants to regrow on fresh SM. Regeneration was recorded as the percentage of the total number of shoot tips that formed normal shoots 8 wk after plating.
For ex vitro transfer, 12-wk-old plantlets were transferred to pots for hardening as described by Sharma et al. (2007). The plants were hardened in plastic pots (7.5-cm diameter) containing autoclaved (20 min at 121°C and 1.06 kg cm−2 for 15 min) vermiculite (Glassil, New Delhi, India) moistened with 10–15-ml 1/2 MS major salt solution (Hi-Media®) and allowed to grow at 25 ± 2°C. The plantlets were covered with transparent perforated polythene bags (Ambay Biotech, Delhi, India) for 2–3 wk to maintain high humidity (75 to 85%) and subsequently exposed to ex vitro conditions by removing the polythene bags after 3 wk. The 3-wk-old hardened plantlets were transplanted to garden soil [10:1 sandy soil:FYM (locally produced farm yard manure)] in earthen pots (30-cm diameter) and allowed to grow in a net house (50 mesh in−2,~50% shade) under natural light.
Genetic stability assessment
For assessment of genetic stability, plants from four different treatments were used. These included (i) plantlets obtained after post-thaw regrowth, (ii) plantlets obtained from in vitro multiplied material (controls) and (iii) and (iv) 6-mo-old plants grown in pots after transfer of hardened plantlets from treatment as described above (i) and (ii), respectively. The age of plantlets regenerated from control and cryopreserved shoot tips was the same at the time of the ex vitro transplanting.
The genetic stability of in vitro-cryopreserved plants was studied using molecular (random amplified polymorphic DNA (RAPD) markers) and biochemical methods (bacoside A estimation) (Sharma et al. 2012).
RAPD analyses Genomic DNA extraction
Total genomic DNA was extracted from 1 g leaves each of the in vitro cultures and ex vitro grown regenerants using the CTAB (cetyl-trimethyl ammonium bromide) (Amresco®, Solon, OH) method described by Saghai-Maroof et al. (1984). After RNAse (Thermo Fisher Scientific®, Waltham, MA) treatment, the DNA concentrations were estimated using NanoDrop® 1000 (Thermo Fisher Scientific®). The DNA samples were stored at −20°C until used. Working solutions of genomic DNA (5 ng μl−1) were prepared after dilution with Tris–EDTA (Amresco®) buffer (0.1 mM Tris–HCl, 0.01 mM EDTA at pH 8.0) and stored at −20°C for subsequent use in RAPD analyses.
PCR reactions were carried out in a DNA thermal cycler (G-Storm™ Thermal Cycler Systems, Somerset, UK). Each 25μl reaction mix contained 1× PCR reaction buffer (10 mM Tris–HCl, 50 mM KCl, at pH 8.8); 1.5 mM MgCl2; 1 U of Taq DNA polymerase; 200 μM each of dATP, dTTP, dCTP, and dGTP (Thermo Fisher Scientific®); 0.4 μM of primer (Qiagen® Operon, Inc., Alameda, CA), and approximately 25 ng of template DNA.
RAPD analyses
A total of 30 primers (Qiagen® Operon, Inc.) were screened for RAPD analysis. Of these, ten primers showing good amplification were used for further analyses (Table 1). The PCR conditions were as follows: initial extended step of denaturation at 94°C for 4 min followed by 39 cycles of denaturation at 94°C for 1 min, primer annealing at 37°C for 1 min, primer elongation at 72°C for 2 min, followed by an extended elongation step at 72°C for 7 min. PCR products were stored at 4 °C until electrophoresis.
Reaction products were mixed with 5 μl of 6× loading dye (0.03% (w/v) bromophenol blue, 0.03% (w/v) xylene cyanol FF, and 60% (w/v) glycerol) (Sigma-Aldrich®) using a micropipette. The mixture was electrophoresed on 1.4% (w/v) agarose (Lonza, Rockland, ME) gels (prepared in 1× TBE buffer) at 80 V (BIO RAD® subcell GT, Hercules, CA) for 3 h. A 100-bp DNA ladder plus (Thermo Fisher Scientific®) was run next to the amplified products to determine their approximate size. The amplified fragments were visualized using a gel-documentation system (Gel Doc Mega, Biosystematica, Llandysul, Wales, UK).
For genetic stability assessment, in each treatment, 48 in vitro plantlets and 12 ex vitro-transferred plantlets were analysed. For each marker system, the strong amplified fragments were scored for their presence (1), absence (0), and missing data (9). Genetic similarity values were computed based on Jaccard’s coefficient (Jaccard 1908) using the NTSYS-PC version 2.11a software (Rohlf 2002). Cluster analyses were performed based on similarity matrices for combined scores of RAPD using unweighted pair group method with arithmetical averages (UPGMA) (Sneath and Sokal 1973).
Biochemical analysis
For the estimation of bacoside A, shoots of in vitro grown plantlets and ex vitro grown plants (same as those used for molecular analysis) were harvested and lyophilized. The secondary metabolite content was assessed using HPLC following the modified procedure of Deepak et al. (2005). For plotting of the standard curve, bacosides standard was obtained from ChromaDex® (Irvine, CA).
Approximately 1 g of lyophilized sample was pulverized in a grinder (FOSS, Hamburg, Germany). The powdered sample (100 mg) was extracted with methanol (E Merck, Darmstadt, Germany) by reflux (3 × 2 ml, 10 min each) at 70°C. The extracts obtained after each extraction were pooled. The solvent was dried on a rotary evaporator and then the volume was made up to 2 ml using HPLC grade methanol (E Merck). The sample was injected into a HPLC column (Phenomenex, Torrance, CA) after filtering through a 0.22μm Millipore filter (E. Merck). Ten microliter of the standard per sample was injected into a Waters HPLC system (Waters® Corporation, Milford, MA) consisting of a M-600 E quaternary gradient pump, 717 autosampler, Nova-Pak® Reverse phase C-18 (3.9 × 150 mm) column, and photo diode array (PDA) detector. The separation of the bacosides was obtained on a C-18 column using an isocratic run of water (A) and acetonitrile (E Merck) (B) in the ratio of 67.5:32.5 at 1.6 ml min−1 as mobile phase. The peaks were monitored at 200 nm. The peak identity was confirmed by comparing the retention time of the peaks and also by PDA spectrum.
Results and Discussion
Cryopreservation is the most appropriate strategy for long-term conservation of vegetatively propagated plants and those with recalcitrant seeds. It is evident from the literature (Chandel et al. 1996; Sarasan et al. 2006; Sharma and Pandey 2013) that most of the work focused on in vitro propagation has resulted in varying degrees of success in a large number of medicinal plants. However, in medicinal plants, there are limited publications related to in vitro conservation and few reports on in vitro cryopreservation (Sharma and Pandey 2013). Over the last two decades, although some success has been achieved with cryopreservation and plantlet regeneration from in vitro shoot tips of B. monnieri, Dioscorea spp., Holostemma ada-kodien, Picrorhiza kurroa, and Rauvolfia serpentina (Mandal and Dixit 2000; Sharma and Sharma 2003; Ray and Bhattacharya 2008; Decruse et al. 2004; Sharma et al. 2011), genetic stability after cryopreservation is reported only in Dioscorea deltoidea (Dixit-Sharma et al. 2005).
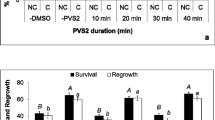
Experiments were conducted to test the feasibility of a previously developed method of cryopreservation (Sharma et al. 2011) on three more accessions of B. monnieri (BCM, BCJ, and BC7) collected from different ecogeographic regions. In all the four accessions tested (including the earlier reported BC1), shoot tips isolated from 24-wk-old cultures and precultured on 0.3-M sucrose medium exhibited 0 to 20% post-thaw regeneration following PVS2 dehydration at 4°C (Fig. 1 A, B). As evident from Fig. 1 A, shoot tips failed to exhibit post-thaw survival in BC7, one of the accessions tested. The non-frozen controls exhibited 80 to 100% regrowth. Upon transfer to SM medium, multiple shoots were produced and normal growth was observed (Fig. 1 C). The morphology of the cryopreserved shoots was comparable to that of non-frozen in vitro shoots. Subsequent multiplication of cryopreserved shoots resulted in shoot multiplication with an average of 18 to 22 shoots explant−1 in 8 wk. Plantlets regenerated from cryopreserved shoot tips were successfully transferred to pots with 100% plant survival (Fig. 1 D).

Cryopreservation of shoot tips in four accessions of Bacopa monnieri (A–D). (A, B) Post-thaw regrowth, (C) shoot multiplication of cryopreserved shoot tips, and (D) ex vitro establishment of regenerated shoot tips.
Studies have shown that genotype-dependent differences with respect to post-thaw recovery are common (Agrawal et al. 2011; Senula and Kellar 2011). In the present study, regrowth of cryopreserved shoot tips occurred without transitory callus formation (Fig. 1 B), which is similar to earlier results (Sharma et al. 2011). It is an important observation while developing a cryopreservation method as direct regeneration suggests a low risk of genetic instability. All the regenerated plants appeared similar to the control plants. To further confirm the genetic stability of the conserved plants, molecular and biochemical analyses were also conducted.
On the basis of RAPD analysis, there was no variation among controls and plants regenerated from cryopreserved shoot tips in all the three accessions tested (BC1, BCM, and BCJ). Representative RAPD profiles of plantlets derived from cryopreserved shoot tips and ex vitro transferred plants after cryopreservation and controls are shown in Fig. 2. On the basis of RAPD analyses, there was no variation observed among controls and plants regenerated from cultures. A minimum number of five bands were amplified by primers OPA-8 (Fig. 2) and OPA-07, and a maximum number of seven bands were amplified by primer OPA-03. The average number of products amplified per primer was 5.3, and out of the total amplicons generated (53 bands), 43 were found to be monomorphic. The size of products varied from 200 to 1200 bp. A total of 1316 amplification products were produced by RAPD markers computed on the basis of (the number of plants) × (the number of bands produced by ten primers). Of these 1316 bands, 1204 were monomorphic (91.14%), indicating not a very significant variation was observed. Ten RAPD primers used in this study generated 53 scorable amplification analyses and indicated the maintenance of genetic stability in conserved cultures. RAPD and ISSR markers have been used to assess genetic stability of micropropagated plants (Ceasar et al. 2010) and in vitro conserved plants of B. monnieri (Sharma et al. 2012). In the present study, RAPD marker analyses indicated maintenance of genetic stability in cryopreserved cultures.

Representative RAPD profiles generated with OPA-08 primer generated from cultures and ex vitro transferred plantlets regenerated from controls and cryopreserved shoot tips in three accessions of Bacopa monnieri. Cultures—BC1 (1–3 control, 5–7 cryopreserved); BCM (9–11 control, 13–15 cryopreserved) and BCJ (17–19 control, 21–23 cryopreserved). Ex vitro plants—BC1 (4 control, 8 cryopreserved); BCM (12 control, 16 cryopreserved) and BCJ (20 control, 24 cryopreserved). M denotes Gene Ruler™ 100-bp DNA ladder.
Retention of biosynthetic ability, especially bacoside A in cryopreserved plants is extremely important in B. monnieri, as bacoside A is shown to be responsible for the pharmacological properties including the memory-facilitating action of ‘brahmi’. Analysis of bacoside A content in cultures after cryopreservation showed that it remained similar to respective controls (Fig. 3), thereby indicating maintenance of genetic stability in the cryopreserved cultures in all the three responding accessions. In one of the accessions tested (BCM), in vitro cultures exhibited a higher content of bacoside compared to ex vitro transferred plants (Fig. 3). This result was similar to the earlier report on in vitro conserved cultures (Sharma et al. 2012). On the basis of morphological, RAPD markers, and bacoside A analyses, it was clear that the genetic stability of cryopreserved cultures was maintained in all three responding accessions.

Bacopa monnieri: Bacoside A content in plantlets obtained after post-thaw regrowth of shoot tips and their controls in different genotypes and ex vitro transferred plantlets (6-mo-old) regenerated from cryopreserved and control shoot tips in genotype BCM.
Conclusions
Due to the low post-thaw recovery, further research is required to standardize various steps of cryopreservation, which generally is genotype-specific. Cryopreservation, though with low recovery, has been proven feasible in three new accessions of Bacopa. No difference in RAPD profiles and bacoside A content of control and cryopreserved regenerants confirmed the genetic stability after cryopreservation. Research continues to improve the recovery rates. However, applicability of the developed protocol in three accessions is important research towards reaching a high multiplication rate in surviving shoot tips.
References
Agrawal A, Tyagi RK, Goswami R, Uma S, Saraswathi MS, Durai P (2011) Cryobanking of banana (Musa sp.) germplasm in India: evaluation of agronomic and molecular traits of cryopreserved plants. Acta Hortic 908:129–138
Anonymous (1988) The wealth of India: raw materials, Vol 2. Council of Scientific and Industrial Research, New Delhi
Ceasar AS, Maxwell SL, Prasad KB, Karthigan M, Ignacimuthu S (2010) Highly efficient shoot regeneration of Bacopa monnieri (L.) using a two stage culture procedure and assessment of genetic integrity of micropropagated plants by RAPD. Acta Physiol Plant 32:442–452
Chandel KPS, Shukla G, Sharma N (1996) Biodiversity in medicinal and aromatic plants in India. National Bureau of Plant Genetic Resources, New Delhi
Decruse SW, Seeni S, Nair GM (2004) Preparative procedures and culture media effect on the success of cryostorage of Holostemma anmulare shoot tips. Plant Cell Tissue Organ Cult 76:179–182
Deepak M, Sangli GK, Arun PC, Amit A (2005) Quantitative determination of major saponin mixture Bacoside A in Bacopa monnieri. Phytochem Anal 16:24–29
Dixit-Sharma S, Ahuja-Ghosh S, Mandal BB, Srivastava PS (2005) Metabolic stability of plants regenerated from cryopreserved shoot tips of Dioscorea deltoidea—an endangered medicinal plant. Scientia Hortic 105:513–517
Jaccard P (1908) Nouvelles recherches sur la distribution florale. Bull Soc Vaudoise Sci Nat 44:223–270
Mandal BB, Dixit S (2000) Cryopreservation of shoot-tips of Dioscorea deltoidea Wall.—an endangered medicinal yam, for long-term conservation. IPGRI Newsl Asia Pac Ocean 33:23
Murashige T, Skoog F (1962) A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15:473–497
Ray A, Bhattacharya S (2008) Cryopreservation of in vitro grown nodal segments of Rauvolfia serpentina by PVS2 vitrification. CryoLetters 29:321–328
Rohlf FJ (2002) NTSYSpc: numerical taxonomy system, ver. 2.1. Exeter Publishing Ltd., Setauket
Saghai-Maroof MA, Soliman KM, Jorgensen RA, Allard RW (1984) Ribosomal DNA spacer-length polymorphisms in barley: Mendelian inheritance, chromosomal location, and population dynamics. Proc Natl Acad Sci U S A 81:8014–8018
Sakai A, Kobayashi S, Oiyama I (1990) Cryopreservation of nucellar cells of navel orange (Citrus sinensis Osb. Var. brasiliensis Tanaka) by vitrification. Plant Cell Rep 9:30–33
Sarasan V, Cripps R, Ramsay MM, Atherton C, McMichen M, Prendergast G, Rowntree JK (2006) Conservation in vitro of threatened plants—progress in the past decade. In vitro Cell Dev Biol Plant 42:206–214
Senula A, Kellar ERJ (2011) Cryopreservation of mints—routine application in a genebank, experience and problems. Acta Hortic 908:467–472
Sharma N, Pandey R (2013) Conservation of medicinal plants in tropics. In: Normah MN, Chin HF, Reed BM (eds) Conservation of tropical plant species. Springer, New York, pp 437–487
Sharma N, Satsangi R, Pandey R (2011) Cryopreservation of shoot tips of Bacopa monnieri (L.) Wettst. by vitrification technique. Acta Hortic 908:283–288
Sharma N, Satsangi R, Pandey R, Singh R, Kaushik N, Tyagi RK (2012) In vitro conservation of Bacopa monnieri (L.) using mineral oil. Plant Cell Tissue Organ Cult 11:291–301
Sharma N, Satsangi R, Pandey R, Vimala Devi S (2007) In vitro clonal propagation and medium term conservation of Brahmi [Bacopa monnieri (L.) Wettst.] J Plant Biochem Biotech 16:139–144
Sharma N, Sharma B (2003) Cryopreservation of shoot tips of Picrorrhiza kurroa Royle ex Benth., an indigenous endangered medicinal plant through vitrification. CryoLetters 24:181–190
Sharma N, Singh R, Pandey R (2016) In vitro propagation and conservation of Bacopa monnieri L. In: Jain M (ed) Protocols for in vitro cultures and secondary metabolite analysis of medicinal and aromatic plants, second edition, methods in molecular biology, Vol 1391. Springer Science + Buisness Media, New York, pp 153–171. doi:10.1007/978-1-4939-3332-7_11
Sneath PHA, Sokal RR (1973) Numerical taxonomy: the principles and practice of numerical classification. WH Freeman, San Francisco
Acknowledgements
We thank the Director, ICAR-NBPGR, New Delhi, for facilities and encouragement. Special thanks to Dr. Barbara Reed, Oregon State University, for academic advice.
Author information
Authors and Affiliations
Corresponding author
Additional information
Editor: Barbara Reed
Rights and permissions
About this article

Cite this article
Sharma, N., Singh, R., Pandey, R. et al. Genetic and biochemical stability assessment of plants regenerated from cryopreserved shoot tips of a commercially valuable medicinal herb Bacopa monnieri (L.) Wettst. In Vitro Cell.Dev.Biol.-Plant 53, 346–351 (2017). https://doi.org/10.1007/s11627-017-9826-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11627-017-9826-5