
Abstract
This study demonstrated that somatic embryogenesis and de novo shoot organogenesis-based systems of root-derived Lisianthus (Eustoma grandiflorum) explants can be alternatively induced by exogenous supply of auxin or cytokinin. Somatic embryogenesis was observed when root explants were cultured in the dark on Murashige and Skoog-based medium supplemented with 10 μM 2,4-dichlorophenoxyacetic acid (2,4-D). Somatic embryos were differentiated by transferring embryonic calluses to an embryo conversion phase medium containing 2 μM 6-benzyladenine (BA) to promote full plantlet development. Regarding de novo shoot organogenesis, the addition of 4 μM of either BA or zeatin was the most effective treatment for inducing adventitious shoot buds. A detailed histological characterization of somatic embryogenesis and de novo shoot organogenesis showed that both morphogenetic processes shared the same cellular origin. The formation of somatic embryos and adventitious shoot buds occurred through the reactivation of pericycle and vascular parenchyma cells into proembryos and meristemoids, respectively, which consisted of meristematic cells with similar characteristics. These results provide further evidence of optimization of in vitro propagation as a useful approach to improve this important ornamental species.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Cut flowers constitute the main segment in the global ornamental plant market. Based on a remarkable geographic expansion and recent scientific and technological advances in cultivation and logistics, this segment has been growing sharply in the floriculture trade (Rabobank 2015). As one of the major ornamental cut flowers, Lisianthus (E. grandiflorum) has been increasingly commercialized over the years as well as gaining popularity due to its eye-catching “rose-like” flowers, long post-harvest duration, and diversity of cultivars exhibiting an attractive range of colors (Harbaugh 2007).
Tissue culture-based techniques applied to ornamentals have become a growing trend relevant for commercial laboratories and biofactories. Micropropagation and regeneration systems have been established and constantly improved through long-term research on in vitro culture of Lisianthus (Semeniuk and Griesbach 1987; Ruffoni et al. 1990; Nhut et al. 2006; Wan et al. 2009). Among the in vitro regeneration strategies, systems that rely on de novo shoot organogenesis and somatic embryogenesis have been widely used (Ruffoni and Bassolino 2016). In both cases, the morphogenetic pathways depend primarily on plant growth regulators present in the culture medium and on the explant source.
Molecular signaling by plant growth regulators added to culture media triggers the regeneration process, which can occur through two distinct cellular mechanisms: (i) reactivation of plant body somatic cells or relatively undifferentiated cells or (ii) reprogramming differentiated somatic cells (Ikeuchi et al. 2016). Manipulation of the in vitro conditions, together with the totipotency of plant cells, can also allow both regeneration routes to become induced alternatively or simultaneously from the same explant source. A system in which both regeneration pathways can be induced from the explants provides several opportunities to better understand plant development and to apply it in breeding programs.
The background literature on other ornamentals (Mikuła and Rybczyński 2001; Fiuk and Rybczyński 2007a; Parveen and Shahzad 2011; Wu et al. 2011; Sahai et al. 2010) shows that the use of root segments as explant sources has often been reported and suggested because of the high regeneration potential, easy maintenance, and in vitro manipulation (Vinocur et al. 2000). In the model species Arabidopsis, root explants have been used to elucidate the cellular and molecular mechanisms involved in in vitro morphogenesis, primarily due to the high regeneration capacity of the explants (Ogas 1997; Atta et al. 2009; Sugimoto and Meyerowitz 2013; Motte et al. 2014; Shemer et al. 2015). In vitro regeneration from Lisianthus root explants has been reported previously (Furukawa et al. 1990), but only the neoformation of shoot meristems has been described.
The present study describes a new, efficient method to induce somatic embryogenesis and de novo shoot organogenesis by reactivating pericycle cells in Lisianthus root explants. This method was demonstrated to be useful for rapid Lisianthus micropropagation and could facilitate the development of biotechnological tools to study such species.
Materials and Methods
Plant material—obtaining root explants
Seeds of E. grandiflorum cv. ABC 2-3 Rose (Ball Horticultural do Brasil, Holambra, Brazil) were aseptically disinfected by immersion in 70% (v/v) ethanol for 60 s followed by immersion for 15 min in a non-diluted commercial sodium hypochlorite solution (2.5% active chlorine; Super Globo Química®, Contagem, MG, Brazil) containing 0.01% (v/v) Tween® 20 (Sigma-Aldrich®, St. Louis, MO). The disinfected seeds then were rinsed for 5 min three times with autoclaved deionized water. The seeds were then placed in test tubes (25 mm × 150 mm, Vidrolabor, Paulínia, Brazil) containing 20 mL of half-strength Murashige and Skoog (1962) basal salt solution (PhytoTechnology Laboratories®, Lenexa, KS), MS vitamin complex (Sigma-Aldrich®), 20 g L−1 sucrose (PhytoTechnology Laboratories®), 100 mg L−1 myo-inositol (Sigma Aldrich®), and 6 g L−1 granulated agar (Merck®, Darmstadt, Germany). The pH of the medium was adjusted to 5.7 ± 0.1 prior to autoclaving at 121°C and 152 kPa. The cultures were placed in a growth chamber at 27 ± 2°C, with a 16-h photoperiod, and an irradiance of 36 μmol m−2 s−1 provided by two fluorescent lamps (20 W, HO, Osram®, São Paulo, Brazil) for 30 d.
Somatic embryogenesis induction
To induce embryonic culture, 30-d-old root segments isolated from previously in vitro-germinated seedlings were used. The roots were excised and divided into fragments of approximately 10 mm in length. Neither the root apex nor the segment adjacent to the hypocotyl was used. Five root segments were placed in 90 × 15 mm polystyrene Petri dishes (J. Prolab®, São José dos Pinhais, Brazil) containing basal MS salts, 20 g L−1 sucrose, and different 2,4-dichlorophenoxyacetic acid (2,4-D) concentrations (i.e., 0, 10, 20, 30, and 40 μM). Unless otherwise stated, all plant growth regulators were purchased from Sigma-Aldrich®. During the embryonic callus induction phase, the material was kept in the dark inside a growth chamber at 27 ± 2°C for 40 d. A completely randomized design comprising five treatments (0, 10, 20, 30, and 40 μM 2,4-D) and eight replicates was used. Both induction frequency (percentage of explants with embryonic callus) and oxidation rate (percentage of explant oxidation) were evaluated. The acquired data were submitted to a linear regression analysis model, and data that presented a significant effect as indicated by F test at 1% and 5% significance were included in the equation.
Differentiation and conversion of somatic embryos
After 40 d in the induction phase, the calluses obtained from MS medium supplemented with 10 μM 2,4-D (the treatment that showed the best results after the embryonic culture induction phase) were transferred to somatic embryo differentiation medium. This medium consisted of MS medium supplemented with 20 g L−1 sucrose and different 6-benzyladenine (BA, 2 or 4 μM) or gibberellic acid (GA3, 1.5 or 3.0 μM) concentrations. Except for GA3, all other plant growth regulators used were added to the medium pre-autoclaving. Embryonic cultures were kept in the same growth chamber conditions as described above. In this phase, cultures were evaluated 40 d after the embryonic calluses were transferred to the embryo conversion medium. A randomized block design comprising five treatments [control (i.e., without plant growth regulator), 2 μM BA, 4 μM BA, 1.5 μM GA3, and 3 μM GA3] was used, for a total of ten treatments with four replicates (Petri dishes with four embryonic calluses). The following parameters were evaluated: conversion rate (percentage of explants with embryos), the number of formed embryos (globular stage, torpedo, cotyledon, and germinated embryos), and the total weight of somatic embryo biomasses (fresh and dry weights).
De novo shoot organogenesis induction
Root segments (10–20 mm long) obtained from 30-d-old in vitro-germinated seedlings were placed horizontally in 90 mm × 15 mm Petri dishes (J. Prolab®) containing 25 mL basal MS salts supplemented with different BA, Zeatin (ZEA), or Thidiazuron (TDZ) (0, 4, 8, or 12 μM) concentrations, at pH 5.7 ± 0.1. Plates were sealed with transparent PVC film (Goodyear®, São Paulo, Brazil) and then kept in the same growth chamber conditions as described above for 40 d. The experimental design was completely randomized and consisted of 13 treatments: four cytokinins in the de novo shoot organogenesis induction phase (BA, KIN, TDZ, and ZEA) at four different concentrations of 0, 4, 8, and 12 μM. Each treatment was composed of three replicates (Petri dishes with four explants). The following parameters were evaluated: maturation rate (percentage of explants with mature embryos) and number of formed shoots.
After 40 d, shoots were separated from the initial explants and transferred to flasks (four shoots per flask) containing MS medium without plant growth regulators. Flasks were sealed with rigid polyethylene lids with two holes covered with a home-made porous membrane (as described by Saldanha et al. 2012), and remained under these conditions for an additional 50 d. After this period, rooted plants were washed in running water to remove excess culture medium and transferred to 300-cm3 plastic cups (one shoot per cup) filled with a commercial substrate (Plantmax®, Paulínia, Brazil). Each cup was covered with a 10 × 20 cm transparent polyethylene bag (PSG Embalagens, Porto Alegre, Brazil) to prevent excessive water loss from the plantlets after their transfer to the ex vitro environment. Relative humidity was gradually reduced to ambient levels by means of lateral cuts (average 1 cm) made in the polyethylene bag, on a weekly basis. Bags were completely removed by the end of the third week. Plants were kept under greenhouse conditions throughout the acclimatization phase.
Microscopy
For structural characterization, embryonic calluses obtained from the treatment using induction and conversion media supplemented with 10 μM 2,4-D and 2 μM BA, respectively, were collected every 10 d during the induction process and at the end of the conversion cycle. For structural characterization of de novo shoot organogenesis, samples from the treatment with 4 μM ZEA were collected at the fifth day of culture and then every 5 d until the 30th day.
Samples were fixed in Karnovsky’s solution (Karnovsky 1965) and dehydrated with increasing serial ethanol concentrations [50, 70, 80, 90, and 2 × 100% (v/v)], with incubation periods of 10 min. The samples were then embedded in methacrylate resin (Historesin®, Leica Instruments, Heidelberg, Germany) according to the manufacturer’s recommendations. Five-micrometer-thick cross and longitudinal sections were produced using an automatic advance rotary microtome (RM2155, Leica Microsystems Inc., Buffalo Grove, IL) and stained with toluidine blue (pH 3.2) (O’Brien and McCully 1981). Images were captured using an Olympus AX70TRF microscope (Olympus Optical, Tokyo, Japan) equipped with a U-Photo Camera System (Spot Insight Color 3.2.0, Diagnostic Instruments Inc., Sterling Heights, MI).
For micromorphological characterization, fixed samples were dehydrated in an acetone series and dried with CO2 (Bozzola and Russell 1992) using a critical point-drier device (CPD model 030, BalTec Inc., Balzers, Liechtenstein). Next, samples were covered with a 10-nm colloidal gold layer in a metallizer (Model FDU 010, BalTec Inc.), coupled to sputter coating equipment (Model SCA-010, BalTec Inc.). The samples were observed and images taken using a scanning electron microscope (LEO 1430 VP, Zeiss, Cambridge, UK), at 5 kV accelerating voltage.
Statistical analysis
The data were submitted to analysis of variance followed by Tukey’s test at a significance level of 5% to compare mean values. Analyses were performed in the R software, version 3.0.3 (R Core Team 2014), with the assistance of ExpDes package, version 1.1.2 (Ferreira et al. 2013) and Easyanova package (Arnhold 2014).
Results
Low 2,4-D concentrations led to somatic embryogenesis pathway in root-derived Lisianthus explants
Root explants showed a response gradient when cultured in semi-solid medium with increasing 2,4-D concentrations (Fig. 1 a–d). Explants cultured on MS medium devoid of plant growth regulators showed no morphogenetic responses (data not shown). In 2,4-D-supplemented treatments, the percentages of explants with induced somatic embryogenesis were fitted to a decreasing linear model (Fig. 1 e). The highest induction percentage (97.5%) was observed for the medium supplemented with 10 μM 2,4-D (Fig. 1 e). The whole explants showed typical globular, smooth embryonic-like structures (Fig. 1 a).

Effect of 2,4-dichlorophenoxyacetic (2,4-D) concentration on somatic embryogenesis induction from Lisianthus root explants. General view of root explants cultured in induction media supplemented with 10 μM (a), 20 μM (b), 30 μM (c), or 40 μM (d) of 2,4-D. Arrowheads indicate the presence of pro-embryonic structures. Somatic embryogenesis induction frequency (e) and oxidation rate (f). Bars = 0.5 cm.
Embryonic structures were also observed for 20 and 30 μM 2,4-D, although less frequently and primarily in explant sectioning regions (Fig. 1 b, c, respectively). However, at 40 μM 2,4-D, embryonic structures were rarely observed (Fig. 1 d). The explant oxidation rates were fitted to a rising linear model, proportional to 2,4-D concentration in the induction medium (Fig. 1 f).
Use of GA and BA to promote differentiation of Lisianthus somatic embryos
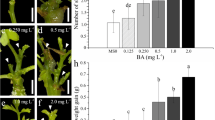
For somatic embryo differentiation, root explants previously cultured on induction medium supplemented with 10 μM 2,4-D were transferred to media either lacking plant growth regulators or supplemented with GA3 or BA and exposed to light. Once transferred, the yellowish-white embryonic structures in the explants turned green (Fig. 2 a, b). Somatic embryo differentiation was observed only in the BA- or GA3-supplemented treatments. Somatic embryos at different developmental stages were observed in the explants (Fig. 2 c–e), with no significant difference in the conversion rates between the treatments. The largest numbers of somatic embryos (35 and 33) were observed in treatments supplemented with 4 μM BA or 1.5 μM GA3, respectively, although the latter was not significantly different from that supplemented with 2 μM BA (Fig. 2 f). The highest accumulation of somatic embryo fresh and dry weights (Fig. 2 g, h) was obtained for 2 or 4 μM BA. In the treatment supplemented with 3 μM GA3, the average number of somatic embryos and the embryo mass accumulation were significantly reduced (Fig. 2 f–h).

Somatic embryo differentiation obtained from Lisianthus root explants. General view of embryonic calluses obtained on embryo conversion medium supplemented with 4 μM BA (a) and 1.5 μM GA3 (b). Somatic embryos at globular (c), torpedo (d), and cotyledonary stages (e). Number of somatic embryos (f), total somatic embryo fresh weight (g), and total somatic embryo dry weight (h). Bars = 0.1 cm.
De novo shoot organogenesis from root-derived Lisianthus explants was influenced by the type and concentration of cytokinin
Both induction and regeneration of adventitious buds were affected by the type and concentration of cytokinins present in the culture medium. Root explants cultured on plant growth regulator-free MS medium and medium supplemented with kinetin or the highest ZEA concentration (12 μM) showed no morphogenetic responses after 30 d of culture (Table 1). The highest response by root explants was observed at 4 or 8 μM BA, 4 μM ZEA, or higher TDZ concentrations (8 to 12 μM), with no significant difference among these treatments. Organogenic responses were observed in the sectioned area and over the whole explant (Fig. 3). Most root segments showed slight swelling in the first week of culture, which subsequently resulted in the disruption of the explant outer layers (Fig. 3 a, b), with no significant difference observed among the treatments. After 25–30 d of culture, adventitious buds emerged directly from the explants (Fig. 3 c, d). The largest number of shoots was obtained in explants cultured at the lowest cytokinin (BA, ZEA, or TDZ) concentration (4 μM). However, in explants cultured in the presence of higher BA or TDZ concentrations (8 or 12 μM), the root segments also showed regeneration of adventitious buds, but at significantly lower rates (Table 1). Buds regenerated through de novo shoot organogenesis (Fig. 3 e) were excised and transferred to a medium containing the standard concentrations of MS salts and vitamins described by Murashige and Skoog (1962). Plantlets were transferred to Plantmax® substrate and successfully acclimatized (Fig. 3 f).

De novo shoot organogenesis from Lisianthus root explants. Explants at 0 (a) and 15 d (b) of induction. Formation of organogenic structures after 25 d (c) and 30 d (d) of culture. Root explants with several buds after 40 d of culture (e). Regenerated plant acclimatized (e). Bars = 0.5 (a–e) and 1 cm (f).
Somatic embryogenesis and de novo shoot organogenesis have the same histological origin in Lisianthus
Histological characterization of regeneration systems obtained from Lisianthus root explants was performed to determine the origin and the temporal development of the morphogenetic responses obtained via somatic embryogenesis (Fig. 4) and de novo shoot organogenesis (Fig. 5). In both morphogenetic pathways, histological changes were first observed in the vascular cylinder. Root segments initially showed a vascular cylinder with a typical polyarch structure (Fig. 4 a) enclosed by a single pericycle layer (Fig. 4 a, b).

Ontogenesis of somatic embryogenesis pathway obtained from Lisianthus root explants. Light microscopy cross (a, g) and longitudinal sections (b–f, h, i, m) and scanning electron microscopy images (j–l). Histological organization of initial root explants (a, b). Cellular divisions of pericycle and vascular parenchyma cells after 10 d (c, d) and 15 d (e, f) of culture. Images d and f show details of cellular divisions observed in c and e, respectively. Cellular proliferation around the vascular cylinder in root explants after 20 d of culture (g). Initial stages of somatic pro-embryo formation (h). Meristematic features of these cells (asterisks). Exposure of pro-embryos in the periphery of root explants (i, j). Somatic embryos developed after root explant transfer to the conversion medium (k–m). Developed somatic embryo. Note the presence of all primary meristems defined. co cortex, e endodermis, g globular embryo, gm ground meristem, lp leaf primordium, m shoot meristem, p pericycle, pc procambium, pl phloem, pt protodermis, se somatic embryos, and x xylem. Bars = 200 μM (a–c, e, g–n) and 100 μM (d, f).

Ontogenesis of de novo shoot organogenesis pathway obtained from Lisianthus root explants. Light microscopy of longitudinal sections (a–f) and scanning electron microscopy images (g). Root explant used as initial explant (a). Cellular divisions (asterisk) in pericycle and vascular parenchyma cells after 1 wk of culture (b). Formation and development of meristemoid (c). Note the meristematic cellular features (asterisk). Disruption of cortex and epidermis caused by the initial development of organogenic structures (d). Developed adventitious buds showing the shoot apical meristem (m) and leaf primordial (lp) (e–g). co cortex, e endodermis, lp leaf primordium, m shoot meristem, p pericycle, and vt vascular tissue. Bars = 300 μM.
Early in the somatic embryogenesis process, both anticlinal and periclinal cell division planes were observed in pericycle cultured for 10 d (Fig. 4 c, d). These divisions intensified over time, and many dividing cell clusters occurred in the pericycle and parenchyma associated with the vascular tissue (Fig. 4 e, f), creating a proliferation area throughout the vascular cylinder (Fig. 4 g). The proliferation zone was composed of several layers of cells with meristematic features, including small size, isodiametric shape, dense cytoplasm, large nuclei, and evident nucleoli (Fig. 4 h). After 40 d of culture, the continuous development of the proliferation area led to clusters of meristematic cells, being interpreted as pro-embryonic areas (Fig. 1 a and Fig.4 i). Pro-embryo differentiation into somatic embryos was gradual and asynchronous.
Twenty days after being transferred to the conversion medium, somatic embryos at different stages were present on the surface of the root explants (Fig. 2 j–g). In the early stages of development, the somatic embryos were well-defined and presented a typical bipolarized structure, a clear protoderm, and no vascular connection with the initial explant (Fig. 4 m). Procambia, leaf primordia, and apical domes were identified at the late development stages of somatic embryos after 40 d on conversion medium (Fig. 4 n).
In de novo shoot organogenesis, the induction process also started from anticlinal and periclinal cell divisions from pericycle cells and vascular parenchyma (Fig. 5 a, b). However, after 10 d of cultivation, cell divisions were prolific and there was no obvious preferred division plane (Fig. 5 c). Cell proliferation led to the formation of organogenic structures (Fig. 5 c). Unlike the proliferation observed in the embryonic zone process, only the apical cells of meristematic organogenic structures had dense cytoplasm and large nuclei constituting the meristems (Fig. 5 c). The continued development of these meristematic regions eventually led to the disruption of the cortex and root epidermis (Fig. 5 d). After 25–30 d of culture, meristem development resulted in the formation of adventitious buds (Fig. 5 e–g).
Discussion
In the present study, an in vitro regeneration system was developed to alternatively induce different morphogenetic pathways, somatic embryogenesis, or de novo shoot organogenesis in Lisianthus root explants. Somatic embryogenesis was obtained when root explants were cultured on induction medium supplemented with 2,4-D, whereas de novo shoot organogenesis was observed on medium containing BA and/or ZEA. Neither somatic embryogenesis nor de novo shoot organogenesis occurred on MS medium devoid of one of these plant growth regulators. Recent data suggested that plant growth regulators may specify cell identity from gene expression reprogramming required for the cell-fate transition in morphogenetic pathway induction (Xu and Huang 2014). Therefore, in some plant species, somatic embryogenesis or de novo shoot organogenesis may be induced in the same explant source, depending on the culture conditions (Dodsworth 2009; Kraut et al. 2011; Wang et al. 2011; Rocha et al. 2015).
The presence of 2,4-D in the culture medium was essential for somatic embryogenesis induction. The key roles that 2,4-D plays in embryonic responses and tissue formation have been widely reported (Paim-Pinto et al. 2011; Krishna Kumar and Thomas 2012; Pathak et al. 2012; You et al. 2012; Stanišić et al. 2015; Ruffoni and Bassolino 2016). However, the present data showed that increasing 2,4-D concentration in the culture medium had a deleterious effect on embryonic induction frequency and increased the explant oxidation rate. Appropriate auxin concentration in the culture medium is crucial for somatic embryogenesis induction and may vary with explant species and type (Fehér et al. 2003; Parimalan et al. 2011; You et al. 2011). Considering that 2,4-D supplementation in culture medium may affect the endogenous auxin level and its distribution and biosynthesis (Kaminek et al. 1997), and that roots are generally known to be sensitive to high auxin concentrations, such factors could explain the observed results.
Some studies have reported successful GA3 and BA supplementation in the culture medium for conversion of somatic embryos in Gentianaceae (Mikuła and Rybczyński 2001; Fiuk and Rybczyński 2007b; Fiuk and Rybczyński 2008). These plant growth regulators have also been used for conversion of somatic embryos obtained from root explants (Komai et al. 1995; Twyford and Mantell 1996; Akashi et al. 1998; Yang et al. 2009). In the present study, BA treatments were more effective than GA3 treatments for Lisianthus somatic embryo maturation. The use of 4 μM BA generated the highest number of somatic embryos per root explant. However, the use of 2 μM BA generated somatic embryos with improved vigor, as evidenced by higher fresh and dry weights and percentages of fresh and dry embryo biomasses per explant. The addition of BA in the conversion medium of Lisianthus somatic embryos has previously been proposed by Nhut et al. (2006), who also observed an increased number of embryos regenerated on media supplemented with lower BA concentrations.
De novo shoot organogenesis induction was influenced by both the type and concentration of cytokinin in the culture medium, possibly due to variations in translocation rate (Blakesley 1991). BA and ZEA promoted the development of more adventitious buds than the treatments supplemented with KIN and TDZ. In addition, the lowest BA and ZEA concentration (4 μM) was more effective in forming and developing morphologically normal buds. Similarly, BA and ZEA were reported as the most effective cytokinins for inducing the development of adventitious buds on Bixa orellana (Cruz et al. 2014). The ability of BA to induce the formation of adventitious buds in Lisianthus had been previously reported for leaf discs (Semeniuk and Griesbach 1987; Barrueto Cid and Teixeira 2006; Ördögh et al. 2006; Esizad et al. 2012). The supplementation of culture media with KIN has also been proposed for Lisianthus micropropagation from meristematic explants (Kaviani 2014). However, the same effect on root segments was not observed in this study.
The morphogenetic responses obtained from both somatic embryogenesis and de novo shoot organogenesis pathways showed the same histological origin. Somatic embryos and adventitious shoots were formed through the proliferation of pericycle and pericycle-like cells (vascular parenchyma), suggesting that pericycle cells have high plasticity and are able to acquire different cell fates depending on the conditions to which they are subjected. Several studies have reported the involvement of pericycle cells in the regeneration of embryos and/or buds from root explants (Vinocur et al. 2000; Lombardi et al. 2007; Rocha et al. 2012; Vieira et al. 2014; Cruz et al. 2014). This is consistent with recent studies on the molecular mechanisms involved in in vitro morphogenesis. Several lines of evidence support the understanding that pericycle and vascular parenchyma cells are intrinsically prone to undertake different morphogenetic pathways (De Smet et al. 2006; Sugimoto et al. 2010; Pulianmackal et al. 2014).
The formation of somatic embryos and adventitious buds occurred through the differentiation of pericycle cells into proembryos and meristemoids, respectively, which consisted of cells featuring similar characteristics, such as small size, dense cytoplasm, large nucleus, and evident nucleolus. These cellular features are considered as meristematic competence markers (Verdeil et al. 2007; Kurczyńska et al. 2012) and highlights the potential of tissues to proliferate and differentiate based on the signaling carried out by the plant growth regulator present in the medium. In this context, the origins of both somatic embryos and adventitious buds were interpreted as multicellular, starting from founder clusters of similar meristematic-like cells. This observation has also been reported for in vitro embryonic and organogenic responses in Cyclamen persicum (Savona et al. 2012) and Passiflora edulis (Rocha et al. 2015), which originated from small groups of cells that exhibited similar characteristics.
In summary, these results demonstrate that both somatic embryogenesis and de novo shoot organogenesis pathways in E. grandiflorum can be induced from root pericycle cells. The best growth regulator concentrations for the establishment of embryonic callus from root segments (10 μM 2,4-D), for the embryo conversion (2 μM BA), and for induction of de novo shoot organogenesis (4 μM BA or 4 μM ZEA) were also demonstrated. These findings provide evidence for micropropagation and biotechnological processes (e.g., selection and genetic breeding) to be optimized for this species.
References
Akashi R, Hoffmann-Tsay S-S, Hoffmann F (1998) Selection of a super-growing legume root culture that permits controlled switching between root cloning and direct embryogenesis. Theor Appl Genet 96:758–764
Arnhold E (2014) Easyanova: Analysis of variance and other important complementary analyzes. R package version 4
Atta R, Laurens L, Boucheron-Dubuisson E, Guivarc'h A, Carnero E, Giraudat-Pautot V, Rech P, Chriqui D (2009) Pluripotency of Arabidopsis xylem pericycle underlies shoot regeneration from root and hypocotyl explants grown in vitro. Plant J 57:626–644
Barrueto Cid LP, Teixeira JB (2006) Indução de organogênese em Lisianthus, Eustoma grandiflorum, a partir de fragmentos foliares cultivados in vitro. Boletim de Pesquisa e Desenvolvimento/Embrapa Recursos Genéticos e Biotecnologia, Brasilia-DF
Blakesley D (1991) Uptake and metabolism of 6-benzyladenine in shoot cultures of Musa and Rhododendron. Plant Cell Tissue Organ Cult 25:69–74
Bozzola JJ, Russell LD (1992) Electron Microscopy: Principles and Techniques for Biologists, 2nd edn. Jones and Barlett, Boston
Cruz ACF, Rocha DI, Iarema L, Ventrella MC, Costa MGC, Paiva Neto VB, Otoni WC (2014) In vitro organogenesis from root culture segments of Bixa orellana L. (Bixaceae). In Vitro Cell Dev Biol - Plant 50:76–83
De Smet I, Vanneste S, Inzé D, Beeckman T (2006) Lateral root initiation or the birth of a new meristem. Plant Mol Biol 60:871–887
Dodsworth S (2009) A diverse and intricate signalling network regulates stem cell fate in the shoot apical meristem. Dev Biol 336:1–9
Esizad SG, Kaviani B, Tarang A, Zanjani SB (2012) Micropropagation of Lisianthus (Eustoma grandiflorum), an ornamental plant. Plant Omics J 5:314–319
Fehér A, Pasternak TP, Dudits D (2003) Transition of somatic plant cells to an embryogenic state. Plant Cell Tissue Organ Cult 74:201–228
Ferreira EB, Cavalcanti PP, Nogueira DA (2013) ExpDes: Experimental Designs package versão:1.1.2
Fiuk A, Rybczyński JJ (2007a) Morphogenic capability of Gentiana kurroo Royle seedling and leaf explants. Acta Physiol Plant 30:157–166
Fiuk A, Rybczyński JJ (2007b) The effect of several factors on somatic embryogenesis and plant regeneration in protoplast cultures of Gentiana kurroo (Royle). Plant Cell Tissue Organ Cult 91:263–271
Fiuk A, Rybczyński JJ (2008) Factors influencing efficiency of somatic embryogenesis of Gentiana kurroo (Royle) cell suspension. Plant Biotechnol Rep 2:33–39
Furukawa H, Matsubara C, Shigematsu N (1990) Shoot regeneration from the roots of prairie gentian (Eustoma grandiflorum (Griseb.) Schinners). Plant Tissue Cult Lett 7:11–13
Harbaugh BK (2007) Lisianthus Eustoma grandiflorum. In: Anderson NO (ed) Flower breeding and genetics issues, challenges and opportunities for the 21st century, 1st edn. Springer, Netherlands, pp 645–663
Ikeuchi M, Ogawa Y, Iwase A, Sugimoto K (2016) Plant regeneration: cellular origins and molecular mechanisms. Development 143:1442–1451
Karnovsky MJ (1965) A formaldehyde-glutaraldehyde fixative of high osmolality for use in electron microscopy. J Cell Biol 27:137–138
Kaminek M, Motyka V, Vankova R (1997) Regulation of cytokinin content in plant cells. Physiol Plant 101:689–700
Kaviani B (2014) Micropropagation of ten weeks (Matthiola incana) and Lisianthus (Eustoma grandiflorum) (two ornamental plants) by using kinetin (kin), naphthalene acetic acid (NAA) and 2,4-dichlorophenoxyacetic acid (2,4-D). Acta Sci Pol Hortorum Cultus 13:141–154
Komai F, Okuse I, Harada T (1995) Histological identification of somatic embryogenesis from excised root tissues of spinach (Spinacia oleracea L.). Plant Tissue Cult Lett 12:313–315
Kraut M, Wójcikowska B, Ledwoń A, Gaj MD (2011) Immature zygotic embryo cultures of Arabidopsis. A model system for molecular studies on morphogenic pathways induced in vitro. Acta Biol Crac Ser Bot 53:59–67
Krishna Kumar G, Thomas TD (2012) High frequency somatic embryogenesis and synthetic seed production in Clitoria ternatea Linn. Plant Cell Tissue Organ Cult 110:141–151
Kurczyńska EU, Potocka I, Dobrowolska I, Kulinska-Lukaszek K, Sala K, Wrobel J (2012) Cellular Markers for Somatic Embryogenesis. In: Sato K-I (ed) Embryogenesis. InTech, Rijeka, pp 307–332
Lombardi SP, Ribeiro I, Nogueira Stolf MC, Appezzato-da-Glória B (2007) In vitro shoot regeneration from roots and leaf discs of Passiflora cincinnata Mast. Braz Arch Biol Technol 50:239–247
Mikuła A, Rybczyński JJ (2001) Somatic embryogenesis of Gentiana genus I. The effect of the preculture treatment and primary explant origin on somatic embryogenesis of Gentiana cruciata (L.), G. pannonica (Scop.), and G. tibetica (King). Acta Physiol Plant 23:15–25
Motte H, Vereecke D, Geelen D, Werbrouck S (2014) The molecular path to in vitro shoot regeneration. Biotechnol Adv 32:107–121
Murashige T, Skoog F (1962) A revised medium for rapid growth and bio assays with tobacco tissue cultures. Physiol Plant 15:473–497
Nhut DT, Tuan NS, Ngoc HM, Uyen PN, Don NT, Mai NT, Teixeira da Silva JA (2006) Somatic embryogenesis induction from in vitro leaf cultures of Lisianthus (Eustoma grandiflorum (Raf.) Shinn.). Propag Ornam Plant 6:121–127
O’Brien T, McCully M (1981) The study of plant structure principles and selected methods, 1st ed. Termarcarphi Pty Ltda., Melbourne, Australia
Ogas J (1997) Cellular differentiation regulated by gibberellin in the Arabidopsis thaliana pickle mutant. Science 277:91–94
Ördögh M, Jámbor-Benczúr E, Tilly-Mándy A (2006) Micropropagation of “Echo” cultivars of Eustoma grandilorum. Acta Hortic 725:457–460
Paim-Pinto LD, Rocha AAM, Monteiro RM, Lemes SM, Jardim OE, Otoni WC (2011) Somatic embryogenesis from mature zygotic embryos of commercial passionfruit (Passiflora edulis Sims) commercial populations. Plant Cell Tissue Organ Cult 107:521–530
Parimalan R, Venugopalan A, Giridhar P, Ravishankar GA (2011) Somatic embryogenesis and Agrobacterium-mediated transformation in Bixa orellana L. Plant Cell Tissue Organ Cult 105:317–328
Parveen S, Shahzad A (2011) A micropropagation protocol for Cassia angustifolia Vahl. from root explants. Acta Physiol Plant 33:789–796
Pathak S, Mishra BK, Misra P, Misra P, Joshi VK, Shukla S, Trivedi PK (2012) High frequency somatic embryogenesis, regeneration and correlation of alkaloid biosynthesis with gene expression in Papaver somniferum. Plant Growth Regul 68:17–25
Pulianmackal AJ, Kareem AVK, Durgaprasad K, Trivedi ZB, Prasad K (2014) Competence and regulatory interactions during regeneration in plants. Front Plant Sci 5:142
R Core Team (2014) Software R: A language and environment for statistical computing version:3.0.3
Rabobank (2015) Floriculture Map 2015. Rabobank Industry Note 475
Rocha DI, Monte-Bello CC, Dornelas MC (2015) Alternative induction of de novo shoot organogenesis or somatic embryogenesis from in vitro cultures of mature zygotic embryos of passion fruit (Passiflora edulis Sims) is modulated by the ratio between auxin and cytokinin in the medium. Plant Cell Tissue Organ Cult 120:1087–1098
Rocha DI, Vieira LM, Tanaka FAO, Silva LC, Otoni WC (2012) Anatomical and ultrastructural analyses of in vitro organogenesis from root explants of commercial passion fruit (Passiflora edulis Sims). Plant Cell Tissue Organ Cult 111:69–78
Ruffoni B, Bassolino L (2016) Somatic Embryogenesis in Lisianthus (Eustoma russellianum Griseb.). In: Maria AG, Maurizio L (eds) In Vitro Embryogenesis in Higher Plants. Springer New York, New York, pp 359–370
Ruffoni B, Damiano C, Massabò F, Esposito P (1990) Organogenesis and embryogenesis in Lisianthus russellianus Hook. Acta Hortic 280:83–88
Saldanha CW, Otoni CG, Azevedo JLF, Dias LLC, Rêgo MM, Otoni WC (2012) A low-cost alternative membrane system that promotes growth in nodal cultures of Brazilian ginseng [Pfaffia glomerata (Spreng.) Pedersen]. Plant Cell Tissue Organ Cult 110:413–422
Semeniuk P, Griesbach RJ (1987) In vitro propagation of prairie gentian. Plant Cell Tissue Organ Cult 8:249–253
Sahai A, Shahzad A, Sharma S (2010) Histology of organogenesis and somatic embryogenesis in excised root cultures of an endangered species Tylophora indica (Asclepiadaceae). Aust J Bot 58:198–205
Savona M, Mattioli R, Nigro S, Falasca G, Della RF, Costantino P, De Vries S, Ruffoni B, Trovato M, Altamura MM (2012) Two SERK genes are markers of pluripotency in Cyclamen persicum Mill. J Exp Bot 63:471–488
Shemer O, Landau U, Candela H, Zemach A, Eshed L (2015) Competency for shoot regeneration from Arabidopsis root explants is regulated by DNA methylation. Plant Sci 238:251–261
Stanišić M, Raspor M, Ninković S, Milošević S, Ćalić D, Bohanec B, Trifunović M, Petrić M, Subotić A, Jevremović S (2015) Clonal fidelity of Iris sibirica plants regenerated by somatic embryogenesis and organogenesis in leaf-base culture — RAPD and flow cytometer analyses. S Afr J Bot 96:42–52
Sugimoto K, Meyerowitz EM (2013) Regeneration in Arabidopsis tissue culture. In: De Smet I (ed) Plant Organogenesis: Methods and Protocols. Methods in Molecular Biology. Humana Press, Totowa, New Jersey, pp 265–275
Sugimoto K, Jiao Y, Meyerowitz EM (2010) Arabidopsis regeneration from multiple tissues occurs via a root development pathway. Dev Cell 18:463–471
Twyford CT, Mantell SH (1996) Production of somatic embryos and plantlets from root cells of the Greater Yam. Plant Cell Tissue Organ Cult 46:17–26
Verdeil J-L, Alemanno L, Niemenak N, Tranbarger TJ (2007) Pluripotent versus totipotent plant stem cells: dependence versus autonomy? Trends Plant Sci 12:245–252
Vieira LM, Rocha DI, Taquetti MF, da Silva LC, de Campos JMS, Viccini LF, Otoni WC (2014) In vitro plant regeneration of Passiflora setacea D.C. (Passifloraceae): the influence of explant type, growth regulators, and incubation conditions. In Vitro Cell Dev Biol - Plant 50:738–745
Vinocur B, Carmi T, Altman A, Ziv M (2000) Enhanced bud regeneration in aspen (Populus tremula L.) roots cultured in liquid media. Plant Cell Rep 19:1146–1154
Wan L, Yang X, Gui M, Qu S, Su Y, Zhou X (2009) In vitro anther culture and plant induction of Eustoma russellianum. Southwest China J Agric Sci 22:1424–1427
Wang XD, Nolan KE, Irwanto RR, Sheahan MB, Rose RJ (2011) Ontogeny of embryogenic callus in Medicago truncatula: The fate of the pluripotent and totipotent stem cells. Ann Bot 107:599–609
Wu HJ, Wang XX, Li Y, Zhang DG, Zhang BW, Xin Y (2011) Propagation of Gentiana macrophylla (Pall) from hairy root explants via indirect somatic embryogenesis and gentiopicroside content in obtained plants. Acta Physiol Plant 33:2229–2237
Xu L, Huang H (2014) Genetic and epigenetic controls of plant regeneration. In: Galliot B (ed) Current Topics in Developmental Biology, 1st edn. Elsevier Inc., USA, pp 1–33
Yang JL, Seong ES, Kim MJ, Ghimire BK, Kang WH, Yu CY, Li CH (2009) Direct somatic embryogenesis from pericycle cells of broccoli (Brassica oleracea L. var. italica) root explants. Plant Cell Tissue Organ Cult 100:49–58
You CR, Fan TJ, Gong XQ, Bian FH, Liang LK, Qu FN (2011) A high-frequency cyclic secondary somatic embryogenesis system for Cyclamen persicum Mill. Plant Cell Tissue Organ Cult 107:233–242
You XL, Tan X, Dai JL, Li YH, Choi YE (2012) Large-scale somatic embryogenesis and regeneration of Panax notoginseng. Plant Cell Tissue Organ Cult 108:333–338
Acknowledgements
The authors would like to thank the Microscopy and Microanalysis Center of the Federal University of Viçosa (Viçosa, MG, Brazil) and the Brazilian funding agencies Conselho Nacional de Desenvolvimento Científico e Tecnológico (CNPq), Fundação de Amparo à Pesquisa do Estado de Minas Gerais (FAPEMIG), and Coordenação de Aperfeiçoamento de Pessoal de Ensino Superior (CAPES) for providing financial support.
Authors’ contributions
MY-O, ACFC, MVMP, DIR, DSB, JGB, ADK, and WCO conceived, designed, and performed the experiments; MYO raised the in vitro plants used in the experiments; MY-O, ACFC, and MVMP carried out histological analysis; MY-O, DIR, DSB, JGB, and WCO interpreted the acquired data and drafted the paper.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflicts of interest.
Additional information
Editor: Jayasankar Subramanian
Rights and permissions
About this article

Cite this article
Yumbla-Orbes, M., da Cruz, A.C.F., Pinheiro, M.V.M. et al. Somatic embryogenesis and de novo shoot organogenesis can be alternatively induced by reactivating pericycle cells in Lisianthus (Eustoma grandiflorum (Raf.) Shinners) root explants. In Vitro Cell.Dev.Biol.-Plant 53, 209–218 (2017). https://doi.org/10.1007/s11627-017-9800-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11627-017-9800-2