
Abstract
In banana and plantain research, it is essential to establish embryogenic cell suspensions together with a highly efficient regeneration and transformation system. This article describes the development of an embryogenic cell suspension (ECS), regeneration, and transformation for plantain cv. “Gonja manjaya”. ECS was established using highly proliferative multiple buds. The frequency of embryogenic friable callus formation was 56.8% of the cultured explants. Friable embryogenic calli with many translucent proembryos were transferred to liquid medium and homogenous cell suspensions were established within 3–4 mo. Approximately 25,000 to 30,000 plants per 1.0 ml of settled cell volume were regenerated in approximately 13–14 mo. ECSs were transformed using Agrobacterium strain EHA 105 harboring the binary vector pBI121. About 50–60 transgenic plants per 0.5 ml settled cell volume were regenerated on selective medium containing 100 mg l−1 kanamycin. Histochemical GUS assays using different tissues of putatively transformed plants demonstrated stable expression of uidA gene. The presence and integration of the uidA gene were confirmed by PCR and Southern blot analysis, respectively. This is the first report showing establishment of embryogenic cell suspension cultures and Agrobacterium-mediated transformation of an important plantain cultivar, “Gonja manjaya”. This study shows the huge potential for genetic transformation of plantains for disease or pest resistance, as well as tolerance to abiotic factors such as drought stress using this robust regeneration and transformation protocol.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Bananas and plantains (Musa spp.) are important staple food for rural and urban consumers and provide a source of income for resource poor farmers in the humid tropics of sub-Saharan Africa. They are cultivated in over 100 countries covering about 10 million ha, with an annual production of 130 million tons (FAOSTAT 2009). Plantains account for about 32% of total Musa production, from mostly Central and West Africa (Lescot 2008). In Africa, bananas and plantains provide more than 25% of the food energy requirements for more than 100 million people, of whom 20 million are from East Africa alone (FAOSTAT 2004). Despite the importance of bananas and plantains, the crop is threatened by many pests and diseases (Viljoen 2010). The application of classical methods of breeding for both disease and pest resistance has resulted in only limited success due to the long generation times, lack of genetic variability, and the high sterility and triploidy of most cultivated bananas (Vuylsteke 2000; Lorenzen et al. 2010). In view of the constraints surrounding conventional breeding of banana and plantain, the case for using transgenic approaches to improve these crops is particularly compelling. However, to be successful, these applications require efficient plant regeneration and transformation protocols for both banana and plantain.
Relative success in genetic engineering of bananas and plantains has been achieved to enable the transfer of foreign genes into plant cells of these species. Genetic transformation using microprojectile bombardment of embryogenic cell suspensions (ECSs) has been reported for banana (Sagi et al. 1995; Côte et al. 1996; Becker et al. 2000). However, Agrobacterium-mediated transformation offers several advantages over direct gene transfer methodologies, such as the possibility to transfer only one or few copies of DNA fragments carrying the genes of interest at higher efficiencies, with lower costs, and the transfer of large DNA fragments with minimal rearrangement (Gheysen et al. 1998; Hansen and Wright 1999; Shibata and Liu 2000). Transformation efficiency is also higher in Agrobacterium-mediated transformation in comparison to microprojectile bombardment of banana ECSs. Becker et al. (2000) obtained only ten individual transformation events from 90 bombardments (approximately 3 ml of settled cell volume), whereas Khanna et al. (2004) generated up to 65 plants per 50 mg settled cell volume through Agrobacterium-mediated transformation of banana. Arinaitwe et al. (2004) also reported significantly higher expression of transgenes in banana cultivars using an Agrobacterium-based method.
Transformation of plants by Agrobacterium-mediated DNA transfer is currently the most commonly used method to accomplish plant gene transfer (Lindsey 1992; Gelvin 2003). Protocols have been developed for Agrobacterium-mediated transformation of ECSs (Ganapathi et al. 2001; Khanna et al. 2004; Kosky et al. 2010; Tripathi et al. 2010) and meristematic tissues of various cultivars of banana and plantain (May et al. 1995; Tripathi et al. 2005, 2008). At present, most of the reported transformation protocols use cell suspensions as the target tissue for transgene integration. However, establishing such cell suspensions is a lengthy process and cultivar dependent. Optimization of cell suspension and transformation protocols for each particular cultivar therefore becomes a prerequisite for agronomic improvement in that particular genetic background. ECS cultures have been obtained for different genotypes of banana using basal leaf sheaths and corm section explants (Novak et al. 1989), highly proliferating shoot tip cultures (Dheda et al. 1991; Strosse et al. 2006), zygotic embryos (Marroquin et al. 1993), and immature male flowers (Escalant et al. 1994; Côte et al. 1996; Navarro et al. 1997; Becker et al. 2000; Grapin et al. 2000).
“Gonja manjaya” is a false horn plantain (AAB) commonly grown in Central and East Africa. In terms of production, in the Democratic Republic of Congo, the roasting variety “Gonja manjaya” is primary followed by cooking bananas. However, its production is severely affected by Banana Xanthomonas Wilt (caused by Xanthomonas campestris pv. musacearum), Fusarium wilt (causal agent Fusarium oxysporum f. sp. cubense), and various nematodes (Viljoen 2010). Developing resistant varieties through genetic engineering potentially is the most cost-effective and sustainable method of controlling diseases and pests. Such improvement initiatives demand efficient transformation frequencies and a standard, rapid, and reproducible protocol that can be used to transform all banana genomic groups. To date, there has been no report describing the establishment of cell suspension cultures and transformation of plantain cv. “Gonja manjaya”. For these reasons, the present study was undertaken to establish a rapid, reproducible protocol for production of ECS and regeneration of plantain cv. “Gonja manjaya” genetic transformation capacity for this variety.
Materials and Methods
Plant materials and explants preparation.
The in vitro plantlets of plantain cv. “Gonja manjaya” (AAB) were regenerated through micropropagation using apical shoot tips as explant material (Tripathi et al. 2003). The in vitro shoots were maintained on Proliferation Medium (PM, Table 1), at 26 ± 2°C under 16 h photoperiod furnished with fluorescent tube providing light of 94 μmol m−2 s−1. Clusters of small shoots and white buds were observed at the base of the explants (suckers) after 1 mo of culture. These buds were cultured on Multiple Buds Induction medium (MBI, Table 1) and incubated in the dark at 26 ± 2°C. The multiple buds were subcultured every 4 wk on MBI medium until clusters of tiny white buds surrounded by a few small leaves were obtained (Fig. 1A ).

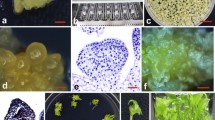
Regeneration of plantain cv. “Gonja manjaya” using embryogenic cell suspensions. (A) Cluster of tiny multiple buds; (B) Friable embryogenic callus; (C) Embryogenic cell suspension; (D) Embryo development; (E) Germination of embryos; (F) Regenerated plants hardened in pots.
Callus induction.
Clusters of multiple buds were cut vertically into 1–2-mm-thick slices and used as explant material for callus induction on Callus Induction Medium (CIM, Table 1). About five to six explants were cultured per 90 mm Petri dish, and a total of 850 explants were cultured in three experiments. The cultures were incubated in the dark at 26 ± 2°C for 3 to 6 mo, without subculturing to fresh CIM, for the development of embryogenic friable calli.
Development of embryogenic cell suspension.
The embryogenic friable calli were transferred to liquid CIM in 25-ml conical flasks containing 5–6 ml of medium and incubated in the dark at 26 ± 2°C, with agitation at 90 rpm. One embryogenic callus was transferred to each flask, and in total, 15 embryogenic calli were used for inoculum in two experiments. During the first month after initiation, 50% of the culture medium was refreshed every 7–10 d, and from the second month after initiation, the culture medium was refreshed every 14 d, retaining approximately 20% of old medium. Once a fine cell suspension started appearing in the flask, brown tissues and large cell clumps were removed with the help of forceps and fine cells were transferred to 100-ml conical flasks containing 15–20 ml CIM liquid medium. The fine light yellow-colored granular cells were transferred to 250-ml conical flasks containing 50 ml of CIM when the settled cell volume exceeded 1–1.5 ml. The successfully established cell suspensions were subcultured every 2 wk to 250-ml conical flasks using 50 ml of medium for proliferation, maintaining 3% settled cell volume. The regeneration ability of these embryogenic cells was checked as described below before use in transformation experiments.
Regeneration of embryogenic cell suspension.
The density of proliferating ECS was adjusted to 3% settled cell volume by diluting with liquid CIM medium. About 1 ml of the diluted ECS was transferred onto filter paper placed in a 90-mm Petri dish containing semisolid Embryo Development Medium (EDM, Table 1). The embryos developed on EDM were transferred to Embryo Maturation Medium (EMM, Table 1) for 1 mo, after which the matured embryos were transferred to Germination Medium (GM, Table 1). Petri dishes with regenerating cell cultures were incubated in the dark at 26 ± 2°C. The regenerated shoots were transferred to PM followed by transfer of individual shoot to Rooting Medium (RM, Table 1) for 2–4 wk at 26 ± 2°C under 16 h photoperiod furnished with fluorescent tube lighting at 94 μmol m−2 s−1. The rooted plantlets were transferred to pots (7 × 9 cm) containing sterile soil for hardening. The plants were maintained in a humid and shady environment for 3 wk and then transferred to larger pots (26 × 24 cm).
Agrobacterium and plasmid.
The pBI121 vector (Clontech, www.clontech.com) containing the β-glucuronidase (uidA) gene was under the control of the CaMV35S promoter, and the neomycin phosphotransferase (nptII) gene as the selection marker was used in this study. A schematic representation of the pBI121 vector is shown in Fig. 2. The vector was transformed into Agrobacterium tumefaciens super virulent strain EHA 105 (Hood et al. 1993) through electroporation. EHA 105 harboring pBI121 was maintained on solid YEB medium (Table 1) with 1.5% agar supplemented with kanamycin (50 mg l−1) and rifampicin (50 mg l−1). Bacterial cultures were grown in liquid YEB medium supplemented with kanamycin (50 mg l−1) and rifampicin (50 mg l−1) with shaking at 150 rpm and 28°C until the optical density (OD600) reached 0.8. The bacterial cells were harvested by centrifugation at 5,000×g for 10 min at 4°C and resuspended in 25 ml of TMA1 medium (Table 1) supplemented with 100 μM acetosyringone. The bacterial suspension was incubated at 28°C for 3 h with shaking at 150 rpm. The optical density (OD600) of the bacterial culture was adjusted to 0.6–0.8 with TMA1 medium.

Schematic representation of the T-DNA construct of binary vector pBI121 used for genetic transformation of plantain. RB right border, NosP nopaline synthase promoter, nptII neomycin phosphotransferase gene, Nos PolyA nopaline synthase terminator, 35SP CaMV35S promoter, uidA β-glucuronidase gene, LB left border.
Transformation, selection, and regeneration.
A settled cell volume of 1 ml of ECS was subcultured into 50 ml of liquid CIM and cultured at reduced density for 5 d to increase cell competence and transformation efficiency. After 5 d, ECSs were collected in 50-ml Falcon tubes and transformed with Agrobacterium strain EHA 105 harboring pBI121 using the procedure described by Khanna et al. (2004). ECSs were co-cultivated for 3 d with Agrobacterium in the dark at 22°C, followed by washing of Agrobacterium-infected ECSs with liquid CIM supplemented with timentin (200 mg l−1), and transferred to selective EDM supplemented with timentin (200 mg l−1) and kanamycin (100 mg l−1) for 2–3 mo, with fortnightly transfer onto fresh medium of the same type. Putatively transformed embryos developed on EDM were transferred to selective EMM supplemented with cefotaxime (300 mg l−1) and kanamycin (100 mg l−1). Mature embryos were transferred to selective GM supplemented with cefotaxime (300 mg l−1) and kanamycin (100 mg l−1). Shoots developing on GM were transferred to PM without selection for maintenance and multiplication of shoots. Regenerated putative transgenic shoots were transferred to RM without selection. Rooted plantlets were then transferred to sterile soil in pots and maintained in a containment facility.
Histochemical GUS analysis.
GUS histochemical assays for transient gene expression was performed 3 d after co-cultivation according to the modified procedure of Jefferson (1987), as described by Tripathi et al. (2005) for banana. Stable expression of the uidA gene was determined using pieces of leaves and roots excised from kanamycin-resistant regenerated plantlets.
PCR analysis.
Genomic DNA (gDNA) was isolated from all putative transgenic plantlets generated using DNAeasy plant mini kit (Qiagen, GmbH, Germany). The presence of uidA and nptII genes in the plant genome of the randomly selected putatively transgenic lines was confirmed by PCR analysis using gene-specific primers. The primer sequences for the uidA gene were: forward 5′-TTTAACTATGCCGGGATCCATCGC-3′ and reverse 5′-CCAGTCGAGCATCTCTTCAGCGTA-3′ and the nptII gene: forward 5′-CCTTATCCGCAACTTCTTTACCTA-3′ and reverse 5′-ACACCCAGCCGGCCACAGTCG-3′. Plasmid DNA of pBI121 was used as a positive control and non-transformed plant gDNA as a negative control. The PCR reaction (25 μl) contained 1x Buffer, 200 μM dNTPs, 1.5 mM MgCl2, 0.5 μM of each primer, 0.5 U of Taq DNA polymerase, and 2 μl (1 μg) of template gDNA. The PCR reactions had an initial denaturation at 95°C for 5 min, followed by 35 cycles of amplification: denaturing for 1 min at 94°C, annealing for 1 min at 55°C, and extension of 1 min at 72°C. The final extension phase was prolonged to 7 min at 72°C. The PCR products were separated by electrophoresis in a 1% agarose gel and visualized under UV transluminator after ethidium bromide staining.
Southern blot analysis.
Integration of the uidA gene into the plant genome was analyzed using Southern blot hybridization. Genomic DNA was isolated from eight randomly selected PCR-positive transgenic plantlets using a modified CTAB (hexadecyltrimethylammonium bromide) extraction method for Musa as described by Gawel and Jarret (1991). The pBI121 plasmid (10 pg) and gDNA (10 μg) of transgenic plants were digested with HindIII, which cuts the plasmid vector DNA only at one site, as shown in Fig. 2. When integrated into genomic DNA, HindIII cut once at one end of the promoter (outside the probe region) and again in the gDNA beyond the T-DNA border. The restricted DNA was resolved on 0.8% agarose gels and blotted onto positively charged nylon membrane (Roche Diagnostics, West Sussex, UK). The blots were hybridized with digoxigenin (DIG)-labeled uidA-specific probe generated using a PCR DIG Probe Synthesis Kit (Roche Diagnostics). Hybridization and detection were carried out using a DIG Luminescent Detection Kit for Nucleic Acids (Roche Diagnostics), according to the manufacturer’s instructions.
Results and Discussion
Callus induction and development of embryogenic cell suspensions.
Clusters of white buds surrounded by very few small or no leaves were obtained (Fig. 1A ) after two to three subculture on MBI. Plantain cv. “Gonja manjaya” has high proliferation capacity under these conditions and only two to three subcultures were required to obtain multiple buds; in comparison to 7–11 subcultures for other plantain cultivars reported by Strosse et al. (2006).
The sectioned tissues derived from multiple buds were cultured on CIM. About 89.6% explants responded positively to callus induction. The frequency of embryogenic callus formation was about 56.8% of the cultured explants, with yellow-colored embryogenic friable callus forming from explants after 2.5–3 mo (Fig. 1B ). Embryogenic callus induction was dependent upon the size and number of meristems within the explant tissue. For maximum response, explants were thinly sliced (1–2 mm thick), free from leaf tissue, with a high ratio of meristematic tissue to the proportion of corm. Larger explants with too much corm tended to swell and turn into “cotton wool” like structures. The frequency of embryogenic callus (56.8%) obtained in this study was much higher than previous reports of only 1.8% in other cultivars of plantain (Strosse et al. 2006). Success rates for initiation of good quality embryogenic cell suspensions depended largely upon the quality of the selected embryogenic calli. Friable embryogenic calli with many translucent proembryos were transferred to liquid CIM, after which, fine cell suspensions appeared after 3–4 wk of culture. These fine cell suspensions increased and reached a settled volume of 1–2 ml in 8–9 wk after initiation. The homogenous cell suspension was established 3–4 mo after initiation (Fig. 1C ). The cells were light yellowish in color with the yellow-pigmented cytoplasm, indicating the embryogenic characteristic of the cell suspensions (Ganapathi et al. 2001; Jalil et al. 2003). Embryogenic cell suspensions were successfully established at a frequency of 70.3% (11 out of 15), whereas previous reports stated only 30.4% as an establishment rate for plantains (Strosse et al. 2006).
Three different cells lines were further maintained for 1 yr by subculturing every 2 wk. The number of cells doubled with every subculture, but the quality of ECS decreased with the number of subcultures. To prevent loss of culture viability, ECSs were cryopreserved for long-term storage using the protocol described by Panis et al. (1990).
Plant regeneration from embryogenic cell suspension.
The regeneration of plants from ECSs was determined for all the three cell lines established. Embryos developed 4–6 wk after subculture to EDM (Fig. 1D ), after which, the embryos were transferred to Maturation Medium for 4 wk. Mature embryos were then transferred to Germination Medium, with germination starting in 3–4 wk (Fig. 1E ). Regenerated shoots were transferred to Proliferation Medium for maintenance and multiplication. All the shoots transferred onto Rooting Medium developed roots in 2–3 wk, and well-rooted plants were transferred to soil in small pots (Fig. 1F ). About 25,000–30,000 plants per 1 ml of settled cell volume were obtained in this manner, with the regeneration efficiency observed to be same for all three cell lines. This compares favorably with Strosse et al. (2006), who reported estimates of 22,000 regenerated plants per 1 ml of packed cell volume of plantain cultivars; whereas, Morais-Lino et al. (2008) reported an average number of 558 plants per milliliter of 5% settled cell volume for Brazilian plantain.
The development of cell suspensions and plant regeneration is time consuming and cultivar-specific. Usually, it takes 14–42 mo for banana cultivars and 18–27 mo for plantains to produce a cell suspension from callus induction to rooted plant regeneration (Strosse et al. 2006). In contrast, this protocol for regeneration of plantain using ECS allowed us to obtain rooted plants in just 13–14 mo.
Transformation, selection, and regeneration of transgenic banana.
ECS cultures were transformed with A. tumefaciens strain EHA 105 containing the pBI121 plasmid in which the uidA gene is driven by the CaMV35S promoter, and the neomycin phosphotransferase (nptII) gene is used as a selection marker. Cells subjected to Agrobacterium-mediated transformation multiplied and proliferated on kanamycin containing selection medium, whereas the untransformed control cells turned black (Fig. 3A ). Transgenic embryogenic cells developed into embryos and germinated on selective medium (Fig. 3B–D ). The regenerated shoots proliferated and were transferred to Rooting Medium without antibiotics. All the shoots developed roots within 2–3 wk (Fig. 2F ). The inhibitory effect of kanamycin in the regeneration of plant transformation has been previously reported for other crops (Yao et al. 1995; Bretagne-Sagnard and Chupeau 1996). Thus, kanamycin was omitted from proliferation and Rooting Medium. The regenerated putatively transformed plantlets were further validated for the presence of transgene using PCR.

Agrobacterium-mediated transformation of plantain cv. “Gonja manjaya” using embryogenic cell suspensions. (A) Agrobacterium-infected cells proliferating on selective medium; (B) Embryos on selective Embryo Development Medium; (C) Embryos on selective Embryo Maturation Medium; (D) Embryo germination on selective medium; (E) Transient expression of uidA gene in cells 3 d after co-cultivation with Agrobacterium; (F) Transgenic plantlets regenerated on selective medium; (G) Stable expression of uidA in leaf segments from transgenic plants; (H) Stable expression of uidA gene in roots excised from transgenic plants.
Approximately 50–60 transgenic plants per 0.5 ml settled cell volume were regenerated for plantain cv. “Gonja manjaya” in about 4–5 mo from the time of Agrobacterium inoculation of ECS. Ganapathi et al. (2001) reported production of up to 40 plants per 0.5 ml packed cell volume after Agrobacterium-mediated transformation of shoot apex derived ECS of cv. “Rasthali” (AAB). Khanna et al. (2004) reported 25–65 plants per 50 mg of settled cell volume of embryogenic suspension cells of banana cvs. Cavendish and Lady Finger. Ghosh et al. (2009) obtained 30 transgenic plants per 50 mg of settled cell volume of embryogenic suspension cells of Cavendish cultivar.
In this study, 95–96% transgenic plantlets survived when transferred to pots with soil in a containment facility. There were no apparent phenotypic alterations observed during vegetative growth of hardened plants. Morphologically normal transgenic plants obtained through this system established the usefulness of the present protocol for introducing important economic traits, such as disease and pest resistance into plantain. Transformation experiments were repeated three times using different cell lines and similar results were obtained suggesting that this protocol is consistently reproducible.
Histochemical GUS analysis.
Transient GUS expression assay 3 d after co-cultivation of explants showed blue coloration confirming transient expression of the reporter gene in embryogenic cells (Fig. 3E ). A uniform blue coloration was observed in all the leaf segments and roots tested from regenerated transgenic plants, confirming stable expression of uidA gene throughout the plant indicating uniform transformation had occurred (Fig. 3G–H ). No blue coloration was observed in leaves and roots of control, non-transformed regenerated plants.
Molecular analysis.
The amplified product of about 500 bp corresponding to the internal fragment of uidA gene was observed from genomic DNA of all tested transgenic plants using uidA gene-specific primers (Fig. 4A ), confirming the presence uidA transgene in transgenic plants. An amplified fragment of ∼700 bp was also observed from all tested transgenic plants using nptII-specific primers (Fig. 4B ), confirming the presence of both uidA and nptII genes. The amplified products were observed in all the plants tested, confirming the presence of transgenes, with no plant escapes. No amplified product was observed in case of non-transgenic control plants.

(A) PCR analysis of genomic DNA from different transgenic lines using uidA-specific primers. (B) PCR analysis of different transgenic lines using nptII-specific primers. Lanes 1–12 Transgenic plant lines, M molecular weight marker, P plasmid DNA, C non-transgenic plant controls. (C) Southern blot analysis of genomic DNA of transgenic lines digested with HindIII. Lanes 1–8 Transgenic plant lines, C control non-transgenic plant, M DIG-labeled molecular weight marker, P plasmid construct DNA digested with HindIII.
Southern blot analysis was performed with HindIII-digested gDNA from transgenic and non-transgenic control plants in order to confirm the transgene integration and to determine the number of inserted copies. This enzyme has a single restriction site in the pBI121 construct used in plant transformation (Fig. 2). Positive bands were observed from all eight independent transgenic lines tested, whereas no band was observed for the non-transgenic control (Fig. 4C ). The copy number of integrated transgenes was estimated to be between 1 and 3 for most samples.
Conclusions
We report here the development of an efficient system for Agrobacterium-mediated transformation and regeneration of an important plantain cv. “Gonja manjaya” using embryogenic cell suspensions. The protocol incorporates steps using embryogenic cell suspensions developed from fine proliferating multiple buds and recovery of regenerated plants in 13–14 mo. Transformed ECS generated plants with high efficiency, at 50–60 transgenic plants per 0.5 ml of settled cell volume. This study shows enormous potential for genetic manipulation of plantain by incorporating agronomically important traits such as those conferring disease or pest resistance, as well as tolerance to abiotic stress factors, using an efficient transformation and regeneration system. We have already generated hundreds of transgenic lines of plantain cv. “Gonja manjaya” as a step towards bacterial disease and nematode resistance using this protocol in our laboratory.
References
Arinaitwe G.; Remy S.; Strosse H.; Swennen R.; Sági L. Agrobacterium- and particle bombardment-mediated transformation of a wide range of banana cultivars. In: Jain S. M.; Swennen R. (eds) Banana Improvement, cellular, molecular biology and induced mutations. Science Publishers Inc., Enfield, pp 351–358; 2004.
Becker D. K.; Dugdale B.; Smith M. K.; Harding R. M.; Dale J. L. Genetic transformation of Cavendish banana (Musa spp. AAA group) cv. ‘Grand Nain’ via microprojectile bombardment. Plant Cell Rep 19: 229–234; 2000.
Bretagne-Sagnard B.; Chupeau Y. Selection of transgenic flax plants facilitated by spectinomycin. Transgene Res 5: 131–137; 1996.
Côte F. X.; Domergue R.; Monmarson S.; Schwendiman J.; Teisson C.; Escalant J.-V. Embryogenic cell suspensions from the male flower of Musa AAA cv. Grand Nain. Physiol Plant 97: 285–290; 1996.
Dheda D.; Dumortier F.; Panis B.; Vuylsteke D.; Langhe E. Plant regeneration in cell suspension cultures of the cooking banana cv. Bluggoe (Musa spp. ABB group). Fruits 46: 125–135; 1991.
Escalant J.-V.; Teisson C.; Cote F. Amplified somatic embryogenesis from male flowers of triploid banana and plantain cultivars (Musa spp.). In Vitro Cell Dev Biol 30: 181–186; 1994.
FAOSTAT (Food and Agriculture Organization of the United Nations). FAOSTAT Agriculture Data http://faostat.fao.org; Cited 2004.
FAOSTAT. FAOSTAT Agriculture Data. http://faostat.fao.org; Cited 2009.
Ganapathi T. R.; Higgs N. S.; Balint-Kurti P. J.; Arntzen C. J.; May G. D.; Van Eck J. M. Agrobacterium-mediated transformation of the embryogenic cell suspensions of the banana cultivars Rasthali (AAB). Plant Cell Rep 20: 157–162; 2001.
Gawel N. J.; Jarret R. L. A modified CTAB DNA extraction procedure for Musa and Ipomoea. Plant Mol Biol Rep 9: 262–266; 1991.
Gelvin S. B. Agrobacterium-Mediated Plant Transformation: the Biology behind the “Gene-Jockeying” Tool. Microbiol Mol Biol Rev 67: 16–37; 2003.
Gheysen G.; Angenon G.; Montague M. V. Agrobacterium-mediated plant transformation: a scientifically intriguing story with significant application. In: Lindsey K. (ed) Transgenic plant research. Harwood Academic, Amsterdam, pp 1–33; 1998.
Ghosh A.; Ganapathi T. R.; Nath P.; Bapat V. A. Establishment of embryogenic cell suspension cultures and Agrobacterium-mediated transformation in an important Cavendish banana cv. Robusta (AAA). Plant Cell Tissue Organ Cult 97: 131–139; 2009.
Grapin A.; Ortíz J.-L.; Lescot T.; Ferrière N.; Côte F. X. Recovery and regeneration of embryogenic cultures from female flowers of False Horn Plantain. Plant Cell Tissue Organ Cult 61: 237–244; 2000.
Hansen G.; Wright M. S. Recent advances in transformation of plants. Trends Plant Sci 4: 226–231; 1999.
Hood E. E.; Gelvin S. B.; Melchers S.; Hoekema A. New Agrobacterium plasmids for gene transfer to plants (EHA 105). Trans Res 2: 208–218; 1993.
Jalil M.; Khalid N.; Othman R. Y. Plant regeneration from embryogenic suspension cultures of Musa acuminata cv. Mas (AA). Plant Cell Tissue Organ Cult 75: 209–214; 2003.
Jefferson R. A. Assaying chimeric genes in plants: The GUS gene fusion system. Plant Mol Biol Rep 5: 387–405; 1987.
Khanna H.; Becker D.; Kleidon J.; Dale J. Centrifugation Assisted Agrobacterium tumefaciens-mediated Transformation (CAAT) of embryogenic cell suspensions of banana (Musa spp. Cavendish AAA and Lady Finger AAB). Mol Breed 14: 239–252; 2004.
Kosky R. G.; Chong-Pérez B.; López-Torres J.; Reyes M.; Bermúdez-Caraballoso I.; Martín N. M.; Machado-Rodriguez J. M.; Portal O.; Ocaña B.; Alvarado-Capó Y.; Leiva-Mora M.; Acosta-Suárez M.; Cruz-Martin M.; Roque B.; Hernández L. Plantain (Musa spp. cv. ‘Navolean’ AAB) transgenic plants from Agrobacterium tumefaciens-mediated transformation of embryogenic cell suspensions. Biotecnología Vegetal 10: 209–218; 2010.
Lescot T. Genetic diversity of banana in figures. FruiTrop 155: 29–33; 2008.
Lindsey K. Genetic manipulation of crop plants. J Biotechnol 26: 1–28; 1992.
Lorenzen J.; Tenkouano A.; Bandyopadhyay R.; Vroh B.; Coyne D.; Tripathi L. Overview of Banana and Plantain (Musa spp.) Improvement in Africa: Past and Future. Acta Hortic 879: 595–603; 2010.
Marroquin C. G.; Paduscheck C.; Escalant J. V.; Teisson C. Somatic embryogenesis and plant regeneration through cell suspensions in Musa acuminate. In Vitro Cell Dev Biol 29: 43–46; 1993.
May G. D.; Rownak A.; Mason H.; Wiecko A.; Novak F. J.; Arntzen C. J. Generation of transgenic banana (Musa acuminata) plants via Agrobacterium-mediated transformation. Bio/Technol 13: 486–492; 1995.
Morais-Lino L. S.; Santos-Serejo J. A.; Oliveira e Silva S.; Ferreira de Santana J. R.; Kobayashi A. K. Cell suspension culture and plant regeneration of a Brazilian plantain, cultivar Terra. Pesqui Agropecu Bras 43: 1325–1330; 2008.
Morel G.; Wetmore R. H. Tissue culture of monocotyledons. Amer J Bot 38: 138–140; 1951.
Murashige T.; Skoog F. A revised medium for rapid growth and bioassays with tobacco tissue cultures. Physiol Plant 15: 473–497; 1962.
Navarro C.; Escobedo R. M.; Mayo A. In vitro plant regeneration from embryogenic cultures of a diploid and a triploid. Cavendish banana. Plant Cell Tissue Organ Cult 51: 17–25; 1997.
Novak F. J.; Afza R.; Van Duren M.; Perea-Dallos M.; Conger B. V.; Xiaolang T. Somatic embryogenesis and plant regeneration in suspension cultures of dessert (AA and AAA) and cooking (ABB) bananas (Musa spp.). Bio/Technol 7: 147–158; 1989.
Panis B.; Withers L. A.; De Langhe E. Cryopreservation of Musa suspension cultures and subsequent regeneration of plants. Cryo-Letters 11: 337–350; 1990.
Sagi L.; Panis B.; Remy S.; Schoofs H.; De Smet K.; Swennen R.; Cammus B. Genetic transformation of banana (Musa spp.) via particle bombardment. Bio/Technol 13: 481–485; 1995.
Schenk R. U.; Hildebrandt A. C. Medium and techniques for induction and growth of monocotyledonous and dicotyledonous plant cell cultures. Can J Bot 50: 199–204; 1972.
Shibata D.; Liu Y.-G. Agrobacterium-mediated plant transformation with large DNA fragments. Trends Plant Sci 5: 354–357; 2000.
Strosse H.; Schoofs H.; Panis B.; Andre E.; Reyniers K.; Swennen R. Development of embryogenic cell suspensions from shoot meristematic tissue bananas and plantains (Musa spp.). Plant Sci 170: 104–112; 2006.
Tripathi L.; Mwaka H.; Tripathi J. N.; Tushemereirwe W. K. Expression of sweet pepper Hrap gene in banana enhances resistance to Xanthomonas campestris pv. musacearum. Mol Plant Pathol 11: 721–731; 2010.
Tripathi L.; Tripathi J. N.; Hughes J. d’ A. Agrobacterium-mediated transformation of plantain (Musa spp.) cultivar Agbagba. African J Biotechnol 4: 1378–1383; 2005.
Tripathi L.; Tripathi J. N.; Oso R. T.; Hughes J. d’ A; Keese P. Regeneration and transient gene expression of African Musa species with diverse genomic constitution and ploidy levels. Tropical Agr 80: 182–187; 2003.
Tripathi L.; Tripathi J. N.; Tushemereirwe W. K. Rapid and efficient production of transgenic East African Highland Banana (Musa spp.) using intercalary meristematic tissues. African J Biotechnol 7: 1438–1445; 2008.
Viljoen A. Protecting the African Banana (Musa spp.): Prospects and Challenges. Acta Hortic 879: 305–314; 2010.
Vuylsteke D. Breeding bananas and plantains: from intractability to feasibility. Acta Hortic 540: 453–459; 2000.
Yao J.-L.; Cohen D.; Atkinson R.; Richardson K.; Morris B. Regeneration of transgenic plants from the commercial apple cultivar Royal Gala. Plant Cell Rep 14: 407–412; 1995.
Acknowledgments
The authors would like to thank the African Agriculture Technology Foundation (AATF) and Gatsby Charitable Foundation for funding support for this work and the National Agriculture Research Laboratories, Uganda for providing the laboratory facilities.
Author information
Authors and Affiliations
Corresponding author
Additional information
Editor: John W. Forster
Rights and permissions
About this article
Cite this article
Tripathi, J.N., Muwonge, A. & Tripathi, L. Efficient regeneration and transformation of plantain cv. “Gonja manjaya” (Musa spp. AAB) using embryogenic cell suspensions. In Vitro Cell.Dev.Biol.-Plant 48, 216–224 (2012). https://doi.org/10.1007/s11627-011-9422-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11627-011-9422-z