
Abstract
Adipose stem cell (ASC) transplantation is a promising therapeutic strategy for diabetic renal fibrosis. Hypoxia-inducible factor 1α (HIF1α) is a negative regulatory factor of mitochondrial function. In the current study, we aimed to explore if HIF1α deletion protects against hyperglycemia-induced ASC damage and enhances the therapeutic efficiency of ASCs in diabetic renal fibrosis. Our data indicated that HIF1α was upregulated in ASCs in response to high glucose stimulation. Higher HIF1α expression was associated with ASC apoptosis and proliferation arrest. Loss of HIF1α activated mitophagy protecting ASCs against high glucose-induced apoptosis via preserving mitochondrial function. Transplanting HIF1α-deleted ASCs in db/db mice improved the abnormalities in glucose metabolic parameters, including the levels of glucose, insulin, C-peptide, HbA1c, and inflammatory markers. In addition, the engraftment of HIF1α-modified ASCs also reversed renal function, decreased renal hypertrophy, and ameliorated renal histological changes in db/db mice. Functional studies confirmed that HIF1α-modified ASCs reduced renal fibrosis. Collectively, our results demonstrate that ASCs may be a promising therapeutic treatment for ameliorating diabetes and the development of renal fibrosis and that the loss of HIF1α in ASCs may further increase the efficiency of stem cell-based therapy. These findings provide a new understanding about the protective effects of HIF1α silencing on ASCs and offer a new strategy for promoting the therapeutic efficacy of ASCs in diabetic renal fibrosis.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Over the last few decades, type 2 diabetes mellitus (T2DM) has been a leading cause of death worldwide. Notably, it has been proposed that 40% of T2DM patients may develop renal fibrosis, which is a major cause of end-stage renal disease (ESRD) (Lu et al. 2017). The hallmarks of renal fibrosis include glomerular cell apoptosis due to high glucose-induced stress and renal interstitial fibrosis owing to the accumulation of extracellular matrix (ECM), which contribute to the irreversible decline in renal function (Haller et al. 2017; Azushima et al. 2018). Unfortunately, the molecular mechanisms underlying renal fibrosis development are still unclear, and thus, no effective drugs are available to control renal fibrosis. Recently, stem cell transplantation-based therapy has emerged as a novel therapeutic strategy for renal fibrosis (Paulini et al. 2016; Jiang et al. 2017). This regenerative medicine offers a new method for replenishing damaged cells and promoting the recovery of renal function via differentiation into glomeruli or the activation of paracrine cytokine signaling (Baban et al. 2016; Nagaishi et al. 2016). Recent data have shown that adipose-derived mesenchymal stem cell (ASC) treatment may ameliorate the development of diabetes in a rodent model (Liu and Tang 2016). For renal fibrosis, although both animal and clinical studies have provided information that stem cell transplantation improves renal function through possible glomerular formation and/or angiogenesis, few studies have investigated the effects of ASC treatment on renal fibrosis progression.
In addition, the major dilemma in stem cell therapy for renal fibrosis is the low survival of transplanted cells in response to high glucose-induced stress (Abdel Aziz et al. 2014; Pan et al. 2014). Endogenous factors, such as inflammatory and immune responses, may contribute to cell death via excessive oxidative stress and mitochondrial damage. Thus, improving the resistance of grafted cells to oxidative stress and increasing cellular survival are critical for enhancing the efficacy of stem cell therapy. In oxidative stress-mediated cellular apoptosis, mitochondria have been documented to transmit apoptotic signals through the release of cytochrome c (cyt-c) into cytoplasm (Fuhrmann and Brune 2017; Griffiths et al. 2017). Recently, several researchers have demonstrated that mitochondria use mitophagy, a mitochondrial autophagy system (Jin et al. 2018), to remove damaged mitochondria and inhibit mitochondrial apoptosis (Zhou et al. 2017a, b, c, d, e, 2018). However, whether mitophagy contributes to stem cell survival is unclear.
Several researchers have found that hypoxia-inducible factor 1α (HIF1α) is a negative factor for stem cells in response to stress attack (Bakker et al. 2007). Excessive HIF1α activation would induce the p53 (Zhao et al. 2016) or Bnip3 (Macher-Goeppinger et al. 2017) upregulation promoting the mitochondrial potential collapse and cellular apoptosis (Feng et al. 2014). Interestingly, HIF1α is also a regulator of mitophagy (Martinez-Outschoorn et al. 2010; Capparelli et al. 2012). Based on this information, we wondered whether modulating HIF1α may enhance stem cell survival after engraftment via modifying mitophagy (Han et al. 2017; Tamura et al. 2017). Accordingly, the present study was designed to evaluate the possible effect of HIF1α-modified ASCs on the development of renal fibrosis. The results demonstrated that HIF1α deletion protected ASCs against high glucose-induced apoptosis via the preservation of mitochondrial homeostasis by activating mitophagy. Subsequently, the transplantation of HIF1α-deleted ASCs attenuated the glucose metabolic abnormalities and ameliorated the renal structural and functional injuries via reducing renal fibrosis.
Methods
Isolation and characterization of ASCs for transplantation
ASCs were isolated from inguinal adipose tissue of mice with a C57 background (as previously described (Zhou et al. 2015a, b)). Cells were cultured in StemPro MSC SFM (serum-free medium) supplemented with 10% fetal bovine serum (Invitrogen, Carlsbad, CA) and were incubated at 37°C with 5% carbon dioxide. Culture medium was changed every 3–4 d. For cellular identification, ASCs at passage 3 were collected to assess surface antigens by flow cytometric analysis on a BD FACSCalibur flow cytometer. After being washed in PBS, ASCs were incubated with the following anti-mouse monoclonal fluorescent antibodies at the concentrations recommended by the manufacturer for 30 min in the dark: FITC-labeled CD29, CD31, CD34, CD45, CD90 (BD Biosciences, Franklin Lakes, NJ), or IgG.
For adipogenic and osteogenic differentiation, StemPro® adipogenesis and osteogenesis differentiation media (Invitrogen, Carlsbad, CA) were used according to the manufacturer’s protocol. Cultures were refreshed every 2 to 3 d. After 3–4 wk of culture, adipogenesis was assessed by incubating cells with Oil Red O solution to stain the neutral lipids in the cytoplasm (de Luxan-Delgado et al. 2016). Alizarin Red S (Sigma-Aldrich, St. Louis, MO) was used to evaluate osteogenic differentiation. To assess ASC viability, normal glucose medium (5.5 mmol/L) and high glucose medium (25 mmol/L) were used.
Animals and treatment
Our study was carried out in accordance with the guidelines of the Ethics Committee of Hunan University of Chinese Medicine, Changsha, China. The experimental protocol was approved by Ethics Committee of Hunan University of Chinese Medicine, Changsha, China. Male db/m mice (wild type) and C57BL/KsJ db/db obese (db/db) mice were obtained from Jackson Laboratory (Bar Harbor, ME). Eight-week-old db/db mice were engrafted with ASCs (1 ml of ASCs, 3 × 106 cells/ml, was slowly injected into the mouse kidney). The db/db mice were randomized into three groups (n = 20 for each group, matched for weight): the (i) control group (ctrl), (ii) db/db mice + ASCs transfected with shRNA against HIF1α (sh-HIF1α), and (iii) db/db mice + ASCs transfected with control shRNA group (sh-ctrl). Before each local injection, the syringe was pushed backwards to confirm that the needle was not present inside the vessel. All mice were maintained on a 12-h/12-h light/dark cycle with free access to tap water and laboratory chow. The animal experiments were performed for a 12-wk period (Ho et al. 2016). In the end, blood pressures were measured on conscious, acclimatized mice using the tail-cuff method. Blood samples were collected, and kidney tissues were harvested; one part of the kidney was snap-frozen in liquid nitrogen, and the other part was fixed with 10% phosphate-buffered saline (PBS)-buffered formalin, processed and embedded in paraffin (Smith et al. 2016).
shRNA against HIF1α
To evaluate the functional role of HIF1α, shRNA was used to reduce its expression. The selective shRNA duplex and a nonspecific control duplex were obtained from Yangzhou Ruibo Biotech Co., Ltd. (Yangzhou, China). Transfection of shRNA into cells was performed using Lipofectamine 2000 (Thermo Fisher Scientific, Waltham, MA) transfection reagent according to the manufacturer’s protocol (Lee and Back 2017). Briefly, cultured cells were washed with Opti-Minimal Essential Medium without serum or antibiotics and seeded in 6-well plates to 30–40% confluence. The transfection reagent and shRNA were diluted separately in serum-free media, mixed and incubated for 10 min at room temperature to form the shRNA/lipid complex. This complex was then added to each well at a final concentration of 70 nM/well of shRNA. At 48 h after transfection, cells were collected to determine HIF1α protein expression levels by western blot analysis (Yu et al. 2017).
Sample preparation and histological analysis
The kidneys were excised and rapidly frozen in optimal cutting temperature medium at room temperature (Agar Scientific Ltd., Stansted, UK) for the preparation of frozen sections (4-μm thickness). Hematoxylin-eosin stain, Masson trichrome stain, and periodic Schiff-methenamine (PASM) staining were performed at room temperature and were observed with an inverted microscope (magnification, ×40; BX51; Olympus Corp., Tokyo, Japan) (Mailloux et al. 2016).
Immunofluorescence staining
The samples were washed in PBS and permeabilized for 10 min at 4°C in a solution of 0.1% Triton X-100. Then, the samples were blocked with 10% goat serum albumin (Invitrogen, Carlsbad, CA) for 1 h at room temperature and subsequently incubated with primary antibodies overnight at 4°C (Xu et al. 2016). After three rinses in PBS, secondary antibodies were added to the samples for 1 h at room temperature (Zhu et al. 2016). The following primary antibodies were used in this study: mitochondrial import receptor subunit TOM20 homolog (Abcam, Cambridge, UK; cat. no. ab78547), lysosome-associated membrane glycoprotein 1 (Abcam; cat. no. ab24170), cytochrome-c (cyt-c; Abcam; cat. no. ab133504), and caspase-3 (Cell Signaling Technology, Inc., Danvers, MA; cat. no. 9664). Images were observed with an inverted microscope (magnification, ×40; BX51; Olympus Corp., Tokyo, Japan).
Western blotting
A mass of 40–60 μg proteins was loaded on a 12–15% SDS-PAGE gel. Following electrophoresis, proteins were transferred to a polyvinylidene fluoride western blotting membrane (Roche Applied Science, Penzberg, Germany) (Han et al. 2016). Bands were detected using an enhanced chemiluminescence substrate (Applygen Technologies, Inc., Beijing, China). Band intensities were normalized to the respective internal standard signal intensity (β-actin, 1:2000; Abcam; cat. no. ab8224) (Zhou et al. 2017a, b, c, d, e). The experiment was repeated three times. The primaries antibodies used in the present study were against the following proteins: Pro-caspase3 (1:1000; Cell Signaling Technology, #9662), cleaved caspase3 (1:1000; Cell Signaling Technology, #9664), caspase9 (1:1000; Abcam; #ab32539), microtubule-associated protein light chain (LC)3II (1:1000; Cell Signaling Technology, #3868), sequestome-1 (p62; 1:1000; Abcam; #ab56416), Beclin1 (1:1000; Cell Signaling Technology, #3495), autophagy protein 5 (Atg5; 1:1000; Cell Signaling Technology, #12994), transforming growth factor (TGF)β (1:1000; Cell Signaling Technology, #3711), matrix metalloproteinase (MMP)9 (1:1000; Cell Signaling Technology, #13667), collagen I (1:1000; Abcam; #ab34710), collagen I (1:1000; Abcam; #ab7778), and Bax (1:1000; Abcam; #ab32503).
Mitochondrial reactive oxygen species detection, JC-1 staining, and adenosine 5′-triphosphate detection
Mitochondrial reactive oxygen species (mROS) measurements were conducted using the MitoSOX red mitochondrial superoxide indicator (Molecular Probes, Eugene, OR). Mitochondrial potential was assessed using a JC-1 probe, a sensitive fluorescent dye used to detect alterations in mitochondrial potential (Zhou et al. 2014). Following treatment, cells were incubated with 10 mg/ml JC-1 for 10 min at 37°C in the dark and monitored with a fluorescence microscope (magnification, ×100; BX51; Olympus Corp., Tokyo, Japan) (Zhou et al. 2015a, b). Red-orange fluorescence was attributable to potential-dependent dye aggregation in the mitochondria. Green fluorescence, reflecting the monomeric form of JC-1, appeared in the cytosol following mitochondrial membrane depolarization (Liu et al. 2017a, b).
The ATP level in cells was determined using an ATP Bioluminescence Assay kit (Beyotime, Haimen, China) (Bhatia et al. 2016). Cells were lysed with PBS (Sigma-Aldrich, St. Louis, MO) lysis buffer, followed by centrifugation at 10,000×g for 2 min at 4°C. The ATP level was determined by mixing 50 μl of supernatant with 50 μl of luciferase reagent, which catalyzed the light production from ATP and luciferin. The emitted light was measured using a microplate luminometer and was linearly compared with the ATP concentration (Lin et al. 2016).
Mitochondrial respiratory function, mPTP opening measurement, and mitochondrial calcium [Ca2+] overload detection
Mitochondrial respiratory function was measured polarographically at 30°C using a Biological Oxygen Monitor System (Hansatech Instruments, King’s Lynn, UK) and a Clark-type oxygen electrode (Hansatech DW1, Norfolk, UK). Mitochondrial respiration was initiated by adding glutamate and malate to the reaction mixture at a final concentration of 5 and 2.5 mmol/L, respectively (Dong et al. 2017). State 3 respiration was initiated by adding ADP (150 nmol/L); state 4 was measured as the rate of oxygen consumption after ADP phosphorylation. The mitochondrial respiratory function (state 3/state 4) was calculated as previously described (Zhou et al. 2017a, b, c, d, e).
The mPTP opening assay was performed via the established calcein-cobalt loading procedure by incubating cells with calcein-acetoxymethyl ester (calcein-AM) according to a previous study (Zhou et al. 2017a, b, c, d, e). The immunofluorescence intensity of calcein-AM was measured as an indicator of mPTP opening.
To assess the [Ca2+]m, Rhod-2 (Molecular Probes) was used, and the images were captured by confocal microscopy. The fluorescence intensity of Rhod-2 was measured at the excitation wavelengths and emission wavelengths of 550 and 570 nm, respectively (Zhu et al. 2017).
BrdU assay
To evaluate cellular proliferation, the BrdU assay (RiboBio Co., Guangzhou, China) was used according to a previous study (Yang et al. 2017). First, cells with or without HIF1α deletion were fixed via 4% paraformaldehyde at 4°C, followed by permeabilization with 0.5% Triton X-100 for approximately 20 min at room temperature. Subsequently, samples were incubated in 2N HCl solution for 30 min at 37°C to unmask the antigen, followed by a neutralization step with 0.1 M sodium tetraborate. Then, BrdU antibody (1:200; Abcam, #ab8152) was used to incubate with samples overnight. After three rinses in PBS, secondary antibodies were added to the samples for 1 h at room temperature (Iggena et al. 2017). At last, the cells were stained with DAPI (Sigma-Aldrich) for 5 min to distinguish the nuclei. Subsequently, the samples were viewed under a fluorescence microscope. Photos were captured, and the number of BrdU positive cells was measured via counting at least three random separate fields.
MTT, TUNEL assays, caspase-3, and MMP9 activities detection
MTT experiments were performed in 96-well plates. Samples were washed three times with PBS, and 50 μl of MTT reagent was added to each well. The samples were subsequently incubated for 4 h at 37°C in a humid atmosphere containing 5% CO2. The MTT solution was removed, 200 μl of dimethyl sulfoxide was added to each sample, and the samples were incubated for 10 min (Han et al. 2017; Xu et al. 2017). Following the addition of Sorensen’s buffer, the absorbance at a wavelength of 570 nm was determined. A TUNEL assay to detect DNA fragmentation in the cell nuclei (a marker for apoptosis in testicular tissue) was performed using an In Situ Cell Death Detection kit (Roche Diagnostics GmbH, Mannheim, Germany) according to the manufacturer’s protocol. DAPI was used to label the nuclei (at room temperature for approximately 30 min). Caspase-3 and MMP9 activities kit (Beyotime Institute of Biotechnology) were used according to the manufacturer’s protocols (Iggena et al. 2017; Sigala et al. 2017). The relative caspase-3 and MMP9 activities were calculated from the ratio of treated cells to untreated cells. The assays were repeated three times.
Measurement of biochemical parameters
After 12 h of fasting, the blood glucose level of venous blood from the tail vein was measured using a glucometer (Roche, Mannheim, Germany). Blood and urine samples were collected. Kidney tissues were homogenized for further biochemical analysis (Peleli et al. 2016). Glycated hemoglobin (HbA1c) was measured using the in2it A1C system (Bio-Rad, Hercules, CA). The triglyceride and cholesterol levels in the serum and kidney and the TBARS, GSH, and GSSG levels in the kidney were determined using commercial assay kits (Nanjing Jiancheng Company, Shanghai, China) (Mailloux and Treberg 2016).
To collect morning spot urine samples, animals were placed in metabolic cages at the beginning of the light cycle and were kept for 2 h with water but without food. To obtain the 24-h urine samples, animals were placed in metabolic cages at the beginning of the light cycle and were kept for 24 h with free access to water and a standard laboratory diet. The levels of creatinine and blood urea nitrogen (BUN) were determined using a Cobas® C311 Autoanalyzer (Roche Diagnostics, Indianapolis, IN) as per the manufacturer’s protocols (Carloni et al. 2016). The urinary albumin concentration was determined using an ELISA kit obtained from Bio-Medical Assays (Beijing, China). The levels of insulin, glucagon, C-reactive protein, C-Peptide, TNFα, MCP-1, and IL-6 were determined using ELISA kits obtained from Cusabio Technology (Wuhan, China). The lipid hydroperoxides (LPOs) in kidney homogenates were determined using an LPO assay kit (Cayman Chemical, Ann Arbor, MI).
RNA isolation qPCR analysis
Total RNA was isolated from cells using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.). A total of 1 μg RNA from each sample was reverse-transcribed into cDNA using an RT kit (Eurogentec, Liege, Belgium) (Yang et al. 2017). The qPCR was performed with primers and matched probes from the Universal Fluorescence-labeled Probe Library (Roche Diagnostics GmbH) (Heppner and van der Vliet 2016). Quantification of gene expression was performed using an ABI PRISM 7500 Sequence Detection system (Applied Biosystems Life Technologies, Foster City, CA) with SYBR® Green (TransGen Biotech Co., Ltd., Beijing, China). The relative mRNA expression levels were normalized to that of β-actin using the 2−ΔΔCT method (Tan et al. 2016). The experiments were repeated three times with triplicates.
Statistical analysis
All data are expressed as the mean ± standard deviation. Statistical analyses were performed with SPSS software (version 17.0; SPSS, Inc., Chicago, IL). Results for more than two groups were evaluated by one-way analysis of variance with the least significant difference test. P < 0.05 was considered to indicate a statistically significant difference.
Results
The identification of isolated ASCs
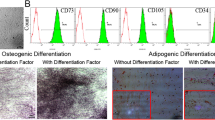
As indicated in Fig. 1a, the majority of ASCs in the third passage were adherent, in a fibrocyte-like form, and had a long shuttle shape. To ensure their multipotency, ASCs were transdifferentiated into adipocytes, which was confirmed by Oil Red O staining; red oil drops were observed in the ASCs (Fig. 1b). Similarly, ASC osteogenesis was identified with Alizarin Red staining of calcified nodules; red calcium nodules were identified after ASC differentiation (Fig. 1c). Third generation ASCs were harvested, and the cell phenotypes were detected using flow cytometry. Most of the ASCs expressed the specific stem cell markers CD29 and CD90 (Fig. 1d), which is consistent with the phenotypical characteristics of mesenchymal stem cells. In contrast, the ASCs seldom expressed the hematopoietic markers CD34 and CD45. These cells were also negative for the expression of the vascular endothelial cell marker CD31.

The identification of ASCs. a The morphology of ASCs under a microscope. b The adipogenic differentiation of ASCs, as detected by Oil Red O staining. c The osteogenic differentiation of ASCs, as detected by Alizarin Red staining. d Flow cytometry results determining the ASC phenotype.
High glucose reduces cellular viability and proliferation via the upregulation of HIF1α
Next, we investigated the role of HIF1α in ASC viability under high glucose stimulation. First, our results showed that HIF1α expression was progressively upregulated under high glucose treatment (25 mM) in a time-dependent manner and that HIF1α expression was highest after 8 h of treatment with high glucose (Fig. 2a, b). Therefore, 25 mM high glucose stimulation for 8 h was used in following experiments. To explain the causal role of HIF1α in regulating ASC viability, we silenced HIF1α in ASCs using shRNA-based method (sh/HIF1α). The transfection efficiency is shown in Fig. 2c. Meanwhile, the knockdown efficiency was confirmed by western blotting (Fig. 2d, e). Subsequently, the ability of HIF1α to affect ASC viability and proliferation was tested. MTT assay results showed that sh/HIF1α significantly increased cellular viability under high glucose treatment (Fig. 2f). The change in caspase-3 activity (Fig. 2g) was also similar to the MTT assay results, suggesting that loss of HIF1α sustains the adipose stem cell viability under high glucose-induced stress. Then, BrdU incorporation assay was used to detect cell duplication. As shown in Fig. 2h, i, high glucose stimulation reduced the number of BrdU positive cells. However, HIF1α deletion rescued the proliferative capacity of ASCs after exposure to high glucose treatment.

HIF1α is involved in ASC viability and proliferation under high glucose stimulation. a, b The change in the HIF1α level in response to the high glucose treatment. c The transfection efficiency of sh-HIF1α in ASCs. d, e The knockdown efficiency of HIF1α in ASCs was confirmed via western blotting. f The MTT assay was used to detect cellular viability. g Caspase-3 activity was measured to assess cellular apoptosis. h, i The BrdU assay was used to label proliferating cells. *P < 0.05 vs the ctrl + normal medium or sh-ctrl + normal medium group, #P < 0.05 vs the sh-ctrl + high glucose group.
HIF1α deficiency promotes ASC survival via blocking mitochondrial apoptosis
We further determined the effect of HIF1α on ASC apoptosis under high glucose-induced stress. TUNEL assays demonstrated that the number of TUNEL positive cells was decreased after sh/HIF1α transfection in response to high glucose-induced stress (Fig. 3a, b), suggesting that the loss of HIF1α reduces ASC apoptosis under high glucose-induced stress. Considering that HIF1α is a negative factor for mitochondrial function, we wondered whether the pro-apoptotic effect of HIF1α results from mitochondrial damage. First, we found that compared to the control group, the HIF1α deletion group presented with sustained mitochondrial membrane potential (MMP), as evidenced by a higher red fluorescence and lower green fluorescence (Fig. 3c, d). The MMP is derived from the proton gradient (Han et al. 2016). After the mitochondrial permeability transition pore (mPTP) is opened, protons are released into the cytoplasm, leading to MMP collapse. Therefore, we examined changes in the mPTP opening. High glucose increased the mPTP opening rate (Fig. 3e), and this change was reversed by sh/HIF1α after exposure to high glucose.

HIF1α deletion protected ASC mitochondrial function. a, b The TUNEL assay was used to detect cellular apoptosis. c, d Changes in mitochondrial membrane potential were analyzed via JC-1 staining. Red fluorescence represents normal mitochondrial potential, whereas green fluorescence is indicative of a damaged mitochondrial potential. e The mPTP opening rate. f, g The staining of cyt-c in ASCs. In response to mitochondrial damage, cyt-c is released from the mitochondria into the cytoplasm and even into the nuclei. h–l Proteins were isolated from ASCs, and then, western blotting was conducted to measure changes in the levels of proteins related to cellular apoptosis. *P < 0.05 vs the normal group, #P < 0.05 vs the sh-ctrl + high glucose group.
In addition, the opening of the mPTP establishes a channel that links the mitochondria and cytoplasm, promoting the leakage of the mitochondrial pro-apoptotic factor cyt-c from the mitochondria into the cytoplasm, where cyt-c has the ability to initiate mitochondrial apoptosis (Zhang et al. 2016). Using immunofluorescence (Fig. 3f, g), we found that high glucose triggered cyt-c diffusion into the cytoplasm and even into the nucleus, and this change was recused by sh/HIF1α in the presence of high glucose. Next, as a consequence of cyt-c release into the cytoplasm, the expression levels of pro-apoptotic proteins were increased, whereas the levels of anti-apoptotic factors were reduced (Fig. 3h–l). However, HIF1α deletion corrected the imbalance between pro-apoptotic proteins and anti-apoptotic factors. Altogether, these data demonstrated that HIF1α deletion protected ASCs against high glucose-induced stress via maintaining mitochondrial function.
HIF1α deficiency activates mitophagy to sustain mitochondrial function and structure
Mitophagy is vital for mitochondrial homeostasis (Shi et al. 2018). In response to mitochondrial damage, the mitochondria utilize the lysosome to degrade damaged mitochondria in order to sustain the quantity and quality of the mitochondrial mass (Zhou et al. 2017a, b, c, d, e). We wondered whether mitophagy is involved in HIF1α-related mitochondrial protection. Through co-staining of the mitochondria and lysosome, we found that compared to the normal group, the group treated with high glucose had mitochondrial separation from the lysosome (Fig. 4a). However, silencing HIF1α caused fusion of the mitochondria and lysosome (Fig. 4a). These data indicated that mitophagy was actually inhibited by high glucose and that the knockdown of HIF1α reversed mitophagy activity. Next, to quantitatively detect mitophagy activity, western blotting was used to analyze the mitophagy markers. As shown in Fig. 4b–f, high glucose reduced the expression of mitochondrial LC3II (mito-LC3II), Beclin1, and Atg5. In contrast, p62 expression was increased. After transfection with sh/HIF1α, mitophagy parameters including mito-LC3II, Beclin1, and Atg5 were increased, suggesting that mitophagy activity was enhanced by HIF1α deletion under high glucose-induced stress.

HIF1α deletion sustained mitochondrial function via activating mitophagy. a Co-staining of the mitochondria and lysosome. Orange fluorescence represents fusion of the mitochondria and lysosome, indicative of mitophagy. b–f Changes in mitophagy-related parameters. g Caspase-3 activity was used to detect the role of mitophagy in cellular apoptosis. 3-MA was used to inhibit mitophagy. h ATP production was detected. i, j Mitochondrial ROS (mROS) was observed via immunofluorescence. k, l Mitochondrial respiratory function measurements via state 3 and state 4 respiratory function assessment. *P < 0.05 vs the normal group, #P < 0.05 vs the sh-ctrl + high glucose group, @P < 0.05 vs the sh- HIF1α + high glucose group.
To further understand the role of mitophagy in mitochondrial protection, we detected caspase-3 activity. Meanwhile, we inhibited mitophagy activity via 3-MA in sh/HIF1α-treated ASCs. As shown in Fig. 4g, compared to the sh/HIF1α-treated ASCs, the inhibition of mitophagy abrogated the anti-apoptotic effects of HIF1α. In addition to cellular apoptosis, we also observed a change in mitochondrial function when mitophagy was inhibited. Compared to the sh/HIF1α-treated cells, cells in which mitophagy was inhibited had reduced levels of ATP production (Fig. 4h). Moreover, the level of mitochondrial ROS (mROS) was actually decreased after HIF1α deletion but was re-increased once mitophagy was inhibited via 3-MA (Fig. 4i, j). Mitochondrial respiratory function (state 3/4 respiratory activity) was also reduced in cells with mitophagy inhibited compared to sh/HIF1α-treated cells (Fig. 4k, l). In summary, these data indicated that HIF1α deletion protected ASCs against high glucose-induced apoptosis via the preservation of mitochondrial function by mitophagy activation.
HIF1α deletion enhanced ASC transplantation therapy in diabetes
We further explored the effects of HIF1α-deleted ASC engraftment on changes in kidney function in diabetes. The results in Fig. 5a show that, as expected, the body weights of the db/db mice were significantly higher than those of the wild-type mice, and the administration of ASCs significantly reduced the body weights of db/db mice. In addition, the sh/HIF1α-treated ASCs further reduced the body weights. The fasting blood glucose (Fig. 5b) and serum insulin (Fig. 5c) levels in the db/db mice were markedly higher than those of the wild-type mice, and ASCs engraftment substantially decreased fasting blood glucose and serum insulin levels in db/db mice. Notably, sh/HIF1α-treated ASCs further reduced the fasting blood glucose and serum insulin levels compared to the effect of sh/ctrl-treated ASCs (Fig. 5b, c). Moreover, the levels of serum C-peptide (Fig. 5d) and glycosylated hemoglobin A1c (HbA1c, Fig. 5e) in the db/db mice were markedly higher than those in the wild-type mice. The administration of ASCs significantly reduced the levels of serum C-peptide and HbA1c in db/db mice. However, sh/HIF1α-treated ASCs exerted a greater inhibitory effect on serum C-peptide and HbA1c levels than that induced by sh/ctrl-treated ASCs. In addition, the high level of glucagon in the db/db mice was alleviated by ASC transplantation (Fig. 5f), and this effect was enhanced by sh/HIF1α-treated ASCs.

Transplantation of HIF1α-deleted ASCs attenuated glucose metabolic abnormalities in db/db mice. Body weights (a) were measured, and fasting blood glucose (b), serum insulin (c), C-peptide (d), HbA1c (e), and glucagon (f) levels were determined. g Systolic blood pressure was measured. h–j The serum concentrations of IL6, MCP1, and TNFα were detected via ELISA assays. *P < 0.05 vs WT mice, #P < 0.05 vs the control group (db/db mice), @P < 0.05 vs sh-ctrl + db/db mice.
In addition, the increased systolic blood pressure of the db/db mice was also inhibited by ASCs in a HIF1α-dependent manner (Fig. 5g). Furthermore, during the development of diabetes, inflammatory marker levels are increased in the db/db mice compared to those in the wild-type mice (Fig. 5h–j). ASC administration markedly reduced the levels of these cytokines in the db/db mice, and this effect was enhanced by HIF1α deletion. In summary, these data illustrated that ASC engraftment exhibited an anti-diabetic effect, which was improved by HIF1α deletion.
HIF1α-deleted ASCs ameliorate renal structural and functional injury in diabetes
Renal function was evaluated by the determination of the blood urea nitrogen (BUN) (Fig. 6a) and serum creatinine (Fig. 6b) levels at the end of the experiment. We revealed that the levels of BUN and serum creatinine in the db/db mice were markedly higher than those in the wild-type mice (Fig. 6a, b). ASC transplantation significantly reduced the levels of BUN and serum creatinine in the db/db mice. The sh/HIF1α-treated ASCs were also able to decrease the BUN and creatinine levels. Additionally, the urinary albumin content and albumin/creatinine ratio (ACR) were measured to evaluate the progression of diabetic nephropathy. As shown in Fig. 6c, d, compared with the wild-type mice, the db/db mice exhibited a significantly higher urinary albumin content level and ACR. The transplantation of ASCs markedly reduced the urinary albumin content level and ACR in the db/db mice, and this protective effect was enhanced by HIF1α deletion.

Transplantation of HIF1α-deleted ASCs attenuated renal injury in db/db mice. a BUN level. b Serum creatinine level. c Urinary albumin content. d ACR level. e Kidney weights. f H&E stains of kidney sections. g Surface areas of Bowman’s capsule. h Bowman’s space. i Glomerular tuft. j PASM staining of kidney tissues. k–n Oxidative stress markers were examined in the kidney. o–p Lipid accumulation in the kidney was evaluated. *P < 0.05 vs WT mice, #P < 0.05 vs the control group (db/db mice), @P < 0.05 vs sh-ctrl + db/db mice.
To evaluate renal hypertrophy, the kidneys were weighed after the mice were sacrificed. The absolute kidney weights were significantly higher in the db/db mice than in the wild-type mice (Fig. 6e). ASC transplantation resulted in a marked decrease in kidney weight in the db/db mice (Fig. 6e). Notably, sh/HIF1α-treated ASCs further reduced the kidney weights. These results were supported by the histopathological examination of H&E- and PASM-stained kidney tissues (Fig. 6f–j). The histopathological results showed that the db/db mice exhibited moderate of glomeruli atrophy and fragmentation, epithelial desquamation, renal tubule degeneration (Fig. 6f–i), and kidney glomerular basement membrane thickening (Fig. 6j). However, these renal histopathological changes in the db/db mice were attenuated by ASC treatment. Notably, the sh/HIF1α-treated ASCs further improved the renal structure.
The effect of ASCs on kidney oxidative stress in mice was evaluated in this study. We showed that the levels of renal cortical tissue lipid hydroperoxides (LPOs) and urinary 8-isoprostane were notably higher in the db/db mice than in the wild-type mice (Fig. 6k, l). ASC administration markedly reduced the renal LPO and urinary 8-isoprostane content in the db/db mice (Fig. 6k, l). These effects were enhanced by HIF1α deletion in ASCs. In addition, the content of glutathione (GSH) in the renal cortical tissue of the db/db mice was notably lower than that of the wild-type mice (Fig. 6m). ASC administration increased the renal level of GSH in the db/db mice (Fig. 6m), while the content of GSSG (oxidized form of GSH) in the renal cortical tissue of the db/db mice was notably higher than that of the wild-type mice (Fig. 6n). The high level of renal GSSG in the db/db mice was reduced by ASC administration in a HIF1α-dependent manner (Fig. 6n).
Finally, lipid accumulation in the kidney tissues was measured in the animals. We revealed that the renal concentrations of triglycerides (Fig. 6o) and cholesterol (Fig. 6p) were notably higher in the db/db mice than in the wild-type mice. As expected, the high levels of renal triglycerides and cholesterol were reduced by ASCs, and this effect was enhanced by HIF1α deletion. The results illustrated that ASCs exhibited anti-hyperlipidemic activity via HIF1α, which may be involved in the renal protective effect of ASCs in diabetes. These results indicated that ASCs protected against renal dysfunction in db/db mice, and the silencing of HIF1α in ASCs further enhanced the therapeutic effect of ASCs on diabetic kidney injury.
Transplantation of HIF1α-deleted ASCs reduces renal fibrosis
Subsequently, we observed renal fibrosis via Masson staining. As shown in Fig. 7a, compared to the WT mice, the db/db mice presented with severe kidney fibrosis, and this change was partly reversed by ASC engraftment. When HIF1α was knocked down in ASCs, renal fibrosis was further suppressed. In addition, the expression levels of collagen I and collagen III in the kidneys were assessed. Chronic hyperglycemia increased the expression of collagen I/III, which was partly inhibited by ASCs in a sh/HIF1α-dependent manner (Fig. 7b–f). Notably, the signaling factors related to fibrosis including TGFβ and MMP9 were also higher in the kidney tissues of db/db mice than in those of the wild-type mice (Fig. 7b–g). Treatment with ASCs markedly reduced the TGFβ and MMP9 expression levels in the db/db mice, and this effect was enhanced by HIF1α deletion.

Transplantation of HIF1α-deleted ASCs reduces renal fibrosis. a Masson staining for kidney fibrosis. b–f Western blotting was used to detect changes in the levels of proteins related to renal fibrosis. g The MMP9 activity. h–i The serum concentration of laminin and precollagen III via ELISA assay. *P < 0.05 vs WT mice, #P < 0.05 vs the control group (db/db mice), @P < 0.05 vs sh-ctrl + db/db mice.
At last, the serum concentrations of laminin (Fig. 7h) and precollagen III (Fig. 7i) were notably higher in the db/db mice than in the wild-type mice. As expected, the high serum concentration of laminin and precollagen III was reduced by ASC transplantation, and this effect was enhanced by HIF1α knockdown. These results indicated that ASCs regulated renal interstitial fibrosis, and sh/HIF1α-modified ASCs have a better anti-fibrotic effect than normal ASCs.
Discussion
In the current study, in the context of diabetes, we investigated a method for improving the therapeutic efficiency of ASCs transplanted in kidney tissue by HIF1α silencing. We found that sh/HIF1α-modified ASCs exerted a more potent protective effect against the development diabetic renal fibrosis than that of normal ASCs. The transplantation of sh/HIF1α-modified ASCs improved the abnormal changes in glucose metabolic parameters, including the levels of glucose, insulin, C-peptide, and HbA1c, and in the blood pressure of db/db mice. In addition, the engraftment of sh/HIF1α-modified ASCs further improved renal function, decreased renal hypertrophy, and ameliorated the renal histological changes observed in db/db mice.
Diabetic kidney injury is one of the most common and severe microvascular complications in patients with diabetes mellitus and is the leading cause of end-stage renal disease (ESRD). Furthermore, approximately 50% of patients with diabetes develop chronic renal fibrosis (Lu et al. 2017). Renal fibrosis is pathologically characterized by mesangial dilatation, glomerulosclerosis, and renal interstitial fibrosis (Wu et al. 2017). Notably, cellular apoptosis is one of the leading causes of podocyte and renal tubular epithelial cell defects and has a crucial role in renal fibrosis pathogenesis. Although some reports have shown that several drugs slow the progression of renal fibrosis, it is difficult to repair a damaged kidney (Das et al. 2017). In recent years, many studies have demonstrated that stem cell transplantation is effective and safe for slowing the development of renal fibrosis via the regeneration of glomerular tissue (Pan et al. 2014; Jiang et al. 2017). However, transplanted stem cells seem to remain in an oxidative environment, and oxidative stress induces apoptosis and cell cycle arrest, which reduce transplantation efficiency (Zhou et al. 2014). Thus, improving the viability of transplanted cells is the key to successful stem cell-based therapy. In the present study, we found that high glucose treatment clearly triggered cellular apoptosis in ASCs. Interestingly, we found that HIF1α was upregulated in response to high glucose. The deletion of HIF1α into ASC reversed the decrease in cellular viability and promoted cellular proliferation. These results demonstrated that HIF1α is the potential target for enhancing stem cell viability under high glucose-induced stress. Notably, considerable disagreements exist with regard to the functional effects of HIF1α signaling because both pro- and anti-apoptotic regulatory roles have been reported. Numerous studies have demonstrated that overexpression of HIF1α in MSCs either by preconditioning with hypoxia or genetic approaches have many beneficial effects on MSCs, such as increasing their survival ability, the secretion of a panel of soluble factors, the pro-angiogenic effects, and therapeutic effects in various pathological settings (Liu et al. 2017a, b; Matsui et al. 2017). In contrast, it has been well documented that excessive HIF1α activation promoted the cellular apoptosis (Bakker et al. 2007; Capparelli et al. 2012; Chen et al. 2018). One possibility is that the opposite functions in cellular survival are cell-circumstance dependent. Moreover, several researchers also argue that HIF1α activation is a double-edged sword. Like p53 or autophagy (Zhou et al. 2017a, b, c, d, e, 2018; Jin et al. 2018), moderate HIF1α activation provides the survival advantage for the cell whereas excessive HIF1α activation triggers the programmed cellular death. Therefore, how to regulate the HIF1α balance is vital to influence the cellular viability.
At the molecular level, we demonstrated that HIF1α deletion sustains mitochondrial function and structure. Furthermore, this beneficial effect relied on mitophagy activation. HIF1α deletion signals to mitophagy, which delivers the damaged mitochondria to lysosome. As a consequence of mitophagy, mitochondrial apoptosis is inhibited, contributing to stem cell survival under high glucose-induced injury. To the best of our knowledge, this is the first study to explore the role of HIF1α-mediated mitophagy in ASC survival under high glucose stimulation. Notably, this finding also suggests that the activation of mitophagy is a key mechanism for suppressing stem cell apoptosis.
In animal experiments, compared to normal ASCs, we demonstrated that HIF1α-deleted ASC engraftment further reduced diabetic kidney damage. Mechanistically, HIF1α-deleted ASCs further promoted glomerular survival and reduced renal fibrosis. The additional anti-fibrotic effects may contribute to diabetic nephropathy repair, but the underlying mechanism was not fully investigated in this study. However, based on our data, we confirmed that HIF1α-modified ASCs may be more powerful than unmodified ASCs for treating diabetic renal fibrosis. Notably, due to technical complications, we were unable to detect changes in the survival rate of HIF1α-modified ASCs after transplantation. More studies are needed in the future.
In conclusion, we explored the effectiveness of HIF1α-modified ASCs in treating diabetic renal fibrosis. HIF1α deletion promotes ASC survival via mitophagy activation, leading to improved renal function. Our results provide a new understanding about the protective effects of HIF1α silencing on ASCs and offer a new strategy for promoting the therapeutic efficacy of ASCs in diabetic renal fibrosis.
References
Abdel Aziz MT, Wassef MA, Ahmed HH, Rashed L, Mahfouz S, Aly MI, Hussein RE, Abdelaziz M (2014) The role of bone marrow derived-mesenchymal stem cells in attenuation of kidney function in rats with diabetic nephropathy. Diabetol Metab Syndr 6(1):34. https://doi.org/10.1186/1758-5996-6-34
Azushima K, Gurley SB, Coffman TM (2018) Modelling diabetic nephropathy in mice. Nat Rev Nephrol 14(1):48–56. https://doi.org/10.1038/nrneph.2017.142
Baban B, Liu JY, Payne S, Abebe W, Yu JC, Mozaffari MS (2016) Status of stem cells in diabetic nephropathy: predictive and preventive potentials. EPMA J 7:21. https://doi.org/10.1186/s13167-016-0070-6
Bakker WJ, Harris IS, Mak TW (2007) FOXO3a is activated in response to hypoxic stress and inhibits HIF1-induced apoptosis via regulation of CITED2. Mol Cell 28(6):941–953. https://doi.org/10.1016/j.molcel.2007.10.035
Bhatia M, McGrath KL, Di Trapani G, Charoentong P, Shah F, King MM, Clarke FM, Tonissen KF (2016) The thioredoxin system in breast cancer cell invasion and migration. Redox Biol 8:68–78. https://doi.org/10.1016/j.redox.2015.12.004
Capparelli C, Whitaker-Menezes D, Guido C, Balliet R, Pestell TG, Howell A, Sneddon S, Pestell RG, Martinez-Outschoorn U, Lisanti MP, Sotgia F (2012) CTGF drives autophagy, glycolysis and senescence in cancer-associated fibroblasts via HIF1 activation, metabolically promoting tumor growth. Cell Cycle 11(12):2272–2284. https://doi.org/10.4161/cc.20717
Carloni S, Favrais G, Saliba E, Albertini MC, Chalon S, Longini M, Gressens P, Buonocore G, Balduini W (2016) Melatonin modulates neonatal brain inflammation through endoplasmic reticulum stress, autophagy, and miR-34a/silent information regulator 1 pathway. J Pineal Res 61(3):370–380. https://doi.org/10.1111/jpi.12354
Chen YF, Pandey S, Day CH, Chen YF, Jiang AZ, Ho TJ, Chen RJ, Padma VV, Kuo WW, Huang CY (2018) Synergistic effect of HIF-1alpha and FoxO3a trigger cardiomyocyte apoptosis under hyperglycemic ischemia condition. J Cell Physiol 233(4):3660–3671. https://doi.org/10.1002/jcp.26235
Das N, Mandala A, Naaz S, Giri S, Jain M, Bandyopadhyay D, Reiter RJ, Roy SS (2017) Melatonin protects against lipid-induced mitochondrial dysfunction in hepatocytes and inhibits stellate cell activation during hepatic fibrosis in mice. J Pineal Res 62(4):e12404. https://doi.org/10.1111/jpi.12404
de Luxan-Delgado B, Potes Y, Rubio-Gonzalez A, Caballero B, Solano JJ, Fernandez-Fernandez M, Bermudez M, Rodrigues Moreira Guimaraes M, Vega-Naredo I, Boga JA, Coto-Montes A (2016) Melatonin reduces endoplasmic reticulum stress and autophagy in liver of leptin-deficient mice. J Pineal Res 61(1):108–123. https://doi.org/10.1111/jpi.12333
Dong X, Fu J, Yin X, Qu C, Yang C, He H, Ni J (2017) Induction of apoptosis in HepaRG cell line by aloe-emodin through generation of reactive oxygen species and the mitochondrial pathway. Cell Physiol Biochem 42(2):685–696. https://doi.org/10.1159/000477886
Feng L, Tao L, Dawei H, Xuliang L, Xiaodong L (2014) HIF-1alpha expression correlates with cellular apoptosis, angiogenesis and clinical prognosis in rectal carcinoma. Pathol Oncol Res 20:603–610
Fuhrmann DC, Brune B (2017) Mitochondrial composition and function under the control of hypoxia. Redox Biol 12:208–215. https://doi.org/10.1016/j.redox.2017.02.012
Griffiths HR, Gao D, Pararasa C (2017) Redox regulation in metabolic programming and inflammation. Redox Biol 12:50–57
Haller H, Ji L, Stahl K, Bertram A, Menne J (2017) Molecular mechanisms and treatment strategies in diabetic nephropathy: new avenues for calcium dobesilate-free radical scavenger and growth factor inhibition. Biomed Res Int 2017:1909258
Han J, Weisbrod RM, Shao D, Watanabe Y, Yin X, Bachschmid MM, Seta F, Janssen-Heininger YM, Matsui R, Zang M, Hamburg NM, Cohen RA (2016) The redox mechanism for vascular barrier dysfunction associated with metabolic disorders: glutathionylation of Rac1 in endothelial cells. Redox Biol 9:306–319. https://doi.org/10.1016/j.redox.2016.09.003
Han L, Wang H, Li L, Li X, Ge J, Reiter RJ, Wang Q (2017) Melatonin protects against maternal obesity-associated oxidative stress and meiotic defects in oocytes via the SIRT3-SOD2-dependent pathway. J Pineal Res 63(3):e12431. https://doi.org/10.1111/jpi.12431
Heppner DE, van der Vliet A (2016) Redox-dependent regulation of epidermal growth factor receptor signaling. Redox Biol 8:24–27. https://doi.org/10.1016/j.redox.2015.12.002
Ho HY, Lin CW, Chien MH, Reiter RJ, Su SC, Hsieh YH, Yang SF (2016) Melatonin suppresses TPA-induced metastasis by downregulating matrix metalloproteinase-9 expression through JNK/SP-1 signaling in nasopharyngeal carcinoma. J Pineal Res 61(4):479–492. https://doi.org/10.1111/jpi.12365
Iggena D, Winter Y, Steiner B (2017) Melatonin restores hippocampal neural precursor cell proliferation and prevents cognitive deficits induced by jet lag simulation in adult mice. J Pineal Res 62
Jiang Y, Zhang W, Xu S, Lin H, Sui W, Liu H, Peng L, Fang Q, Chen L, Lou J (2017) Transplantation of human fetal pancreatic progenitor cells ameliorates renal injury in streptozotocin-induced diabetic nephropathy. J Transl Med 15(147):147. https://doi.org/10.1186/s12967-017-1253-1
Jin Q, Li R, Hu N, Xin T, Zhu P, Hu S, Ma S, Zhu H, Ren J, Zhou H (2018) DUSP1 alleviates cardiac ischemia/reperfusion injury by suppressing the Mff-required mitochondrial fission and Bnip3-related mitophagy via the JNK pathways. Redox Biol 14:576–587. https://doi.org/10.1016/j.redox.2017.11.004
Lee K, Back K (2017) Overexpression of rice serotonin N-acetyltransferase 1 in transgenic rice plants confers resistance to cadmium and senescence and increases grain yield. J Pineal Res 62(3):e12392. https://doi.org/10.1111/jpi.12392
Lin YW, Lee LM, Lee WJ, Chu CY, Tan P, Yang YC, Chen WY, Yang SF, Hsiao M, Chien MH (2016) Melatonin inhibits MMP-9 transactivation and renal cell carcinoma metastasis by suppressing Akt-MAPKs pathway and NF-kappaB DNA-binding activity. J Pineal Res 60(3):277–290. https://doi.org/10.1111/jpi.12308
Liu DC, Zheng X, Zho Y, Yi WR, Li ZH, Hu X, Yu AX (2017a) HIF-1alpha inhibits IDH-1 expression in osteosarcoma. Oncol Rep 38(1):336–342. https://doi.org/10.3892/or.2017.5655
Liu L, Li H, Cui Y, Li R, Meng F, Ye Z, Zhang X (2017b) Calcium channel opening rather than the release of ATP causes the apoptosis of osteoblasts induced by overloaded mechanical stimulation. Cell Physiol Biochem 42:441–454
Liu Y, Tang SC (2016) Recent progress in stem cell therapy for diabetic nephropathy. Kidney Dis (Basel) 2(1):20–27. https://doi.org/10.1159/000441913
Lu CC, Ma KL, Ruan XZ, Liu BC (2017) The emerging roles of microparticles in diabetic nephropathy. Int J Biol Sci 13(9):1118–1125. https://doi.org/10.7150/ijbs.21140
Macher-Goeppinger S, Keith M, Hatiboglu G, Hohenfellner M, Schirmacher P, Roth W, Tagscherer KE (2017) Expression and functional characterization of the BNIP3 protein in renal cell carcinomas. Transl Oncol 10:869–875
Mailloux RJ, Craig Ayre D, Christian SL (2016) Induction of mitochondrial reactive oxygen species production by GSH mediated S-glutathionylation of 2-oxoglutarate dehydrogenase. Redox Biol 8:285–297
Mailloux RJ, Treberg JR (2016) Protein S-glutathionlyation links energy metabolism to redox signaling in mitochondria. Redox Biol 8:110–118. https://doi.org/10.1016/j.redox.2015.12.010
Martinez-Outschoorn UE, Trimmer C, Lin Z, Whitaker-Menezes D, Chiavarina B, Zhou J, Wang C, Pavlides S, Martinez-Cantarin MP, Capozza F, Witkiewicz AK, Flomenberg N, Howell A, Pestell RG, Caro J, Lisanti MP, Sotgia F (2010) Autophagy in cancer associated fibroblasts promotes tumor cell survival: role of hypoxia, HIF1 induction and NFkappaB activation in the tumor stromal microenvironment. Cell Cycle 9(17):3515–3533. https://doi.org/10.4161/cc.9.17.12928
Matsui R, Watanabe Y, Murdoch CE (2017) Redox regulation of ischemic limb neovascularization—what we have learned from animal studies. Redox Biol 12:1011–1019
Nagaishi K, Mizue Y, Chikenji T, Otani M, Nakano M, Konari N, Fujimiya M (2016) Mesenchymal stem cell therapy ameliorates diabetic nephropathy via the paracrine effect of renal trophic factors including exosomes. Sci Rep 6:34842
Pan XH, Yang XY, Yao X, Sun XM, Zhu L, Wang JX, Pang RQ, Cai XM, Dai JJ, Ruan GP (2014) Bone-marrow mesenchymal stem cell transplantation to treat diabetic nephropathy in tree shrews. Cell Biochem Funct 32(5):453–463. https://doi.org/10.1002/cbf.3037
Paulini J, Higuti E, Bastos RM, Gomes SA, Rangel EB (2016) Mesenchymal stem cells as therapeutic candidates for halting the progression of diabetic nephropathy. Stem Cells Int 2016(9521629):1–16. https://doi.org/10.1155/2016/9521629
Peleli M, Zollbrecht C, Montenegro MF, Hezel M, Zhong J, Persson EG, Holmdahl R, Weitzberg E, Lundberg JO, Carlstrom M (2016) Enhanced XOR activity in eNOS-deficient mice: effects on the nitrate-nitrite-NO pathway and ROS homeostasis. Free Radic Biol Med 99:472–484. https://doi.org/10.1016/j.freeradbiomed.2016.09.004
Shi C, Cai Y, Li Y, Li Y, Hu N, Ma S, Hu S, Zhu P, Wang W, Zhou H (2018) Yap promotes hepatocellular carcinoma metastasis and mobilization via governing cofilin/F-actin/lamellipodium axis by regulation of JNK/Bnip3/SERCA/CaMKII pathways. Redox Biol 14:59–71. https://doi.org/10.1016/j.redox.2017.08.013
Sigala F, Efentakis P, Karageorgiadi D, Filis K, Zampas P, Iliodromitis EK, Zografos G, Papapetropoulos A, Andreadou I (2017) Reciprocal regulation of eNOS, H2S and CO-synthesizing enzymes in human atheroma: correlation with plaque stability and effects of simvastatin. Redox Biol 12:70–81. https://doi.org/10.1016/j.redox.2017.02.006
Smith MR, Vayalil PK, Zhou F, Benavides GA, Beggs RR, Golzarian H, Nijampatnam B, Oliver PG, Smith RA, Murphy MP, Velu SE, Landar A (2016) Mitochondrial thiol modification by a targeted electrophile inhibits metabolism in breast adenocarcinoma cells by inhibiting enzyme activity and protein levels. Redox Biol 8:136–148. https://doi.org/10.1016/j.redox.2016.01.002
Tamura H, Kawamoto M, Sato S, Tamura I, Maekawa R, Taketani T, Aasada H, Takaki E, Nakai A, Reiter RJ, Sugino N (2017) Long-term melatonin treatment delays ovarian aging. J Pineal Res 62(2):e12381. https://doi.org/10.1111/jpi.12381
Tan DX, Hardeland R, Back K, Manchester LC, Alatorre-Jimenez MA, Reiter RJ (2016) On the significance of an alternate pathway of melatonin synthesis via 5-methoxytryptamine: comparisons across species. J Pineal Res 61(1):27–40. https://doi.org/10.1111/jpi.12336
Wu T, Qiao S, Shi C, Wang S, Ji G (2017) Metabolomics window into diabetic complications. J Diabetes Investig. https://doi.org/10.1111/jdi.12723
Xu J, Wu Y, Lu G, Xie S, Ma Z, Chen Z, Shen HM, Xia D (2017) Importance of ROS-mediated autophagy in determining apoptotic cell death induced by physapubescin B. Redox Biol 12:198–207. https://doi.org/10.1016/j.redox.2017.02.017
Xu S, Pi H, Zhang L, Zhang N, Li Y, Zhang H, Tang J, Li H, Feng M, Deng P, Guo P, Tian L, Xie J, He M, Lu Y, Zhong M, Zhang Y, Wang W, Reiter RJ, Yu Z, Zhou Z (2016) Melatonin prevents abnormal mitochondrial dynamics resulting from the neurotoxicity of cadmium by blocking calcium-dependent translocation of Drp1 to the mitochondria. J Pineal Res 60(3):291–302. https://doi.org/10.1111/jpi.12310
Yang HH, Chen Y, Gao CY, Cui ZT, Yao JM (2017) Protective effects of MicroRNA-126 on human cardiac microvascular endothelial cells against hypoxia/reoxygenation-induced injury and inflammatory response by activating PI3K/Akt/eNOS signaling pathway. Cell Physiol Biochem 42(2):506–518. https://doi.org/10.1159/000477597
Yu S, Wang X, Geng P, Tang X, Xiang L, Lu X, Li J, Ruan Z, Chen J, Xie G, Wang Z, Ou J, Peng Y, Luo X, Zhang X, Dong Y, Pang X, Miao H, Chen H, Liang H (2017) Melatonin regulates PARP1 to control the senescence-associated secretory phenotype (SASP) in human fetal lung fibroblast cells. J Pineal Res 63(1):e12405. https://doi.org/10.1111/jpi.12405
Zhang Y, Zhou H, Wu W, Shi C, Hu S, Yin T, Ma Q, Han T, Zhang Y, Tian F, Chen Y (2016) Liraglutide protects cardiac microvascular endothelial cells against hypoxia/reoxygenation injury through the suppression of the SR-Ca(2+)-XO-ROS axis via activation of the GLP-1R/PI3K/Akt/survivin pathways. Free Radic Biol Med 95:278–292. https://doi.org/10.1016/j.freeradbiomed.2016.03.035
Zhao S, Li L, Wang S, Yu C, Xiao B, Lin L, Cong W, Cheng J, Yang W, Sun W, Cui S (2016) H2O2 treatment or serum deprivation induces autophagy and apoptosis in naked mole-rat skin fibroblasts by inhibiting the PI3K/Akt signaling pathway. Oncotarget 7(51):84839–84850. https://doi.org/10.18632/oncotarget.13321
Zhou H, Du W, Li Y, Shi C, Hu N, Ma S, Wang W, Ren J (2018) Effects of melatonin on fatty liver disease: the role of NR4A1/DNA-PKcs/p53 pathway, mitochondrial fission, and mitophagy. J Pineal Res 64(1):e12450. https://doi.org/10.1111/jpi.12450
Zhou H, Hu S, Jin Q, Shi C, Zhang Y, Zhu P, Ma Q, Tian F, Chen Y (2017a) Mff-dependent mitochondrial fission contributes to the pathogenesis of cardiac microvasculature ischemia/reperfusion injury via induction of mROS-mediated cardiolipin oxidation and HK2/VDAC1 disassociation-involved mPTP opening. J Am Heart Assoc 6(3):e005328. https://doi.org/10.1161/JAHA.116.005328
Zhou H, Li D, Shi C, Xin T, Yang J, Zhou Y, Hu S, Tian F, Wang J, Chen Y (2015a) Effects of exendin-4 on bone marrow mesenchymal stem cell proliferation, migration and apoptosis in vitro. Sci Rep 5:12898
Zhou H, Li D, Zhu P, Hu S, Hu N, Ma S, Zhang Y, Han T, Ren J, Cao F, Chen Y (2017b) Melatonin suppresses platelet activation and function against cardiac ischemia/reperfusion injury via PPARgamma/FUNDC1/mitophagy pathways. J Pineal Res 63(4):e12438. https://doi.org/10.1111/jpi.12438
Zhou H, Yang J, Xin T, Li D, Guo J, Hu S, Zhou S, Zhang T, Zhang Y, Han T, Chen Y (2014) Exendin-4 protects adipose-derived mesenchymal stem cells from apoptosis induced by hydrogen peroxide through the PI3K/Akt-Sfrp2 pathways. Free Radic Biol Med 77:363–375. https://doi.org/10.1016/j.freeradbiomed.2014.09.033
Zhou H, Yang J, Xin T, Zhang T, Hu S, Zhou S, Chen G, Chen Y (2015b) Exendin-4 enhances the migration of adipose-derived stem cells to neonatal rat ventricular cardiomyocyte-derived conditioned medium via the phosphoinositide 3-kinase/Akt-stromal cell-derived factor-1alpha/CXC chemokine receptor 4 pathway. Mol Med Rep 11(6):4063–4072. https://doi.org/10.3892/mmr.2015.3243
Zhou H, Zhang Y, Hu S, Shi C, Zhu P, Ma Q, Jin Q, Cao F, Tian F, Chen Y (2017d) Melatonin protects cardiac microvasculature against ischemia/reperfusion injury via suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis. J Pineal Res 63(1):e12413. https://doi.org/10.1111/jpi.12413
Zhou H, Zhu P, Guo J, Hu N, Wang S, Li D, Hu S, Ren J, Cao F, Chen Y (2017e) Ripk3 induces mitochondrial apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury. Redox Biol 13:498–507. https://doi.org/10.1016/j.redox.2017.07.007
Zhou W, Yu L, Fan J, Wan B, Jiang T, Yin J, Huang Y, Li Q, Yin G, Hu Z (2017c) Endogenous parathyroid hormone promotes fracture healing by increasing expression of BMPR2 through cAMP/PKA/CREB pathway in mice. Cell Physiol Biochem 42(2):551–563. https://doi.org/10.1159/000477605
Zhu H, Jin Q, Li Y, Ma Q, Wang J, Li D, Zhou H, Chen Y (2017) Melatonin protected cardiac microvascular endothelial cells against oxidative stress injury via suppression of IP3R-[Ca(2+)]c/VDAC-[Ca(2+)]m axis by activation of MAPK/ERK signaling pathway. Cell Stress Chaperones 23(1):101–113. https://doi.org/10.1007/s12192-017-0827-4
Zhu YW, Yan JK, Li JJ, Ou YM, Yang Q (2016) Knockdown of radixin suppresses gastric cancer metastasis in vitro by up-regulation of E-cadherin via NF-kappaB/snail pathway. Cell Physiol Biochem 39(6):2509–2521. https://doi.org/10.1159/000452518
Funding
This study was supported by project supported by National Natural Science Foundation of People’s Republic of China (No. 81503442, No. 81603470) and research project of Hunan Provincial Department of Education (No. 17B197, No. 16B196).
Author information
Authors and Affiliations
Contributions
QT, CYL, and HW conceived the research; QT, JSL, CY, WFX, and WQL performed the experiments; FY and QT analyzed the data; QT and CYL wrote the manuscript; all authors participated in discussing and revising the manuscript.
Corresponding author
Ethics declarations
Our study was carried out in accordance with the guidelines of the Ethics Committee of Hunan University of Chinese Medicine, Changsha, China. The experimental protocol was approved by Ethics Committee of Hunan University of Chinese Medicine, Changsha, China.
Conflict of interest
The authors declare that they have no conflict of interest.
Disclaimer
The funders had no role in the study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Additional information
Editor: Tetsuji Okamoto
Rights and permissions
About this article

Cite this article
Tang, Q., Wu, H., Lei, J. et al. HIF1α deletion facilitates adipose stem cells to repair renal fibrosis in diabetic mice. In Vitro Cell.Dev.Biol.-Animal 54, 272–286 (2018). https://doi.org/10.1007/s11626-018-0231-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11626-018-0231-0