
Abstract
Clear cell sarcoma (CCS) is an aggressive mesenchymal malignancy characterized by the unique chimeric EWS-ATF1 fusion gene. Patient-derived cancer models are essential tools for the understanding of tumorigenesis and the development of anti-cancer drugs; however, only a limited number of CCS cell lines exist. The objective of this study was to establish patient-derived CCS models. We established patient-derived CCS models from a 43-yr-old female patient. We prepared the patient-derived xenografts (PDXs) from tumor tissues obtained through biopsy or surgery and isolated stable cell lines from PDXs and the original tumor tissue. The presence of gene fusions was examined by RT-PCR, and Sanger sequencing. The established cell lines were characterized by short tandem repeat, viability, colony and spheroid formation, and invasion analyses. Differences in gene enrichment between the primary tumor and cell lines were examined by mass spectrometry and KEGG pathway analysis. The cell lines were maintained for more than 80 passages, and had tumorigenic characteristics such as colony and spheroid formation and invasion. Mass spectrometric proteome analysis demonstrated that the cell lines were enriched for similar but distinct molecular pathways, compared to those in the xenografts and original tumor tissue. Next, tyrosine kinase inhibitors were screened for their suppressive effects on viability. We found that ponatinib, vandetanib, and doxorubicin suppressed the growth of cell lines, and had equivalent IC50 values. Further in-depth investigation and understanding of drug-sensitivity mechanisms will be important for the clinical applications of our cell lines.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Clear cell sarcoma (CCS) is a mesenchymal malignancy of the tendons and aponeuroses that exhibits melanocytic differentiation (Enzinger 1965; Fletcher et al. 2013; JR et al. 2013). This disease arises in the extremities of young adults, appearing as a deep-seated, slowly-growing mass, and results in an aggressive malignancy; approximately 50% of patients develop lung or nodal metastases, and show resistance to conventional chemotherapy and radiation therapy. The prognosis of CCS is dismal, and the 5-year overall survival remains at 30–70% (Sara et al. 1990; el-Naggar et al. 1991; Lucas et al. 1992; EA et al. 1993; Deenik et al. 1999; Finley et al. 2001; Ferrari et al. 2002; Takahira et al. 2004; Kawai et al. 2007). Characteristic genetic aberrations such as the clonal chromosomal translocation, specifically t(12;22)(q13;q12), and EWS-ATF1 gene fusions have been reported (Bridge et al. 1991; Zucman et al. 1993). Although the gene fusion might play an important role in the carcinogenesis of CCS, molecular targeting drugs specific for the associated products have not been developed. Recently, the novel multi-tyrosine kinase inhibitor pazopanib was approved for the treatment of soft-tissue sarcoma (van der Graaf et al. 2012). However, there has been no conclusive report regarding the response of CCS to such treatments. Therefore, a novel therapeutic strategy is required to improve clinical outcome in CCS.
Patient-derived cancer models are valuable tools for studying the molecular backgrounds of malignant phenotypes, examining the roles of genes in carcinogenesis, and investigating the effects and mode of actions of anti-cancer drugs (Iorio et al. 2016). Despite the urgent need for novel therapeutic strategies in CCS, a small number of derived cell lines have been successfully established (Epstein et al. 1984; Sonobe et al. 1993; Takenouchi et al. 1994; Brown et al. 1995; Liao et al. 1996; Hiraga et al. 1997; Crnalic et al. 2002; Moritake et al. 2002; Schaefer et al. 2002; Schaefer et al. 2004; Davis et al. 2006; Outani et al. 2014). Establishing additional CCS cell lines as well as xenograft models is thus required for the development of novel therapeutic strategies for this disease.
In this study, we established two patient-derived xenografts (PDXs) and three stable cell lines from CCS. We then examined their morphological and functional characteristics, protein expression profile, and response to anti-cancer drugs. The use of these preclinical models might help to elucidate the molecular mechanisms underlying the poor prognosis associated with CCS, and facilitate the development of novel therapeutic strategies for this malignancy.
Materials and Methods
Patient background
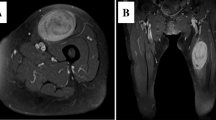
The patient-derived cancer models were generated from a 43-yr-old female patient with CCS in the National Cancer Center Hospital. The tumor was multinodular and located in the left thigh with invasion into the femur (Fig. 1 a). Multiple lymph node metastases were found by computed tomography (Fig. 1 b). RT-PCR suggested the fusion of EWSR1 with ATF1 in the tumor cells from the biopsy sample and surgically resected specimen (Supplementary Fig. 1). Tumor histology is shown in Fig. 1 c. Sanger sequencing confirmed the presence of this fusion in the biopsy sample (Fig. 1 d) and surgically resected specimen (Fig. 1 e). The patient was treated with left hip disarticulation and postoperative radiotherapy targeting lymph node metastases inside the pelvis. The patient died from the disease two mo after surgery. This study was approved by the ethics committee of the National Cancer Center, and written informed consent was obtained from the patient.

Characteristics of original clear cell sarcoma tumor tissue in this study. (a) T2-weighted magnetic resonance image showing a multinodular soft tissue mass in the left thigh (arrow). (b) Commuted tomography showed an enlarged external iliac lymph node (arrow). H&E staining of the surgically resected sample (c). Original magnification is 400×. Sanger sequencing of the gene fusion in surgically resected tumor (d) and biopsied (e) tissues.
Patient-derived xenografts
Several CCS tissue pieces, obtained by biopsy or surgical resection, were subcutaneously engrafted with a 13-gauge transplant needle into the hind bilateral flanks of 6–12-wk-old female severe immunodeficient NOD.Cg-Prkdc scid Il2rg tm1Sug /Jic (also known as NOD/Shi-scid IL-2Rγ null or NOG) mice (Central Institute for Experimental Animals, Tokyo, Japan). When the tumors reached 500–1000 mm3, they were transplanted into another recipient mouse. Tumor size was measured periodically using digital calipers (SuperCaliper, Mitutoyo, Kanagawa, Japan). Tumor volume was calculated as π/6 × length × width × thickness (Tomayko and Reynolds 1989). After two passages, the tumors were cryopreserved using Cellbanker 1 plus (Takara Bio, Shiga, Japan) in liquid nitrogen. All animal experiments were performed in accordance with the guidelines for Animal Experiments of the National Cancer Center and approved by the Institutional Committee for Ethics of Animal Experimentation.
Primary tissue culture
Tumor tissue was minced with scissors and the cells were seeded in a tissue culture plate (Thermo Fisher Scientific, Waltham, MA). The cells were maintained in DMEM medium (Sigma-Aldrich, St. Louis, MO) supplemented with 10% heat-inactivated fetal bovine serum (FBS; Gibco, Grand Island, NY), 100 U penicillin G, and 100 μg/ml streptomycin (Gibco) at 37°C in a humidified atmosphere of 5% CO2. The culture medium was changed once or twice per wk. When cells reached sub-confluence, they were dispersed with 0.1% trypsin-EDTA (Gibco) and seeded in a new culture plate. Cells were maintained for more than 30 mo and 100 passages.
Immunohistochemical analysis of CCS cells
Tumor cells were detached through trypsin treatment, and cell suspensions were solidified using iPGell (Genostaff, Tokyo, Japan) according to the manufacturer’s instructions. Cell masses were fixed with 10% formalin and embedded in paraffin. Cell blocks were cut into sections, which were processed for immunolabeling and H&E staining. Sections were incubated with antibodies against the following proteins: S100 (polyclonal, 1:2000), HMB45 (1:100), and MelanA (1:100) (all from Dako). Immunoreactivity was detected with the EnVision system (Dako).
Authentication and quality control of established cell line
Genomic DNA was extracted from the tumor tissues or cell lines using AllPrep DNA/RNA Mini kits (Qiagen, Hilden, Germany), quantified using a NanoDrop 8000 (Thermo Fisher Scientific), and stored at −80°C until use. To authenticate the cells, we analyzed short tandem repeats (STRs) at 10 loci using STR multiplex assays (GenePrint 10; Promega, Madison, WI), according to the manufacturer’s instructions. Genomic DNA (500 pg) was amplified and then examined using a 3500xL Genetic Analyzer (Applied Biosystems, Foster City, CA). The data were analyzed using PeakScanner software (Applied Biosystems) and the STR profiles were compared with those recorded in public cell banks including the American Type Culture Collection (ATCC), Deutsche Sammlung von Mikroorganismen und Zellkulturen (DSMZ), and the Japanese Collection of Research Bioresources Cell Bank (JCRB) for reference matching.
DNA in the culture medium was examined to rule out the possibility of contamination by mycoplasma. Briefly, DNA was isolated from the medium of cultures at 70–90% confluency, heated at 95°C for 10 min, and then amplified using the e-Myco Mycoplasma PCR Detection Kit (Intron Biotechnology, Gyeonggi-do, Korea). The amplified DNA was separated by 1.5% agarose gel electrophoresis, stained with Midori Green Advanced (Nippon Genetics, Tokyo, Japan), and detected using an Amersham Imager 600 (GE Healthcare Biosciences, Little Chalfont, UK).
Detection of fusion gene product by RT-PCR and sanger sequencing
Total RNA (1 μg) was used for the reverse transcription (RT) reaction with Superscript III reverse transcriptase (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. PCR amplifications were performed using a 25-μl reaction volume containing 1 unit of PlatinumTaq polymerase (Thermo Fisher Scientific, Waltham, MA), 0.5 μM each of the EWS1 forward primer EWS225F, 5′-ACAGTTATCCCCAGGTACCTGGG-3′ and the ATF1 reverse primer ATF803R, 5′-GGGGGTCATCTGTCTTAGTTGTCTG-3′, and 1 μl of cDNA (Panagopoulos et al. 2002). The amplification program consisted of an initial denaturation at 94°C for 2 min and then 40 cycles of denaturation at 94°C for 15 s, annealing at 57°C for 30 s, extension at 68°C for 30 s, and a final extension at 72°C for 5 min using the GeneAmp PCR System 9700 (Applied Biosystems). The sequence data were matched against the EWSR1 (NM_001163285.1) and ATF1 (NM_005171.4) sequences, using BLAST (NIH, Bethesda, MD).
Cell growth analysis
The growth characteristics of the cell line were analyzed as follows. Cells were seeded in triplicate in 96-well culture plates at 2 or 4 × 103 cells/well. Over the next 1–4 days, tumor cell proliferation was evaluated using the Cell Counting Kit (CCK)-8 kit (Dojindo Molecular Technologies, Kumamoto, Japan), according to the manufacturer’s protocol. Briefly, CCK-8 reagent was added to each well, which was followed by incubation at 37°C for 2 h. The absorbance at 450 nm was measured using a microplate reader (Bio-Rad, Hercules, CA) and plotted as a function of days after cell seeding. The PCR products were purified with ExoSAP-IT (Affymetrix) and direct sequencing was performed using BigDye v3.1 Cycle Sequencing Kit (Applied Biosystems) on the Applied Biosystems 3130xL by Eurofin genomics (Japan).
Colony formation assay
For colony formation assays, the bottom of a 35-mm dish was coated with 0.5% low melting agarose (Noble agar; Sigma-Aldrich) with Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with 10% FBS and penicillin/streptomycin (Thermo Fisher Scientific). Wells were then covered with 0.33% agarose containing 5 × 104 cells. The plates were incubated at 37°C and 5% CO2 for 4 weeks, and colonies containing more than 50 cells were identified using a Primo Vert inverted microscope (Keyence). The assay was performed in duplicate.
Spheroid formation assay
For spheroid formation assays, 1 × 105 cells were seeded with DMEM containing 10% FBS in 6-cm plates (Ultra Low Culture Dish, Thermo Fisher Science). The cells were incubated in a humidified atmosphere containing 5% CO2 at 37°C. Two weeks after inoculation, the presence of spheroids was confirmed by microscopic observation (Keyence). All assays were performed in duplicate.
Cell invasion assay
Invasion assays were performed using BD Biocoat Matrigel Invasion Chambers (BD Biosciences, Bedford, MA) according to the manufacturer’s instructions. Briefly, cells were plated with number of 2 × 105, 1.0 × 105, or 0.5 × 105 were plated in serum-free DMEM in the upper chamber. DMEM containing 10% FBS was added to the bottom chamber. After 5 days of culturing, invading cells on the bottom surface were fixed and stained; three separate areas were counted at ×100 magnification. Percent invasion was calculated by subtracting the control chamber ratio from the number of cells in the Matrigel chamber.
Proteomic analysis
Filter-aided sample preparation (FASP) was performed to digest the proteins as previously described (Wisniewski et al. 2009). Briefly, proteins were loaded on microcon-30 kDa filters. Following two washes with urea, proteins were alkylated with 50 mM iodoacetamide. Filters were washed twice with urea and twice with 40 mM ammonium bicarbonate, and digested overnight with trypsin (1∶50; w/w) at 37°C. The resulting peptides were collected by centrifugation. The filters were rinsed twice with 40 μl NH4HCO3 and 0.5 M NaCl, and dried using a SpeedVac concentrator (Thermo Fisher Scientific).
Peptides were separated by reverse-phase chromatography using a 15-cm column (inner diameter 75 μm, 3 μm C18-AQ media), using a nanoflow high performance liquid chromatography (HPLC) system (AMR, Tokyo, Japan). HPLC was coupled on-line, via a nanoelectrospray ion source, to a LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific). Peptides were loaded onto the column with a 170-min gradient from 5% to 45% of 90% (v/v) CH3CN and 0.1% (v/v) formic acid at a flow rate of 250 nl/min. Mass spectra were acquired in the positive ion mode applying a data-dependent automatic switch between survey scan and tandem mass spectra (MS/MS) acquisition. Samples were analyzed using the ‘top 10’ method, acquiring one Orbitrap survey scan in the mass range of m/z 350–1800 followed by MS/MS of the five most intense ions in the LTQ. The target value in the Orbitrap was 500,000 ions for the survey scan at a resolution of 60,000 at m/z 400. Fragmentation in the LTQ was performed by collision-induced dissociation with a target value of 10,000 ions. The ion selection threshold was 500 counts.
MS/MS spectra were searched against the Swiss-Prot database (Homo sapiens, 20,205 sequences in the Swiss Prot_2015_09.fasta file) using the MASCOT search engine (version 2.5.1; Matrix Science, UK). The search included variable modifications of methionine oxidation and serine, threonine, and tyrosine phosphorylations, and fixed modification of cysteine carbamidomethylation. A maximum of one miscleavage was allowed. The maximal precursor ion mass tolerance was set to ±10 ppm, whereas the fragment ion mass tolerance was set to ±0.8 Da. Peptide charge was 2+ and 3 + .
Sensitivity to anti-cancer drugs
Cells were seeded in a 384-well plate at 5 × 103 cells/well in DMEM supplemented with 10% FBS and 1% antibiotic-antimycotic (Thermo Fisher Scientific) and incubated at 37°C in a humidified atmosphere of 5% CO2. The following day, cells were treated with various concentrations of anti-cancer reagents (Selleck Chemicals, Houston, TX). After 72 h, survival rates were assessed using CCK-8, according to the manufacturer’s protocol. Anti-cancer drugs used in this study are listed in Supplementary Table 1.
Kinase activity assay
Each sample was lysed and analyzed independently on a separate array. Kinomic profiling of 10 μg of protein lysate (lysed in M-Per lysis buffer, Pierce Scientific, containing 1:100 Halt’s protease and phosphatase inhibitors (Pierce Scientific)) was conducted on the PamStation 12 platform, manufactured by PamGene (‘s-Hertogenbosch, Netherlands). Protein concentrations were determined by performing a Bradford protein assay (BioRad, Hercules, CA), and the lysates were dissolved in kinase buffer. Subsequently, the lysates were loaded onto the PamChip PTK, on which tyrosine kinase substrate peptides are arrayed. FITC-conjugated phospho-specific antibodies (PamGene) were used for the detection of phospho-tyrosine during and after pumping lysates through the array. The status of peptide phosphorylation was monitored using a computer-controlled CCD camera. The phosphorylation status of substrate peptides was analyzed using Evolve (PamGene) and BioNavigator (PamGene). Peptide spot intensity was captured across multiple exposure times (10, 20, 50,100, 200 ms) and calculated as described previously (Anderson et al. 2015). The intensity of peptide phosphorylation was used for unsupervised hierarchical clustering using heatmap.2 R script (https://cran.r-project.org/web/packages/gplots/index.html). The prediction of upstream kinases was achieved using the Kinexus Kinase Predictor (www.phosphonet.ca). Pathways involving the predicted kinases were predicted by DAVID functional annotation tools (Huang et al. 2009a, b).
Statistical analysis
Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway annotations were inferred using the Database for Annotation, Visualization and Integration Discovery (DAVID) software (http://david.abcc.ncifcrf.gov/) (Altermann and Klaenhammer 2005). Significance is indicated based on the p-value for each category and differences were considered significant at p < 0.05. The R package ‘treemap’ was used for plotting (Tyanova et al. 2016).
Results
The overall work flow for establishing the patient-derived cancer model in this study is illustrated in Fig. 2. We used both biopsy samples and surgically resected tumor tissue. We also established cell lines from biopsy samples and PDXs from biopsy samples or surgically-resected tissue. Although we tried to establish cell lines from surgically resected tumor tissue, the cells did not grow with in vitro tissue culture conditions. The names of the cell lines are provided in Fig. 2. The cell line directly established from the biopsied tumor tissue was named NCC-CCS1B-C1. The cell line derived from xenograft tumor tissue passaged once was named NCC-CCS1B-X2-C1. The xenograft from surgically-resected tumor samples (e.g. the first generation xenograft) was further passaged twice, frozen once in liquid nitrogen, and subcutaneously engrafted. The cell line prepared from the corresponding tumor tissue was named NCC-CCS1-X4-C1.

Overall work flow for cell and xenograft passage from clear cell sarcoma specimen. Xenografts were established from both biopsy and surgically-resected tumor tissues. The cell lines were established from xenografts using biopsy and surgically-resected samples. The names of the cell lines are shown under the pictures of the cell lines.
PDX growth and morphology
The patient-derived biopsy samples and surgically resected tumor tissue were subcutaneously inoculated into immune-deficient mice. After propagation in vivo, they were serially transplanted twice. Xenograft tumor histology of the second generation is shown in Fig. 3 a, and that of the fourth generation of the PDX from surgically resected tissue is shown in Fig. 3 b. Histological features were quite similar between the two samples, and the original tumor tissue (Fig. 1 c). Both in the PDXs from biopsy sample and the surgically-resected ones, the tumors started to increase in size around 20 d after transplantation, and reached the size enough for transplantation (Figs. 3 c and d). Third-generation xenograft tissues of both PDXs were frozen and later inoculated in mice; this confirmed successful tumor propagation after prolonged storage. RT-PCR identified the gene fusion in both PDXs (Supplementary Fig. 1). Sanger sequencing confirmed the presence of the fusion gene transcript, EWSR1-ATF1, in both xenograft tumors (Fig. 3 e and f).

Histological analysis and growth curves of xenograft tumors derived from biopsy or surgical specimens of clear cell sarcoma and their fusion gene analysis the tumor consisted of intersecting fascicles of pleomorphic, spindle-shaped cells exhibiting an eosinophilic cytoplasm. H&E staining of the second generation xenograft tissue derived from biopsy sample (a) and the fourth generation xenograft tissue derived from surgically resected sample (b). Original magnification: 400× (a, b). The xenograft tumors started to increase in size around 20 d after transplantation (c, d). Sanger sequencing revealed the presence of a unique fusion gene in both xenograft tissues (e, f).
Authentication of established cell lines
We examined the status of 10 STRs in the original tumor tissue, PDXs, and cell lines (Supplementary Table 2). All samples exhibited the identical STR pattern, except at the TH01 loci. We therefore concluded that the established PDXs and cell lines were derived from the same patient. A database search confirmed that the obtained STR patterns did not match any STR signatures from cell lines in public cell banks such as ATCC, DSMZ, and JRCB. Therefore, we concluded that the established PDXs and cell lines were novel.
We investigated the presence of mycoplasma DNA in the cell lines by PCR, and found no evidence of contamination (data not shown).
Phenotypic characterization of established cell lines
The presence of unique fusion gene products in the cell lines was confirmed by both RT-PCR (Supplementary Fig. 1) and Sanger sequencing (Supplementary Fig. 2). The three established cell lines were spindle shaped (Fig. 4 a–c), consistent with the morphology of cells in the original tumor tissue. When the cells were seeded on low-attachment tissue culture dishes, cell lines from the biopsied tumor tissues formed spheroids, whereas the cell line from surgically resected tissue, NCC-CCS1-X4-C1, did not (Fig. 4 d–f). Also NCC-CCS1B-C1 and NCC-CCS1B-X2-C1cells formed colonies but NCC-CCS1-X4-C1 cells did not when they were seeded in soft agar (Fig. 4 g–i). These observations might suggest that tumor tissues and xenografts are heterogeneous. We then confirmed the presence of the fusion gene, EWSR1-ATF1, in all three cell lines by Sanger sequencing (Supplementary Fig. 2). In addition, the cells showed continual growth when different numbers were seeded (Supplementary Fig. 3 a–c), indicating that these cells would be useful for cell growth experiments. The population doubling times of individual cell lines were found to be 20, 20, and 17 h for NCC-CCS1B-X2-C1, NCC-CCS1B-C1, and NCC-CCS1-X4-C1 cells, respectively. We found that all three cell lines had equivalent invasion ability (Supplementary Fig. 3 d–g). The expression of typical biomarker gene products was evaluated by immunohistochemistry staining (Supplementary Fig. 4). Positive expression of S100 was observed in NCC-CCS1B-X2-C1, NCC-CCS1B-C1, and NCC-CCS1-X4-C1 cells. Positive but sparse expression of HMB45 was observed in NCC-CCS1B-C1 cells, and 100% of cells exhibited positive expression of HMB45 in NCC-CCS1B-X2-C1 and NCC-CCS1-X4-C1 cell lines. NCC-CCS1-X4-C1 cells showed negative expression of Melan A; however, a subset of NCC-CCS1B-X2-C1 cells exhibited positive expression of this marker. In addition, 70–80% of NCC-CCS1-X4-C1 cells showed positive expression of Melan A.

Phenotypic analysis of the clear cell sarcoma-derived cell lines. Phase-contrast images of NCC-CCS1 (a), NCC-CCS1B-X2-C1 (b), and NCC-CCS1-X4-C1 (c) cells cultured in a serum-containing medium. Phase-contrast images of NCC-CCS1-C1 (d), NCC-CCS1B-X2-C1 (e), and NCC-CCS1-X4-C1 (f) cells cultured in ultra-low attachment culture dishes. Morphology of NCC-CCS1 (g), NCC-CCS1B-X2-C1 (h), and NCC-CCS1-X4-C1 (i) cells in soft agar. NCC-CCS1 and NCC-CCS1B-X2-C1Cells exhibited anchorage-independent growth on soft agar.
Proteomic profiling
Protein expression profiles of the primary tumor and a cell line were obtained by mass spectrometry (Supplementary Table 3–8). The proteins identified in the individual samples were classified according to their possible functions based on data from the KEGG database, and the top 10 most enriched pathways are presented in treemap format (Fig. 5 and Supplementary Table 7), wherein area size is indicative of the number of proteins allocated, and the colors indicate the degree of statistical enrichment. In the primary tumor, proteins in the biosynthesis of antibiotics pathway were predominantly detected and enriched (Fig. 5 a). In the PDX lines, the overall appearance of the treemap was very similar to that of the primary tumor. Proteins from the biosynthesis of antibiotics pathway were mostly detected, and the ribosome pathway was the most significantly enriched pathway (Fig. 5 b–c).

Tree map view of Kyoto encyclopedia of genes and genomes (KEGG) pathway categories for proteins identified by mass spectrometry from clear cell sarcoma-derived cell lines and xenografts. (a–f) KEGG pathway categories for proteins enriched in surgically resected tumor tissue (a), NCC-CCS1B-X2 cells (b), NCC-CCS1-X4 tissue (c), NCC-CCS1B-X2-C1 cells (d), NCC-CCS1B-C1 cells (e), and NCC-CCS1-X4-C1 cells (f) are shown as a tree map. The names of the pathways are shown in each rectangle. The color and size of individual areas correspond to the degree of enrichment and the number of proteins assigned to the category, respectively. P < 0.05 was considered statistically significant for pathway enrichment.
In the cell line, the overall appearance of the treemap was similar to that of PDX lines rather than that of the primary tumor. Proteins from the biosynthesis of antibiotics or metabolic pathway were mostly detected, and the ribosome pathway was the most significantly enriched pathway (Fig. 5 d–f). The enrichment scores for the biosynthesis of antibiotics, carbon metabolism, protein processing in endoplasmic reticulum, and cysteine and methionine metabolism pathways were decreased in the PDX and cell lines compared to those in the original tumor, and the enrichment score for the ribosome pathway was higher in the cell lines (Fig. 5).
A treemap of common pathways among all samples is shown in Fig. 6. Biosynthesis of antibiotics, ribosome, and carbon metabolism pathways were for the most part enriched in all cell lines, indicating representative pathways for these samples (Fig. 6 a). Ribosome, biosynthesis of antibiotics, and biosynthesis of amino acids pathways were enriched in both PDX models (Fig. 6 b). Ribosome, biosynthesis of antibiotics, and carbon metabolism pathways were enriched, when comparing PDX lines and cells (Fig. 6 c). The enriched pathways in the primary and established cell lines, as well as the common ones, are shown in Supplementary Table 9.

Tree map view of Kyoto encyclopedia of genes and genomes (KEGG) pathway categories for proteins identified by mass spectrometry from clear cell sarcoma-derived cell lines and xenografts. (left panel) Venn diagrams showing a comparison among all cell lines (a), among all patient-derived xenograft (PDX) lines (b), and between integrated cell lines and integrated PDX lines (c). (middle panel) KEGG pathway categories for proteins common to all cell lines (a), all PDX lines (b), and both integrated cell lines and integrated PDX lines (b) are shown as a tree map. (right panel) KEGG pathway categories for proteins unique to all cell lines (a), all PDX lines (b), and both integrated cell lines and integrated PDX lines (c) are shown as a tree map. The names of pathways are shown in each rectangle. The color and size of individual areas correspond to the degree of enrichment and the number of proteins assigned to the category, respectively. P < 0.05 was considered statistically significant for pathway enrichment.
Sensitivity of established cell lines to anti-cancer drugs
Sensitivity to treatments with tyrosine kinase inhibitors was assessed in the cells (Fig. 7 a, Supplementary Table 1). The cells were treated with tyrosine kinase inhibitors at a fixed concentration of 10 μM. Among tyrosine kinase inhibitors examined, two reduced the cell viability by more than 90% (Supplementary Table 10). The half-maximal inhibitory concentrations of these tyrosine kinase inhibitors as well as doxorubicin, dacarbazine, ifosfamide, mesna, and cisplatin were calculated by treating the cells with different concentrations, and the results are summarized in Supplementary Table 11. We found that the three cell lines showed sensitivity to ponatinib, vandetanib, and doxorubicin (Fig.7 b–d). The IC50 values were not significantly different among the three cell lines (Supplementary Table 11, Fig. 7 e).

Effects of tyrosine kinase inhibitors on clear cell sarcoma (CCS) cell growth. The effects of tyrosine kinase inhibitors, at fixed concentrations, on cell growth were monitored in established CCS cell lines using CCK-8 assays (a). The anti-cancer drugs examined are listed in supplementary Table 1. Two tyrosine kinase inhibitors reduced cell growth by more than 90% (supplementary Table 10). The half-maximal inhibitory concentration values were monitored for these two tyrosine kinases and commonly used anti-cancer drugs for clear cell sarcomas (b) (supplementary Table 11). The IC50 values for ponatinib, vandetanib, and doxorubicin were calculated, and there was no significant difference among the cell lines (c).
Profiling of tyrosine kinase activity
Phosphorylation activity in each lysate sample was analyzed using immobilized substrate peptide arrays of tyrosine kinases. Each sample was characterized based on the phosphorylation status of 140 substrate peptides using PamChip. The degree of phosphorylation of substrate peptides was considered as the phosphorylation activity of each sample. Similarities in phosphorylation activity between xenograft and cell line samples were examined by measuring correlation coefficients (Fig. 8 a). One versus one comparison between the samples was performed, and the xenografts and cell lines showed obviously high similarity within the same sample groups (Fig. 8 a). Principal component analysis using phosphorylation activity revealed that the xenografts and cell lines had distinct profiles, and that phosphorylation activity among the cell lines was more similar (Fig. 8 b). When the samples were grouped by unsupervised hierarchical clustering analysis based on phosphorylation activity, the xenografts and cell lines were clearly separated (Fig. 8 c). Substrate peptides were grouped into clusters A and B (Fig. 8 c). Cluster A and B comprised 28 and 112 substrate peptides, respectively. Peptides in cluster A were phosphorylated by all samples examined in this study. In addition, the kinase activity for these peptides was preserved among xenografts and cell lines. In contrast, phosphorylation of peptides in cluster B was enhanced when treated with protein lysates from the cell lines. The kinase activities for these peptides were considerably different between xenografts and cell lines. These observations suggested that although all cancer models in this study originated from the same cancer patient, the kinase activities of xenografts and cell lines were substantially different. In addition, among the same model type, the overall kinase activity was similar. This highlights the need to understand the molecular characteristics of cancer models, and select these according to experimental purposes.

Kinase activity was similar in clear cell sarcoma xenografts and cell lines. Similarities in phosphorylation activity among xenograft and cell line samples were examined by measuring correlation coefficients after utilizing a PamChip PTK array (a). Principal component analysis of phosphorylation activity for the xenografts and cell lines (b). Unsupervised hierarchical clustering analysis for the xenografts and cell lines (c).
Discussion
In vitro and in vivo cancer models are inevitably required to investigate the molecular basis of carcinogenesis and cancer progression, and develop novel therapies. Although the number of patients with CCS is quite limited, CCS cell lines were reported in previous studies (Epstein et al. 1984; Sonobe et al. 1990; Takenouchi et al. 1994; Brown et al. 1995; Liao et al. 1996; Hiraga et al. 1997; Crnalic et al. 2002; Moritake et al. 2002; Schaefer et al. 2002; Davis et al. 2006; Outani et al. 2014). Our cell lines are unique because PDXs and cell lines were derived from the same CCS patient. The cell lines and PDXs have unique characters, and their complementary use will be helpful to further our understanding of diseases. For example, the cell lines consistently and indefinitely replicate, and we can easily obtain in vitro results with the expectation that the results can be reproduced by other researchers. Moreover, the functional effects of gene expression and treatments with reagents on cell behavior can be monitored using living cells. Additionally, high throughput drug assays can be performed using cell lines and lab automation. However, conditions for growing cells in culture are different from those of native tumors, and the limitation of cell lines as pre-clinical models has been pointed out (Li et al. 2008; Paul et al. 2010). PDXs can provide more natural experimental conditions (Wang et al. 2017). Although the presence of unique mutations can serve as a predictive biomarker, there remains a considerable number of cases where genetic biomarkers fail to identify effective drugs or applicable clinical trials. The utility of PDXs as a platform to establish predictive biomarkers has been expected, and concordant responses to treatment were reported between PDXs and patients (Byrne et al. 2017). However, drug response monitoring requires increased efforts using PDX models, as opposed to cell lines; however, high throughput drug screening using PDXs might not always be feasible. Thus, the combined use of cell lines and PDXs should be considered. In this sense, paired PDXs and cell lines represent a unique and intriguing model system.
Our proteomic studies suggested that established PDXs and cell lines have similar but distinct proteome profiles, which also differed from those of the original tumor. The proteome data suggested that although the histological appearances of PDXs were maintained, the molecular backgrounds change during the process of tissue implantation. It is natural that the proteomes of cell lines are different from those of original and PDX tumors, because tissue culture conditions are artificial. Novel protocols to establish model systems with more similar molecular backgrounds are desirable. Alternatively, by identifying common molecular backgrounds, we will be able to determine the best use of established models.
This study included only one case of CCS. Considering the heterogeneity of clinical CCS, tumor tissues from more CCS patients should be subjected to the establishment of model systems. Because CCS is a rare cancer, continuous and persistent efforts will be required to obtain sufficient numbers of PDXs and cell lines. However, this is worthwhile to achieve a better understanding of CCS and to improve clinical outcomes in CCS patients.
Conclusions
The present study details the establishment and proteomic characterization of patient-derived cancer models of CCS. Notably, during the process of establishment, the models retained the original characteristics of CCS, including gene fusion expression, morphology, and immunohistochemical features. Proteomic analysis revealed similar, but not identical, protein expression patterns between the original tumor tissue and models. Among tyrosine kinase inhibitors examined in this study, ponatinib and vandetanib exhibited suppressive effects on cell proliferation at significantly low concentrations. These anti-cancer drugs are candidates for the treatment of CCS, and the molecular mechanisms associated with their inhibitory effects are worth investigating in pre-clinical studies. For this, our CCS cell lines will be useful models to investigate the molecular basis of the anti-cancer effects of such tyrosine kinase inhibitors. Accumulating clinical outcome and in vitro drug sensitivity data will be required to evaluate the utility of the established models. Our study included a single CCS case, and conclusive results cannot be obtained from a limited number of cases. The establishment of more cell lines with detailed clinical and pathological data from multiple CCS cases will facilitate translational research for CCS.
Abbreviations
- DAVID:
-
Database for Annotation, Visualization and Integration Discovery
- KEGG:
-
Kyoto Encyclopedia of Genes and Genomes
- LC-MS/MS:
-
liquid-chromatography combined mass spectrometry
References
Altermann E, Klaenhammer TR (2005) PathwayVoyager: pathway mapping using the Kyoto encyclopedia of genes and genomes (KEGG) database. BMC Genomics 6:60
Anderson JC, Willey CD, Mehta A, Welaya K, Chen D, Duarte CW, Ghatalia P, Arafat W, Madan A, Sudarshan S, Naik G, Grizzle WE, Choueiri TK, Sonpavde G (2015) High throughput Kinomic profiling of human clear cell renal cell carcinoma identifies kinase activity dependent molecular subtypes. PLoS One 10:e0139267
Bridge JA, Sreekantaiah C, Neff JR, Sandberg AA (1991) Cytogenetic findings in clear cell sarcoma of tendons and aponeuroses. Malignant melanoma of soft parts. Cancer Genet Cytogenet 52:101–106
Brown AD, Lopez-Terrada D, Denny C, Lee KA (1995) Promoters containing ATF-binding sites are de-regulated in cells that express the EWS/ATF1 oncogene. Oncogene 10:1749–1756
Byrne AT, Alferez DG, Amant F, Annibali D, Arribas J, Biankin AV, Bruna A, Budinska E, Caldas C, Chang DK, Clarke RB, Clevers H, Coukos G, Dangles-Marie V, Eckhardt SG, Gonzalez-Suarez E, Hermans E, Hidalgo M, Jarzabek MA, de Jong S, Jonkers J, Kemper K, Lanfrancone L, Maelandsmo GM, Marangoni E, Marine JC, Medico E, Norum JH, Palmer HG, Peeper DS, Pelicci PG, Piris-Gimenez A, Roman-Roman S, Rueda OM, Seoane J, Serra V, Soucek L, Vanhecke D, Villanueva A, Vinolo E, Bertotti A, Trusolino L (2017) Interrogating open issues in cancer precision medicine with patient-derived xenografts. Nat Rev Cancer 17:254–268
Crnalic S, Panagopoulos I, Boquist L, Mandahl N, Stenling R, Lofvenberg R (2002) Establishment and characterisation of a human clear cell sarcoma model in nude mice. Int J Cancer 101:505–511
Davis IJ, Kim JJ, Ozsolak F, Widlund HR, Rozenblatt-Rosen O, Granter SR, Du J, Fletcher JA, Denny CT, Lessnick SL, Linehan WM, Kung AL, Fisher DE (2006) Oncogenic MITF dysregulation in clear cell sarcoma: defining the MiT family of human cancers. Cancer Cell 9:473–484
Deenik W, Mooi WJ, Rutgers EJ, Peterse JL, Hart AA, Kroon BB (1999) Clear cell sarcoma (malignant melanoma) of soft parts: a clinicopathologic study of 30 cases. Cancer 86:969–975
EA M, JM M, AG R, DM F, KL M (1993) Clear cell sarcoma of tendons and aponuroses: a clinicopathologic study of 58 cases with analysis of prognostic factors. Int J Surg Pathol 1:89–100
el-Naggar AK, Ordonez NG, Sara A, McLemore D, Batsakis JG (1991) Clear cell sarcomas and metastatic soft tissue melanomas. A flow cytometric comparison and prognostic implications. Cancer 67:2173–2179
Enzinger FM (1965) Clear-cell sarcoma of tendons and aponeuroses. An analysis of 21 cases. Cancer 18:1163–1174
Epstein AL, Martin AO, Kempson R (1984) Use of a newly established human cell line (SU-CCS-1) to demonstrate the relationship of clear cell sarcoma to malignant melanoma. Cancer Res 44:1265–1274
Ferrari A, Casanova M, Bisogno G, Mattke A, Meazza C, Gandola L, Sotti G, Cecchetto G, Harms D, Koscielniak E, Treuner J, Carli M (2002) Clear cell sarcoma of tendons and aponeuroses in pediatric patients: a report from the Italian and German soft tissue sarcoma cooperative group. Cancer 94:3269–3276
Finley JW, Hanypsiak B, McGrath B, Kraybill W, Gibbs JF (2001) Clear cell sarcoma: the Roswell Park experience. J Surg Oncol 77:16–20
Fletcher CDM, Bridge JA, Hogendoorn P, Mertens F (2013) WHO classification of Tumours of soft tissue and bone, 4th Edition edn. WHO Press, Geneva
Hiraga H, Nojima T, Abe S, Yamashiro K, Yamawaki S, Kaneda K, Nagashima K (1997) Establishment of a new continuous clear cell sarcoma cell line. Morphological and cytogenetic characterization and detection of chimaeric EWS/ATF-1 transcripts. Virchows Arch 431:45–51
Huang d W, Sherman BT, Lempicki RA (2009a) Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res 37:1–13
Huang d W, Sherman BT, Lempicki RA (2009b) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57
Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, Schubert M, Aben N, Goncalves E, Barthorpe S, Lightfoot H, Cokelaer T, Greninger P, van Dyk E, Chang H, de Silva H, Heyn H, Deng X, Egan RK, Liu Q, Mironenko T, Mitropoulos X, Richardson L, Wang J, Zhang T, Moran S, Sayols S, Soleimani M, Tamborero D, Lopez-Bigas N, Ross-Macdonald P, Esteller M, Gray NS, Haber DA, Stratton MR, Benes CH, Wessels LF, Saez-Rodriguez J, McDermott U, Garnett MJ (2016) A landscape of pharmacogenomic interactions in cancer. Cell 166:740–754
JR G, AL F, SW W (2013) Soft tissue tumors showing melanocytic differentiation, 6th edn. Elsevier, Philadelphia
Kawai A, Hosono A, Nakayama R, Matsumine A, Matsumoto S, Ueda T, Tsuchiya H, Beppu Y, Morioka H, Yabe H (2007) Clear cell sarcoma of tendons and aponeuroses: a study of 75 patients. Cancer 109:109–116
Li A, Walling J, Kotliarov Y, Center A, Steed ME, Ahn SJ, Rosenblum M, Mikkelsen T, Zenklusen JC, Fine HA (2008) Genomic changes and gene expression profiles reveal that established glioma cell lines are poorly representative of primary human gliomas. Mol Cancer Res 6:21–30
Liao SK, Perng YP, Lee LA, Chang KS, Lai GM, Wong E, Ho YS (1996) Newly established MST-1 tumour cell line and tumour-infiltrating lymphocyte culture from a patient with soft tissue melanoma (clear cell sarcoma) and their potential applications to patient immunotherapy. Eur J Cancer 32a:346–356
Lucas DR, Nascimento AG, Sim FH (1992) Clear cell sarcoma of soft tissues. Mayo Clinic experience with 35 cases. Am J Surg Pathol 16:1197–1204
Moritake H, Sugimoto T, Asada Y, Yoshida MA, Maehara Y, Epstein AL, Kuroda H (2002) Newly established clear cell sarcoma (malignant melanoma of soft parts) cell line expressing melanoma-associated Melan-a antigen and overexpressing C-MYC oncogene. Cancer Genet Cytogenet 135:48–56
Outani H, Tanaka T, Wakamatsu T, Imura Y, Hamada K, Araki N, Itoh K, Yoshikawa H, Naka N (2014) Establishment of a novel clear cell sarcoma cell line (Hewga-CCS), and investigation of the antitumor effects of pazopanib on Hewga-CCS. BMC Cancer 14:455
Panagopoulos I, Mertens F, Debiec-Rychter M, Isaksson M, Limon J, Kardas I, Domanski HA, Sciot R, Perek D, Crnalic S, Larsson O, Mandahl N (2002) Molecular genetic characterization of the EWS/ATF1 fusion gene in clear cell sarcoma of tendons and aponeuroses. Int J Cancer 99:560–567
Paul SM, Mytelka DS, Dunwiddie CT, Persinger CC, Munos BH, Lindborg SR, Schacht AL (2010) How to improve R&D productivity: the pharmaceutical industry's grand challenge. Nat Rev Drug Discov 9:203–214
Sara AS, Evans HL, Benjamin RS (1990) Malignant melanoma of soft parts (clear cell sarcoma). A study of 17 cases, with emphasis on prognostic factors. Cancer 65:367–374
Schaefer KL, Brachwitz K, Wai DH, Braun Y, Diallo R, Korsching E, Eisenacher M, Voss R, Van Valen F, Baer C, Selle B, Spahn L, Liao SK, Lee KA, Hogendoorn PC, Reifenberger G, Gabbert HE, Poremba C (2004) Expression profiling of t(12;22) positive clear cell sarcoma of soft tissue cell lines reveals characteristic up-regulation of potential new marker genes including ERBB3. Cancer Res 64:3395–3405
Schaefer KL, Wai DH, Poremba C, Korsching E, van Valen F, Ozaki T, Boecker W, Dockhorn-Dworniczak B (2002) Characterization of the malignant melanoma of soft-parts cell line GG-62 by expression analysis using DNA microarrays. Virchows Arch 440:476–484
Sonobe H, Furihata M, Iwata J, Ohtsuki Y, Mizobuchi H, Yamamoto H, Kumano O (1993) Establishment and characterization of a new human clear-cell sarcoma cell-line, HS-MM. J Pathol 169:317–322
Sonobe H, Manabe Y, Furihata M, Iwata J, Hikita T, Tanimoto T, Kiuna O, Oka T, Ohtsuki Y, Mizobuchi H et al (1990) Establishment and characterization of a human cell line, HS-MM, derived from a case of clear cell sarcoma. Hum Cell 3:352–356
Takahira T, Oda Y, Tamiya S, Yamamoto H, Kawaguchi K, Kobayashi C, Iwamoto Y, Tsuneyoshi M (2004) Alterations of the p16INK4a/p14ARF pathway in clear cell sarcoma. Cancer Sci 95:651–655
Takenouchi T, Ito K, Kazama T, Ito M (1994) Establishment and characterization of a clear-cell sarcoma (malignant melanoma of soft parts) cell line. Arch Dermatol Res 286:254–260
Tomayko MM, Reynolds CP (1989) Determination of subcutaneous tumor size in athymic (nude) mice. Cancer Chemother Pharmacol 24:148–154
Tyanova S, Albrechtsen R, Kronqvist P, Cox J, Mann M, Geiger T (2016) Proteomic maps of breast cancer subtypes. Nat Commun 7:10259
van der Graaf WT, Blay JY, Chawla SP, Kim DW, Bui-Nguyen B, Casali PG, Schoffski P, Aglietta M, Staddon AP, Beppu Y, Le Cesne A, Gelderblom H, Judson IR, Araki N, Ouali M, Marreaud S, Hodge R, Dewji MR, Coens C, Demetri GD, Fletcher CD, Dei Tos AP, Hohenberger P (2012) Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 379:1879–1886
Wang D, Pham NA, Tong J, Sakashita S, Allo G, Kim L, Yanagawa N, Raghavan V, Wei Y, To C, Trinh QM, Starmans MH, Chan-Seng-Yue MA, Chadwick D, Li L, Zhu CQ, Liu N, Li M, Lee S, Ignatchenko V, Strumpf D, Taylor P, Moghal N, Liu G, Boutros PC, Kislinger T, Pintilie M, Jurisica I, Shepherd FA, McPherson JD, Muthuswamy L, Moran MF, Tsao MS (2017) Molecular heterogeneity of non-small cell lung carcinoma patient-derived xenografts closely reflect their primary tumors. Int J Cancer 140:662–673
Wisniewski JR, Zougman A, Nagaraj N, Mann M (2009) Universal sample preparation method for proteome analysis. Nat Methods 6:359–362
Zucman J, Delattre O, Desmaze C, Epstein AL, Stenman G, Speleman F, Fletchers CD, Aurias A, Thomas G (1993) EWS and ATF-1 gene fusion induced by t(12;22) translocation in malignant melanoma of soft parts. Nat Genet 4:341–345
Acknowledgments
We thank Drs. Y. Minami, K. Shimizu, T. Mori, T. Uehara, M. Sugawara, Y. Araki, S. Toki, and Ms. R. Nakano (Division of Musculoskeletal Oncology, National Cancer Center Hospital) for sampling tumor tissue specimens from surgically-resected materials. We would like to thank Editage (www.editage.jp) for English language editing and constructive comments regarding the manuscript.
Funding
This work was supported by the National Cancer Center Research and Development Fund [26-A-3, 26-A-9, and 29-A-2].
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
There is no conflict of interest to declare regarding this study.
Additional information
Editor: Tetsuji Okamoto
Electronic supplementary material
Supplementary Fig 1
(JPEG 35 kb)
Supplementary Fig 2
(JPEG 77 kb)
Supplementary Fig 3
(JPEG 177 kb)
Supplementary Fig 4
(JPEG 117 kb)
Supplementary Table 1
(XLSX 13 kb)
Supplementary Table 2
(DOC 36 kb)
Supplementary Table 3
(XLSX 3570 kb)
Supplementary Table 4
(XLSX 3384 kb)
Supplementary Table 5
(XLSX 3585 kb)
Supplementary Table 6
(XLSX 2935 kb)
Supplementary Table 7
(XLSX 3718 kb)
Supplementary Table 8
(XLSX 3929 kb)
Supplementary Table 9
(XLSX 21 kb)
Supplementary Table 10
(XLSX 14 kb)
Supplementary Table 11
(XLSX 10 kb)
Rights and permissions
About this article

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cite this article
Sakumoto, M., Oyama, R., Takahashi, M. et al. Establishment and proteomic characterization of patient-derived clear cell sarcoma xenografts and cell lines. In Vitro Cell.Dev.Biol.-Animal 54, 163–176 (2018). https://doi.org/10.1007/s11626-017-0207-5
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11626-017-0207-5