
Abstract
Background
A high rate of postoperative recurrence, especially early recurrence (ER) occurring within 1 year, seriously impedes patients with hepatocellular carcinoma (HCC) from achieving long-term survival. This study aimed to establish a genomic-clinicopathologic nomogram for precisely predicting ER in HCC patients after R0 resection.
Methods
Two reliable datasets from The Cancer Genome Atlas (TCGA) and the Gene Expression Omnibus (GEO) databases were selected as the training and validation cohorts, respectively. The prognostic genes related to ER were screened out by univariate Cox regression analysis and differential expression analysis. The gene-based prognostic index was constructed using LASSO and Cox regression analyses, and its independent prognostic value was assessed by Kaplan-Meier and multivariate Cox analyses. Gene set enrichment analysis (GSEA) was performed to explore the biological pathways related to the prognostic index. Finally, the nomogram integrating all the independent prognostic factors was established and comprehensively evaluated by calibration plots, the C-index, receiver operating characteristic curves, and decision curve analysis.
Results
Nine dysregulated and prognostic genes related to ER (ZNF131, TATDN2, TXN, DDX55, KPNA2, ZNF30, TIMELESS, SFRP1, and COLEC11) were identified (all P < 0.05). The prognostic index model based on the 9 genes was successfully constructed using the TCGA cohort and showed a certain capability to discriminate the ER group from the non-ER group (P < 0.05) and good independent prognostic value in terms of predicting poor early recurrence-free survival (P < 0.05). Eight biological pathways significantly related to ER were identified by GSEA, such as “cell cycle”, “homologous recombination” and “p53 signaling pathway.” The genomic-clinicopathologic nomogram integrating the 9-gene-based prognostic index and TNM stage displayed significantly higher predictive accuracy and clinical application value than that of TNM stage model both in the training and validation cohorts (all P < 0.05).
Conclusions
The novel genomic-clinicopathologic nomogram may be a convenient and powerful tool for accurately predicting ER in HCC patients after R0 resection.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Hepatocellular carcinoma (HCC), which ranks as the sixth most common malignancy and the fourth leading cause of cancer death worldwide, is one of the major global public health problems.1 Due to improvements in the early detection and surgical techniques of HCC, numerous curative therapies have been applied to clinical practice, including liver transplantation (LT), hepatic resection, and local ablation.2 However, considering the current organ shortage, strict indications for LT and limited ablation extent, hepatic resection remains the most prevalent curative treatment for early-stage or partial intermediate-stage HCC patients to improve their prognosis.3 Nevertheless, a high rate of postoperative recurrence poses a great challenge for the long-term overall survival of HCC patients.4,5
It is estimated that HCC recurrence rates within 5 years following hepatic resection currently approach 70%.6 Among them, two different types of HCC recurrence are generally recognized using 1 year as the threshold of time to recurrence after curative resection, namely early recurrence (ER) and late recurrence (LR).7 While LR can be “de novo” tumors other from the original one, ER, which occupies the main part of HCC recurrence, is presumed to be “true recurrence” (pre-existing occult dissemination) and is closely related to aggressive tumor characteristics and worse prognosis in HCC patients.4,6,8,9 Thus, recognizing HCC patients with high ER risk after curative hepatectomy is crucial for preoperative decision-making, personalization of follow-up strategies, economization of medical resources, early detection of recurrent tumors, timely postoperative intervention, and overall survival improvement.
By integrating several clinicopathological characteristics of HCC, numerous clinical staging systems and their modified models have been established, among which the TNM staging system proposed by AJCC is one of the most widely accepted tools for assessing the postoperative recurrence and prognosis of HCC patients.10,11 However, in view of the phenotypic and molecular heterogeneity of HCC, the predictive accuracy of the TNM stage model for ER prediction is unsatisfactory, indicating that conventional clinicopathologic evaluations (such as tumor size, number of nodules, vascular invasion, lymph node metastasis, and distant metastasis) regardless of HCC genetic characteristics no longer meet the needs of clinical practice for postoperative ER prediction in the era of precision medicine.4,12,13 To date, gene-based prognostic models specifically designed for ER prediction after curative hepatectomy have rarely been reported. Thus, it is urgent and meaningful to construct a gene-based prognostic model and explore that whether combination of the TNM stage model with a gene-based prognostic model could significantly enhance the predictive accuracy for early HCC recurrence.
In this study, HCC patients with TNM stage I/II/IIIA (7th edition) tumors, who were eligible to receive curative hepatectomy as the initial treatment and were expected to achieve long-term survival, were enrolled from two reliable public datasets (TCGA-LIHC and GSE76427). By comprehensive bioinformatics and mathematical analyses, we aimed to construct a genomic-clinicopathologic nomogram for early HCC recurrence prediction by integrating the TNM stage and a gene-based prognostic index, and further determine its predictive accuracy compared with that of the TNM stage model.
Materials and Methods
Data Preparation and Study Design
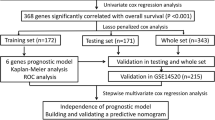
The clinical and mRNA-seq data (level 3) of the TCGA-LIHC dataset (Illumina HiSeq RNA-seq platform), which included 377 primary liver cancer and 50 normal liver tissues, were downloaded from the TCGA database. In addition, the clinical information and mRNA profile data of 115 HCC and 52 normal liver tissues were also downloaded from the GSE76427 dataset (Illumina HT-12 V4.0 platform) in the GEO database. The inclusion criteria of this study were as follows: (1) patients with pathological diagnosis of HCC; (2) patients with complete mRNA profile and follow-up data; (3) patients with TNM stage I (T1N0M0)/II (T2N0M0)/IIIA (T3aN0M0) tumors (7th edition); and (4) patients receiving curative resection with tumor-negative margins (R0 resection) as the initial treatment. Thus, 142 patients in the TCGA dataset and 99 patients in the GSE76427 dataset were enrolled serving as the training and the validation cohorts, respectively. The normalization of the raw data was carried out by the R package “edgeR” (TCGA-LIHC dataset) or R package “lumi” (GSE76427 dataset), and the normalized expression data were log2-transformed. During the modeling process, the Z-score was further calculated as the relative gene expression level in HCC tissues for each selected gene to normalize the datasets obtained from different platforms. As shown in Fig. 1, the study design was briefly described in the flowchart.

The flowchart describing the process of construction and validation of the genomic-clinicopathologic nomogram
Gene Selection and Construction of the Gene-Based Model
First, genes significantly related to recurrence-free survival (RFS) (P < 0.05) in the TCGA training cohort were screened out by univariate Cox regression analysis using the R package “survival.” Meanwhile, genes significantly related to RFS in the GSE76427 dataset were also screened out as a validation cohort. RFS was defined as the time from the date of initial hepatectomy to the date of (intra- or extrahepatic) recurrence or last follow-up. Next, by overlapping analysis, the common prognostic genes of the two cohorts were retained for further differential expression analysis. Besides comparing the mRNA expression levels of the genes between the HCC tissues and normal controls, the gene expressions at the protein levels were also assessed by using the immunohistochemical (IHC) staining data in the Human Protein Atlas (HPA) database (Protein Atlas version 19) (http://www.proteinatlas.org).14 Only those genes that were differentially expressed at both the mRNA and protein levels in the two cohorts were retained for further analysis. Then, by using the R package “glmnet”, LASSO-penalized Cox regression analysis was conducted using the TCGA dataset to select the best genes for modeling. The best gene group for modeling was chosen based on the 10-fold cross-validation and lambda.min. Finally, the best prognostic genes were included in the multivariate Cox regression analysis to obtain their coefficients (β values) using the TCGA dataset. The prognostic index formula was as follows: prognostic index = (β1 × Z-score of gene1) + (β2 × Z-score of gene2) + ... + (βn × Z-score of genen). Thus, a gene-based prognostic index model was successfully constructed using the TCGA dataset.
Establishment and Validation of a Predictive Nomogram
According to the results of multivariate Cox analyses, all the independent risk factors associated with RFS were included to establish a predictive nomogram using the R package “rms.” The performance of the nomogram was comprehensively evaluated. First, calibration plots were generated to visualize the consistency between the actual probability of 1-year RFS and the nomogram-predicted probability of 1-year RFS. In addition, the concordance index (C-index) was calculated to mathematically judge the predictive accuracy. Besides, time-dependent receiver operating characteristic (ROC) curve analysis was conducted to measure the accuracy by calculating the area under the curve (AUC) using the R package “survivalROC.” Moreover, to judge the clinical application value, decision curve analysis (DCA) was also carried out with the R package “rmda.”
Gene Set Enrichment Analysis
To explore the potential cancer-associated pathways significantly related to the high risk of early HCC recurrence as defined by the gene-based prognostic index, gene set enrichment analysis (GSEA) (reference gene sets: c2.cp.kegg.v6.0.symbols.gmt) was performed using mRNA-seq data from the TCGA dataset. The normalized enrichment score was calculated, and the gene set was regarded as significantly enriched unless the normalized p value was < 0.05 and false discovery rate q-value was < 0.05.
Statistical Analysis
Continuous variables in the different groups were compared by the Student’s t test or Mann-Whitney test based on the results of normality test and homogeneity test of variances. Categorical variables in the TCGA cohort and the GSE76427 cohort were assessed by Pearson’s chi-square test. X-tile 3.6.1 was used to calculate the optimal threshold for grouping (high/low group) based on the Kaplan-Meier (KM) method for RFS.15 The RFS between two groups was compared by KM analysis and the log-rank test. The independent prognostic factors for RFS were identified by Cox regression analysis, and hazard ratios (HRs) with 95% confidence intervals (CIs) were obtained. Statistical analyses were conducted with R version 3.4.2 and SPSS 22.0, and a two-tailed p value < 0.05 was considered to be statistically significant.
Results
The Clinicopathological Characteristics of the Training Cohort and the Validation Cohort
According to the strict inclusion criteria mentioned above, a total of 142 patients in the TCGA dataset and 99 patients in the GSE76427 dataset were finally enrolled in this study serving as the training cohort and validation cohort respectively. As shown in Table 1, the median follow-up time was 16.2 months (interquartile range (IQR), 8.7–34.6 months) in the TCGA training cohort and 9.1 months (IQR, 1.8–20 months) in the GSE76427 validation cohort. In addition, except for patient age (P < 0.05), no significant difference was detected between the two cohorts for the other variables, including patient gender, TNM stage, or recurrence status (all P > 0.05), suggesting that GSE76427 cohort could serve as a reliable reference for the TCGA training cohort.
Construction of Gene-Based Prognostic Index Model
Using the methods mentioned above, the optimal prognostic gene group for modeling was obtained, which including ZNF131 (zinc finger protein 131), TATDN2 (TatD DNase domain containing 2), TXN (thioredoxin), DDX55 (DEAD (Asp-Glu-Ala-Asp) box polypeptide 55), KPNA2 (karyopherin alpha 2), ZNF30 (zinc finger protein 30), TIMELESS (timeless circadian clock), SFRP1 (secreted frizzled-related protein 1), and COLEC11 (collectin sub-family member 11) (Fig. 2). Based on the optimal thresholds of the expression of the nine genes at the mRNA level, KM analyses confirmed that HCC patients with high expression of ZNF131, TATDN2, TXN, DDX55, KPNA2, ZNF30, and TIMELESS or with low expression of COLEC11 and SFRP1 were significantly more likely to have poor RFS in both the TCGA cohort and GSE76427 cohort (Fig. 3). Furthermore, differential expression analyses verified that the nine prognostic genes were significantly dysregulated at the mRNA level in HCC tissues relative to normal controls, including the 7 upregulated genes (ZNF131, TATDN2, TXN, DDX55, KPNA2, ZNF30, and TIMELESS) and the 2 downregulated genes (SFRP1 and COLEC11) (all P < 0.05) (Fig. 4). Consistent with the mRNA expression data, the dysregulations of these molecules at the protein level in HCC were also identified using the IHC data in HPA database (Supplementary Fig. 1). In addition, the clinical implications of the nine dysregulated genes were also explored using the TCGA dataset, and the results are shown in Supplementary Table 1. By performing multivariate Cox regression analysis using the TCGA dataset, the coefficients of the nine mRNA signatures were calculated, and the formula of the gene-based prognostic index model was as follows: prognostic index = (0.460 × Z-score of ZNF131) + (0.437 × Z-score of TATDN2) + (0.218 × Z-score of TXN) + (0.119 × Z-score of DDX55) + (0.099 × Z-score of KPNA2) + (0.050 × Z-score of ZNF30) + (0.012 × Z-score of TIMELESS) + (− 0.033 × Z-score of SFRP1) + (− 0.142 × Z-score of COLEC11). We further defined two groups, namely, the early recurrence (ER) group and the non-early recurrence (non-ER) group, in both TCGA cohort and GSE76427 cohort. The ER group represented patients with early HCC recurrence, while the non-ER group included patients without HCC recurrence who were followed up for at least 1 year and patients with late HCC recurrence. As shown in Fig. 5, the prognostic index in the ER group was significantly higher than that in the non-ER group (all P < 0.05), and the prognostic index showed a certain capability to discriminate early HCC recurrence in both the training cohort (AUC = 0.7759, P < 0.05) and validation cohort (AUC = 0.8110, P < 0.05).

Selection of the optimal genes for modeling by LASSO-penalized Cox regression analysis. a The optimal gene group was chosen by 10-fold cross-validation and lambda.min; b LASSO coefficient profile of the genes

Kaplan-Meier survival analyses of the nine genes in both the TCGA training cohort and GSE76427 validation cohort

Differential expression analyses of the nine genes at the mRNA level. 7 upregulated genes (ZNF131, TATDN2, TXN, DDX55, KPNA2, ZNF30, and TIMELESS) (a–g) and 2 downregulated genes (SFRP1 and COLEC11) (h, i) were identified in HCC tissues relative to normal liver tissues (NT)

Comparison of the prognostic index between the ER group and the non-ER group, and validation of its discrimination for ER using the ROC curves in both the training (a, b) cohort and validation cohort (c, d)
Identification of the Prognostic Value of the Gene-Based Prognostic Index
First, the patients enrolled in the TCGA training cohort and the GSE76427 validation cohort were severally split into high or low risk of recurrence groups according to the best cut-off values of the gene-based prognostic index. By generating KM survival curves, we observed that HCC patients in the high-risk group had significantly worse RFS than patients in the low-risk group in both cohorts (all P < 0.05). Importantly, the subgroup analyses based on TNM stage indicated that the prognostic value of the prognostic index was independent of the TNM stage (all P < 0.05) (Fig. 6). To more precisely identify the independent prognostic value of the gene-based prognostic index, Cox regression analyses were conducted using the TCGA dataset with the intact clinical data. As shown in Table 2, the prognostic index, TNM stage and microvascular invasion were significantly associated with RFS of HCC patients in univariate Cox regression analyses. The following multivariate analysis further verified that the prognostic index (HR = 2.820 (2.005, 3.968), P < 0.001) was an independent predictor of poor RFS of HCC patients by fully considering all potential prognostic factors detected in the univariate analyses.

Kaplan-Meier survival analyses of the gene-based prognostic index using the X-tile optimal cut-off points, and its subgroup analyses based on TNM stage both in the training (a–c) and validation cohorts (d–f)
Functional Enrichment Analysis
GSEA was conducted to identify the potential biological pathways related to the nine gene signatures using the TCGA dataset. As shown in Supplementary Fig. 2, a total of 8 KEGG signaling pathways were significantly enriched in the high-risk group, including “cell cycle”, “DNA replication”, “base excision repair”, “spliceosome”, “nucleotide excision repair”, “RNA degradation”, “homologous recombination”, and “p53 signaling pathway” (all P < 0.05).
Establishment and Validation of a Genomic-Clinicopathologic Predictive Nomogram
To develop a model with clinical application value for predicting the ER of HCC after curative resection, a genomic-clinicopathologic predictive nomogram (combined model) was established by integrating the gene-based prognostic index and the TNM stage, which were recognized as independent risk factors of RFS in the multivariate Cox regression model (Table 2 and Fig. 7a). Internal validation was comprehensively performed in the TCGA training cohort. First, calibration plots were generated that exhibited high consistency between the actual proportion of 1-year RFS and the nomogram-predicted probability of 1-year RFS (Fig. 7b). In addition, the C-index of the combined model for RFS prediction was 0.743 (0.673–0.813), which was remarkably improved compared with that of the TNM stage model (C-index, 0.598 (0.533–0.663)) (P < 0.001). In addition, the predictive accuracy of the nomograms was further assessed by time-dependent ROC curves and the combined model had an obviously larger AUC than that of the TNM stage model (1-year AUC, 0.803 vs 0.627) (Fig. 7c). Moreover, to visually evaluate the clinical application value of the nomograms, DCA curves were further generated. The combined model displayed a significantly higher net benefit than that of the TNM stage model (Fig. 7d).

Establishment and validation of the predictive nomogram. The genomic-clinicopathologic nomogram for postoperative ER prediction in HCC patients (a); the calibration curves, the time-dependent ROC curves and the DCA curves of the nomogram for predicting 1-year RFS in both the training cohort (b–d) and validation cohort (e–g)
The following validation was carried out using the GSE76427 validation cohort. In line with the results described above, calibration plots also showed that the prediction of the combined model was in excellent accordance with the actual observations in terms of the probability of 1-year RFS (Fig. 7e). In addition, the C-index of the combined model for RFS prediction was 0.716 (0.632–0.800), which was also superior to that of the TNM stage model (C-index, 0.562 (0.465–0.659)) (P < 0.001). Furthermore, time-dependent ROC curves and DCA curves still showed that the combined model had an obviously larger AUC (1-year AUC, 0.804 vs 0.602) and a higher net benefit than that of the TNM stage model (Fig. 7f, g).
Discussion
Along with the increasing importance of mechanism research on ER after hepatectomy, several molecular biomarkers related to early HCC recurrence have been identified, such as NUF2 (NUF2, NDC80 kinetochore complex component), ECT2 (epithelial cell transforming 2), PRC1 (protein regulator of cytokinesis 1), EDIL3 (EGF-like repeats and discoidin I-like domains 3), FLNA (filamin A, alpha), AKR1B10 (aldo-keto reductase family 1, member B10 (aldose reductase)), CKAP2 (cytoskeleton associated protein 2), and KIAA0101.16,17,18,19,20,21,22,23 Nevertheless, the specificity and sensitivity of the individual biomarkers alone were insufficient to accurately predict postoperative ER in HCC patients, suggesting that it might be necessary to integrate multiple gene signatures into a single model or combine biomarkers with clinicopathologic factors to enhance the ER predictive accuracy. The RFS-related gene sets for HCC identified in some key previous studies have been properly summarized in the Supplementary Table 2, which might contribute to future studies exploring the molecular mechanisms of HCC recurrence.
To the best of our knowledge, multigene-based prognostic models or combined models specifically designed for postoperative ER prediction have rarely been reported. In the last few decades, a 20-gene-based model established by Kurokawa et al., a 172-gene-based model established by Yoshioka et al., and a 6-gene-based model established by Yuan et al. have been reported sequentially as earlier attempts to comprehensively examine the molecular heterogeneity of ER and predict ER in HCC patients following hepatectomy at the molecular level.24,25,26 However, it is undeniable that the limitations of those studies are obvious from the current perspective, such as their relatively small sample sizes, unreasonable or inaccurate selection of the best genes for modeling (by simply selecting the top predictive genes or including all the DEGs), a lack of comprehensive evaluations of the predictive accuracy or clinical application value, and a lack of comparative studies with reference to existing clinical staging systems. Recently, consistent with our views, a 24-mRNA-based predictive model for ER was proposed by Cai et al. by data mining of the GEO and TCGA datasets, which relatively avoided the deficiencies mentioned above and displayed good discrimination and clinical usefulness.5 The achievements of this study should be recognized, as well as its specific limitations. First, the DEGs between HCC tissues and normal controls included in the model were only identified at the mRNA expression level without verification at the protein level. In addition, the sequencing platforms of the 13 datasets were varied and proper normalization of the raw data among the different platforms was not performed. Furthermore, the 24-mRNA-signature-integrated nomogram was just constructed and internally validated using the GSE14520 dataset without external validation in the TCGA dataset. Finally, the inclusion criteria of the HCC patients in the study were disputable, such as the undefined edition of TNM staging system, the patients with advanced stage (TNM IIIB/IIIC) and the patients receiving hepatectomy with tumor-positive margins (R1 resection). As we all know, the HCC patients with portal/hepatic vein invasions (T3b) or adjacent organ invasions (T4) were not highly recommended to receive resection according to AASLD and EASL guidelines because of the high rate of postoperative recurrence and poor long-term survival.27,28,29 And palliative resection with a positive residual tumor margin was an important confounding factor in terms of aiming to specifically explore the molecular mechanisms of ER and construct a prognostic model for ER prediction following curative hepatectomy.30 Therefore, a powerful genomic-clinicopathologic model specifically designed to predict ER for HCC patients who are eligible for receiving curative resection (R0 resection) as initial treatment and who are expected to achieve long-term overall survival is still highly anticipated.
In this study, two reliable datasets (TCGA-LIHC and GSE76427), both with large sample sizes and based on the Illumina platform, were selected for data mining. And 142 patients in TCGA-LIHC dataset and 99 patients in GSE76427 dataset with TNM stage I/II/IIIA tumors and receiving R0 resection were finally included serving as the training and validation cohorts, respectively. Before screening the best genes for modeling, proper normalization of the raw data among the two datasets was performed to eliminate the influence of differences in the platforms. Importantly, the strategy of selecting the optimal genes for modeling was rigorous in this study and the statistical approaches were reasonable and advanced, such as overlapping analysis for mutual confirmation, identification of DEGs at both mRNA and protein levels, LASSO analysis for selecting optimal gene group and multivariate Cox regression analysis for calculating coefficients. Thus, nine dysregulated and prognostic genes related to ER (ZNF131, TATDN2, TXN, DDX55, KPNA2, ZNF30, TIMELESS, SFRP1, and COLEC11) were identified and the 9-gene-based prognostic index model was successfully constructed. The subsequent assessments further showed that the 9-gene-based prognostic index not only had a certain ability to discriminate the ER group from the non-ER group but also was an independent prognostic indicator of poor early-RFS.
It has been reported that the nine genes integrated in the gene-based prognostic index act as either oncogenes or anti-oncogenes involved in the development and progression of several cancers. For example, ZNF131 overexpression in glioblastoma could promote the mitotic spindle integrity and viability of glioblastoma stem-like cells.31 Upregulation of TATDN2 was an independent risk factor of prostate cancer recurrence.32 DDX55 was involved in RNA metabolism and was regarded as a molecular signature associated with liver metastasis in human colon carcinoma.33,34 ZNF30 was identified as an upregulated gene in prostate cancer mainly caused by chromosomal gain (19q13.11).35 Downregulation of COLEC11 was associated with the prognosis of esophageal cancer and the metastasis of ovarian serous papillary carcinomas.36,37 Importantly, in line with our results, the roles of TXN, KPNA2, TIMELESS, and SFRP1 in HCC have been identified in previous studies. TXN, a redox-sensitive protein, was overexpressed in HCC tissues and associated with unfavorable OS in HCC patients, while silencing TXN in HCC cells could significantly induce cell senescence.38,39 KPNA2 upregulation was correlated with carcinogenic progression and unfavorable OS and RFS in HCC, and it could accelerate cell cycle progression by increasing CCNB2/CDK1 expression.40,41,42 Overexpression of TIMELESS might play a protumorigenic role in HCC via interacting with CHEK2 and EEF1A2.43 As a vital antagonist of the Wnt signaling pathway, SFRP1 was frequently downregulated in HCC due to promoter hypermethylation, which was associated with the angiogenesis, tumor growth, and poor prognosis in HCC.44,45,46 In addition, the significant correlations between the expression of the dysregulated genes (such as KPNA2, TIMELESS, TXN, DDX55, and SFRP1) and the underlying liver diseases (such as liver fibrosis/cirrhosis and hepatitis b/c infection) or other important clinicopathological characteristics (such as histologic grade and serum AFP levels) of HCC patients also highlighted the key involvement of these genes in the development and progression of HCC. Moreover, by performing GSEA based on the risk stratification of the prognostic index, the potential biological pathways related to the nine dysregulated and prognostic genes in HCC were further identified in this study, such as “cell cycle”, “DNA replication”, “base excision repair”, “spliceosome”, “nucleotide excision repair”, “RNA degradation”, “homologous recombination”, and “p53 signaling pathway”, which would contribute to the understanding of the underlying biological mechanisms of ER following curative hepatectomy and the development of novel target therapies in the future.
Nomogram is a convenient, visual, and powerful tool widely applied for precise prognosis assessment in cancer patients. In this study, a genomic-clinicopathologic nomogram for ER prediction after curative resection was established by integrating the 9-gene-based prognostic index and the TNM stage, which were identified as prognostic indicators of poor early-RFS using the TCGA dataset. The combined model displayed significantly higher predictive accuracy than that of TNM stage model, which were comprehensively evaluated by generating calibration curves, calculating the C-index (0.743 vs 0.598) and performing time-dependent ROC curve analyses (1-year AUC, 0.803 vs 0.627). Following DCA also confirmed the higher clinical application value of the genomic-clinicopathologic nomogram compared with that of the TNM stage model. Importantly, subsequent validation by using the GSE76427 cohort further verified the good calibration, discrimination (C-index, 0.716 vs 0.562; 1-year AUC, 0.804 vs 0.602), and clinical usefulness of the genomic-clinicopathologic nomogram with reference to the TNM stage model. Thus, in view of the good performance of the genomic-clinicopathologic nomogram for postoperative ER prediction, the combined model might provide necessary guidance for preoperative or postoperative decision-making. On the one hand, the accurate risk stratification of ER based on preoperative needle biopsy and molecular diagnosis could guide the optimal choice of treatments (such as local ablation, resection, transplantation, TACE or systemic therapy). On the other hand, effective risk stratification of ER after surgery would contribute to the optimization of postoperative surveillance strategies (such as the monitoring time interval and appropriate application of imaging examinations), the early detection of tumor recurrence and timely adjuvant treatment (such as re-resection, ablation and systemic therapy), which may effectively improve the long-term overall survival of HCC patients.
Conclusions
In conclusion, the genomic-clinicopathologic nomogram established by integrating the 9-gene-based prognostic index and the TNM stage exhibited higher predictive accuracy and clinical application value for ER prediction after curative hepatectomy (R0 resection) than that of the conventional TNM staging system. This nomogram may be a powerful and convenient tool for ER risk stratification to help clinicians choose the optimal treatments based on preoperative biopsy or develop personalized strategies of postoperative surveillance and adjuvant therapy for patients with resectable HCC.
References
Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018; 68:394–424.
Forner A, Reig M, Bruix J. Hepatocellular carcinoma. Lancet 2018; 391:1301–1314.
Akateh C, Black SM, Conteh L, Miller ED, Noonan A, Elliott E, Pawlik TM, Tsung A, Cloyd JM. Neoadjuvant and adjuvant treatment strategies for hepatocellular carcinoma. World J Gastroenterol 2019; 25:3704–3721.
Chan AWH, Zhong J, Berhane S, Toyoda H, Cucchetti A, Shi K, Tada T, Chong CCN, Xiang BD, Li LQ, Lai PBS, Mazzaferro V, Garcia-Finana M, Kudo M, Kumada T, Roayaie S, Johnson PJ. Development of pre and post-operative models to predict early recurrence of hepatocellular carcinoma after surgical resection. J Hepatol 2018; 69:1284–1293.
Cai J, Tong Y, Huang L, Xia L, Guo H, Wu H, Kong X, Xia Q. Identification and validation of a potent multi-mRNA signature for the prediction of early relapse in hepatocellular carcinoma. Carcinogenesis 2019; 40:840–852.
Kulik L, El-Serag HB. Epidemiology and Management of Hepatocellular Carcinoma. Gastroenterology 2019; 156:477–491.e1.
Zhang XP, Chen ZH, Zhou TF, Li LQ, Chen MS, Wen TF, Shi J, Guo WX, Wu MC, Lau WY, Cheng SQ, Chinese National Research Cooperative Group for D, Treatment of Hepatocellular Carcinoma with Tumour T. A nomogram to predict early postoperative recurrence of hepatocellular carcinoma with portal vein tumour thrombus after R0 liver resection: A large-scale, multicenter study. Eur J Surg Oncol 2019; 45:1644–1651.
Zhang YM, Zhou ZT, Liu GM. Factors predicting early recurrence after surgical resection of hepatocellular carcinoma. J Hepatol 2019; 70:571–572.
Llovet JM, Di Bisceglie AM, Bruix J, Kramer BS, Lencioni R, Zhu AX, Sherman M, Schwartz M, Lotze M, Talwalkar J, Gores GJ, Panel of Experts in HCCDCT. Design and endpoints of clinical trials in hepatocellular carcinoma. J Natl Cancer Inst 2008; 100:698–711.
Chan AC, Fan ST, Poon RT, Cheung TT, Chok KS, Chan SC, Lo CM. Evaluation of the seventh edition of the American Joint Committee on Cancer tumour-node-metastasis (TNM) staging system for patients undergoing curative resection of hepatocellular carcinoma: implications for the development of a refined staging system. HPB (Oxford) 2013; 15:439–448.
Addissie BD, Roberts LR. Classification and staging of hepatocellular carcinoma: an aid to clinical decision-making. Clin Liver Dis 2015; 19:277–294.
Calderaro J, Ziol M, Paradis V, Zucman-Rossi J. Molecular and histological correlations in liver cancer. J Hepatol 2019; 71:616–630.
Nault JC, Galle PR, Marquardt JU. The role of molecular enrichment on future therapies in hepatocellular carcinoma. J Hepatol 2018; 69:237–247.
Uhlen M, Zhang C, Lee S, Sjostedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, Sanli K, von Feilitzen K, Oksvold P, Lundberg E, Hober S, Nilsson P, Mattsson J, Schwenk JM, Brunnstrom H, Glimelius B, Sjoblom T, Edqvist PH, Djureinovic D, Micke P, Lindskog C, Mardinoglu A, Ponten F. A pathology atlas of the human cancer transcriptome. Science 2017; 357.
Camp RL, Dolled-Filhart M, Rimm DL. X-tile: a new bio-informatics tool for biomarker assessment and outcome-based cut-point optimization. Clin Cancer Res 2004; 10:7252–7259.
Wang Y, Tan PY, Handoko YA, Sekar K, Shi M, Xie C, Jiang XD, Dong QZ, Goh BKP, Ooi LL, Gao Z, Hui KM. NUF2 is a valuable prognostic biomarker to predict early recurrence of hepatocellular carcinoma after surgical resection. Int J Cancer 2019; 145:662–670.
Chen J, Xia H, Zhang X, Karthik S, Pratap SV, Ooi LL, Hong W, Hui KM. ECT2 regulates the Rho/ERK signalling axis to promote early recurrence in human hepatocellular carcinoma. J Hepatol 2015; 62:1287–1295.
Chen J, Rajasekaran M, Xia H, Zhang X, Kong SN, Sekar K, Seshachalam VP, Deivasigamani A, Goh BK, Ooi LL, Hong W, Hui KM. The microtubule-associated protein PRC1 promotes early recurrence of hepatocellular carcinoma in association with the Wnt/beta-catenin signalling pathway. Gut 2016; 65:1522–1534.
Xia H, Chen J, Shi M, Gao H, Sekar K, Seshachalam VP, Ooi LL, Hui KM. EDIL3 is a novel regulator of epithelial-mesenchymal transition controlling early recurrence of hepatocellular carcinoma. J Hepatol 2015; 63:863–873.
Donadon M, Di Tommaso L, Soldani C, Franceschini B, Terrone A, Mimmo A, Vitali E, Roncalli M, Lania A, Torzilli G. Filamin A expression predicts early recurrence of hepatocellular carcinoma after hepatectomy. Liver Int 2018; 38:303–311.
Wang YY, Qi LN, Zhong JH, Qin HG, Ye JZ, Lu SD, Ma L, Xiang BD, Li LQ, You XM. High expression of AKR1B10 predicts low risk of early tumor recurrence in patients with hepatitis B virus-related hepatocellular carcinoma. Sci Rep 2017; 7:42199.
Hayashi T, Ohtsuka M, Okamura D, Seki N, Kimura F, Shimizu H, Yoshidome H, Kato A, Yoshitomi H, Furukawa K, Miyazaki M. Cytoskeleton-associated protein 2 is a potential predictive marker for risk of early and extensive recurrence of hepatocellular carcinoma after operative resection. Surgery 2014; 155:114–123.
Yuan RH, Jeng YM, Pan HW, Hu FC, Lai PL, Lee PH, Hsu HC. Overexpression of KIAA0101 predicts high stage, early tumor recurrence, and poor prognosis of hepatocellular carcinoma. Clin Cancer Res 2007; 13:5368–5376.
Kurokawa Y, Matoba R, Takemasa I, Nagano H, Dono K, Nakamori S, Umeshita K, Sakon M, Ueno N, Oba S, Ishii S, Kato K, Monden M. Molecular-based prediction of early recurrence in hepatocellular carcinoma. J Hepatol 2004; 41:284–291.
Yoshioka S, Takemasa I, Nagano H, Kittaka N, Noda T, Wada H, Kobayashi S, Marubashi S, Takeda Y, Umeshita K, Dono K, Matsubara K, Monden M. Molecular prediction of early recurrence after resection of hepatocellular carcinoma. Eur J Cancer 2009; 45:881–889.
Yuan S, Wang J, Yang Y, Zhang J, Liu H, Xiao J, Xu Q, Huang X, Xiang B, Zhu S, Li L, Liu J, Liu L, Zhou W. The Prediction of Clinical Outcome in Hepatocellular Carcinoma Based on a Six-Gene Metastasis Signature. Clin Cancer Res 2017; 23:289–297.
Zhang XY, Ou J, Chen JY, Li WW. Predicting early hepatocellular carcinoma recurrence after resection: A comment for moving forward. J Hepatol 2019; 70:567–568.
European Association for the Study of the Liver. EASL Clinical Practice Guidelines: Management of hepatocellular carcinoma. J Hepatol 2018; 69:182–236.
Heimbach JK, Kulik LM, Finn RS, Sirlin CB, Abecassis MM, Roberts LR, Zhu AX, Murad MH, Marrero JA. AASLD guidelines for the treatment of hepatocellular carcinoma. Hepatology 2018; 67:358–380.
Shim JH, Jun MJ, Han S, Lee YJ, Lee SG, Kim KM, Lim YS, Lee HC. Prognostic nomograms for prediction of recurrence and survival after curative liver resection for hepatocellular carcinoma. Ann Surg 2015; 261:939–946.
Ding Y, Herman JA, Toledo CM, Lang JM, Corrin P, Girard EJ, Basom R, Delrow JJ, Olson JM, Paddison PJ. ZNF131 suppresses centrosome fragmentation in glioblastoma stem-like cells through regulation of HAUS5. Oncotarget 2017; 8:48545–48562.
Jiang Y, Mei W, Gu Y, Lin X, He L, Zeng H, Wei F, Wan X, Yang H, Major P, Tang D. Construction of a set of novel and robust gene expression signatures predicting prostate cancer recurrence. Mol Oncol 2018; 12:1559–1578.
Sarkar M, Ghosh MK. DEAD box RNA helicases: crucial regulators of gene expression and oncogenesis. Front Biosci (Landmark Ed) 2016; 21:225–250.
Liu J, Wang D, Zhang C, Zhang Z, Chen X, Lian J, Liu J, Wang G, Yuan W, Sun Z, Wang W, Song M, Wang Y, Wu Q, Cao L, Wang D, Zhang Y. Identification of liver metastasis-associated genes in human colon carcinoma by mRNA profiling. Chin J Cancer Res 2018; 30:633–646.
Jiang M, Li M, Fu X, Huang Y, Qian H, Sun R, Mao Y, Xie Y, Li Y. Simultaneously detection of genomic and expression alterations in prostate cancer using cDNA microarray. Prostate 2008; 68:1496–1509.
Zhang C, Sun Q. Weighted gene co-expression network analysis of gene modules for the prognosis of esophageal cancer. J Huazhong Univ Sci Technolog Med Sci 2017; 37:319–325.
Bignotti E, Tassi RA, Calza S, Ravaggi A, Bandiera E, Rossi E, Donzelli C, Pasinetti B, Pecorelli S, Santin AD. Gene expression profile of ovarian serous papillary carcinomas: identification of metastasis-associated genes. Am J Obstet Gynecol 2007; 196:245 e1–11.
Cho SY, Kim S, Son MJ, Rou WS, Kim SH, Eun HS, Lee BS. Clinical Significance of the Thioredoxin System and Thioredoxin-Domain-Containing Protein Family in Hepatocellular Carcinoma. Dig Dis Sci 2019; 64:123–136.
Zhang T, Liu H, Zhu C, Briggs K, Kang Y, Fleming JA, Curley SA. Silencing thioredoxin induces liver cancer cell senescence under hypoxia. Hepatol Res 2012; 42:706–713.
Guo X, Wang Z, Zhang J, Xu Q, Hou G, Yang Y, Dong C, Liu G, Liang C, Liu L, Zhou W, Liu H. Upregulated KPNA2 promotes hepatocellular carcinoma progression and indicates prognostic significance across human cancer types. Acta Biochim Biophys Sin (Shanghai) 2019; 51:285–292.
Jiang P, Tang Y, He L, Tang H, Liang M, Mai C, Hu L, Hong J. Aberrant expression of nuclear KPNA2 is correlated with early recurrence and poor prognosis in patients with small hepatocellular carcinoma after hepatectomy. Med Oncol 2014; 31:131.
Gao CL, Wang GW, Yang GQ, Yang H, Zhuang L. Karyopherin subunit-alpha 2 expression accelerates cell cycle progression by upregulating CCNB2 and CDK1 in hepatocellular carcinoma. Oncol Lett 2018; 15:2815–2820.
Elgohary N, Pellegrino R, Neumann O, Elzawahry HM, Saber MM, Zeeneldin AA, Geffers R, Ehemann V, Schemmer P, Schirmacher P, Longerich T. Protumorigenic role of Timeless in hepatocellular carcinoma. Int J Oncol 2015; 46:597–606.
Wu Y, Li J, Sun CY, Zhou Y, Zhao YF, Zhang SJ. Epigenetic inactivation of the canonical Wnt antagonist secreted frizzled-related protein 1 in hepatocellular carcinoma cells. Neoplasma 2012; 59:326–332.
Davaadorj M, Imura S, Saito YU, Morine Y, Ikemoto T, Yamada S, Takasu C, Hiroki T, Yoshikawa M, Shimada M. Loss of SFRP1 Expression Is Associated with Poor Prognosis in Hepatocellular Carcinoma. Anticancer Res 2016; 36:659–664.
Hu J, Dong A, Fernandez-Ruiz V, Shan J, Kawa M, Martinez-Anso E, Prieto J, Qian C. Blockade of Wnt signaling inhibits angiogenesis and tumor growth in hepatocellular carcinoma. Cancer Res 2009; 69:6951–6959.
Funding
This work was supported by the National Natural Science Foundation of China (grant number: 81570079).
Author information
Authors and Affiliations
Contributions
Bin Yu designed this study, analyzed the data, and wrote the paper. Han Liang downloaded the data and prepared the data. Qifa Ye revised the paper. Yanfeng Wang designed this study, revised the paper, and provided supervision.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflicts of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Supplementary Figure 1
Representative IHC images of the nine genes in the HPA database. (a) Protein levels of ZNF131 in normal liver tissues (https://www.proteinatlas.org/ENSG00000172262-ZNF131/tissue/liver#img) and HCC tissues (https://www.proteinatlas.org/ENSG00000172262-ZNF131/ pathology/liver+cancer#img). (b) Protein levels of TATDN2 in normal liver tissues (https://www.proteinatlas.org/ENSG00000157014-TATDN2/tissue/ liver#img) and HCC tissues (https://www.proteinatlas.org/ENSG00000157014- TATDN2/pathology/liver+cancer#img). (c) Protein levels of TXN in normal liver tissues (https://www.proteinatlas.org/ENSG00000136810-TXN/tissue/liver# img) and HCC tissues (https://www.proteinatlas.org/ENSG00000136810- TXN/pathology/liver+cancer#img). (d) Protein levels of DDX55 in normal liver tissues (https://www.proteinatlas.org/ENSG00000111364-DDX55/tissue/liver# img) and HCC tissues (https://www.proteinatlas.org/ENSG00000111364- DDX55/pathology/liver+cancer#img). (e) Protein levels of KPNA2 in normal liver tissues (https://www.proteinatlas.org/ENSG00000182481-KPNA2/tissue/ liver#img) and HCC tissues (https://www.proteinatlas.org/ENSG00000182481- KPNA2/pathology/liver+cancer#img). (f) Protein levels of ZNF30 in normal liver tissues (https://www.proteinatlas.org/ENSG00000168661-ZNF30/ tissue/liver#img) and HCC tissues (https://www.proteinatlas.org/ ENSG00000168661-ZNF30/pathology/liver+cancer#img). (g) Protein levels of TIMELESS in normal liver tissues (https://www.proteinatlas.org/ ENSG00000111602-TIMELESS/tissue/liver#img) and HCC tissues (https://www.proteinatlas.org/ENSG00000111602-TIMELESS/pathology/liver+cancer#img). (h) Protein levels of SFRP1 in normal liver tissues (https://www.proteinatlas.org/ENSG00000104332-SFRP1/tissue/liver#img) and HCC tissues (https://www.proteinatlas.org/ENSG00000104332-SFRP1/ pathology/liver+cancer#img). (i) Protein levels of COLEC11 in normal liver tissues (https://www.proteinatlas.org/ENSG00000118004-COLEC11/tissue /liver#img) and HCC tissues (https://www.proteinatlas.org/ ENSG00000118004-COLEC11/pathology/liver+cancer#img). In the HPA database, the expression level score is based on the staining intensity (negative, weak, moderate or strong) and fraction of stained cells (<25%, 25–75% or >75%), including not detected (N) (negative or weak with <25%), low (L) (weak with 25–75%/>75%, or moderate with <25%), medium (M) (moderate with 25–75%/>75%, or strong with <25%), and high (H) (strong with 25–75%/>75%). (PNG 2618 kb).
Supplementary Figure 2
Gene set enrichment analysis using the TCGA-LIHC dataset. A total of 8 KEGG signaling pathways were significantly enriched in high-risk group defined by gene-based prognostic index, including “cell cycle” (a), “DNA replication” (b), “base excision repair” (c), “spliceosome” (d), “nucleotide excision repair” (e), “RNA degradation” (f), “homologous recombination” (g) and “p53 signaling pathway” (h). NES: normalized enrichment score, NOM p: normalized p-value, FDR q: false discovery rate q-value. (PNG 1043 kb).
ESM 1
(DOCX 27.6 kb).
Rights and permissions
About this article

Cite this article
Yu, B., Liang, H., Ye, Q. et al. Establishment of a Genomic-Clinicopathologic Nomogram for Predicting Early Recurrence of Hepatocellular Carcinoma After R0 Resection. J Gastrointest Surg 25, 112–124 (2021). https://doi.org/10.1007/s11605-020-04554-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11605-020-04554-1