
Abstract
Background
Colorectal cancer remains the most common gastrointestinal cancer. While screening combined with effective surgical treatment has reduced its mortality, we still do not have effective means to prevent recurrence nor to treat metastatic disease. What we know about cancer biology has gone through revolutionary changes in recent decades. The advent of the cancer stem cell theory has accelerated our understanding of the cancer cell. However, there is increasing evidence that cancer cells are influenced by their surrounding microenvironment.
Purpose
This review divides the tumor microenvironment into four functional components—the stem cell niche, cancer stroma, immune cells, and vascular endothelia—and examines their individual and collective influence on the growth and metastasis of the colon cancer stem cell. The discussion will highlight the need to fully exploit the tumor microenvironment when designing future prognostic tools and therapies.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer is the third most common type of cancer in the USA. The incidence has been on the decline for the past two decades, which is largely attributable to the implementation of aggressive screening including combinations of endoscopy and fecal occult blood testing.1 As in most solid tumors, surgical resection of the primary colorectal cancer remains the mainstay of curative treatment. However, our knowledge of basic cancer biology, particularly of cancer cells originating from the lower GI tract, has undergone revolutionary changes where new paradigms have emerged. These changes will aid in the development of new tools for the diagnosis, prognosis, and treatment of colorectal cancer in the foreseeable future, with the potential to drastically enhance the durability of surgical treatment. This review will briefly summarize the current understanding of colon cancer stem cells (CCSC), and will then focus the discussion on the microenvironment in which CCSCs proliferate and metastasize.
Cancer stem cells (CSC) were initially thought to mirror stem cells in normal tissue. In contrast to the stochastic model of cancer, where all cells in a tumor have the capability to reconstitute a tumor, the CSC model offers better explanations to a number of clinical properties of cancer because they have the ability to self-renew, differentiate, and lay dormant for years after irradiation or chemotherapy only to recur, at times, decades later. Additionally, CSCs may migrate to distant organs and cause metastasis. These metastatic foci escape detection by current methodologies, yet are capable of re-expanding, producing multiple aspects of a tumor. Given these reasons, CSCs provide a valuable target for new cancer therapies.2,3
It is important to recognize that CSCs are not strictly abnormal stem cells. For example, they may not represent a rare tumor cell population, especially as tumors become less differentiated.4 In addition, CSCs may not be a homogeneous population. They often originate with particular mutations that initiate the oncogenic process, but they evolve subsequently, accumulating additional mutations in that are effectively evolutionary “branch points,” resulting in clonal heterogeneity.5 These genetic and epigenetic changes make tumors complex and render them increasingly resistant to therapy. Furthermore, other cell types in the tumor and its surrounding tissues, including niche cells, stromal cells, immune cells, and vasculature, also undergo pathological changes, providing fertile “soil” for CSCs and compounding the complexity of tumor biology. English surgeon Stephen Paget first proposed the “seed and soil” hypothesis in 1889. However, it has only been in recent decades that the importance of the tumor microenvironment has gained appreciation.6,7 The complexity of the tumor microenvironment promotes the idea of “tumors as organs” that function with intricate interactions among several tissue types.8 Tackling the CSCs alone will not be sufficient in the search for future cancer therapies; we must also search the tumor microenvironment for additional, synergistic targets. Here, we review the current research on CSC microenvironment components and discuss the clinical applications associated with these discoveries.
Normal Colonic Stem Cell Regulation
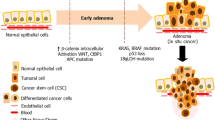
Colon cancer is a prototypical epithelial cancer. The maintenance of normal intestinal mucosa is essential for the prevention of cancer, and it is achieved in large part by a healthy microenvironment (Fig. 1). Fibroblasts synthesize a scaffold containing matrix proteins and adhesion molecules. Niche cells flank stem cells at the bottom of crypts and are responsible for stem cell maintenance and normal crypt architecture. In the small intestine, Paneth cells, in addition to their role in gut immunity, function as the stem cell niche. In the colon, an equivalent cell population may also exist.9 Within the crypt, immune cells help maintain stem cell integrity by removing defective cells. In addition, vascular endothelial cells form extensive vascular networks that shuttle nutrients and waste to and from the mucosa to foster healthy growth and renewal. Homeostasis is maintained by well-orchestrated interactions among several microenvironment components via growth factors and chemokines.

The stem cell niche and other surrounding cell types, including stromal fibroblasts, immune cells, and vascular endothelia, tightly regulate colonic crypt homeostasis. In colon cancer, these cells constitute the tumor microenvironment and play important roles in cancer initiation, progression, and metastasis
In oncogenesis, epithelial stem cells undergo pathological changes. Often, alterations in the microenvironment can trigger or propagate these changes. In other cases, changes in the CSCs can render these cells insensitive to maintenance and suppressive signals from the microenvironment. Consequently, various elements in the tumor microenvironment undergo further adaptive changes that sustain a vicious cycle of uncontrolled epithelial growth and invasion. The key to successful therapeutic intervention, therefore, rests in our ability to intercept the runaway pathological processes in and between various components of the tumor microenvironment and restore normal checks and balances.
The Stem Cell Niche
In order to function as a tissue reserve, stem cells must be sustained and maintained. In a normal colonic crypt, Paneth-cell-equivalent niche cells play a key role in normal stem cell maintenance. During colon cancer development, normal niche cells may become dysfunctional and the microenvironment converts to maintain these CSCs, which function as the tumor-cell reserve.
Paneth cells are a rare epithelial cell type in the small intestine. They normally reside in the base of the crypts, along with stem cells. Although they are better known for their role in gut immunity, it is also known that Paneth cell dysfunction is associated with inflammatory bowel disease (IBD).10 Given that IBD patients have a higher propensity to develop colon cancer, new insights about their function within the stem cell niche provide a major link between inflammation and cancer. Paneth cells provide all essential growth factors and signals for the in vitro growth of intestinal stem cells, including epithelial growth factor (EGF), transforming growth factor (TGF-α), Wnt3, and the Notch ligand Dll4. In addition, they are required for the in vitro formation of crypt-like organoids (Fig. 2), and exogenous administration of Wnt has been shown to substitute for this requirement.9 Defects in the Wnt signaling pathway commonly occur at early stages of sporadic colorectal cancer, which suggests that Paneth cells play a key role in cancer initiation. In addition, studies on colonic CD24+ cells (equivalents of Paneth cells in the small intestine) will further delineate the role of crypt base niche cells in the development of colorectal cancer.

Paneth cells (CD24+ cells in the colon) at the crypt base provide growth and maintenance signals to the adjacent stem cells and the crypt architecture. Alterations in this niche, such as mutations in Wnt signaling pathway genes, cause abnormal growth and loss of normal tissue architecture
Vascular endothelial cells are not only the main structural component in tumor vasculature; they also act as niche cells for CSCs. These cells activate signaling pathways that promote CSC phenotypes, including survival and self-renewal.11,12 Indeed, CSC survival along vasculature could be the key to metastatic seeding.
Stromal Fibroblasts
Cancer stroma differs from normal tissue stroma in both its form and function. Based on observations in breast tumors, cancer stroma is characterized by an abundance of connective tissue and dense arrays of fibroblasts.13,14 Stromal cells are derived from the mesenchymal lineage, and fibroblasts are the most abundant stromal cell type. Fibroblasts in the tumor microenvironment are termed cancer-associated fibroblasts (CAFs), and this cell population is dynamic. CAFs can arise from mesenchymal stem cells, endothelial cells, adipocytes, and even cancer epithelial cells.15–17 CAFs play an important role in tumor progression and invasion. In addition, they also participate in de novo cancer initiation.18,19 Healthy fibroblasts support normal tissue structure and function and participate in inflammation and injury repair. They do so by secreting a host of growth factors and cytokines such as fibroblast growth factor (FGF), epidermal growth factor (EGF), hepatic growth factor (HGF), macrophage stimulating protein (MSP), colony-stimulating factors (CSF), transforming growth factor-β (TGF-β), Wnt, matrix metalloproteinases (MMP), and interleukins. In cancer, many cellular processes mediated by CAFs’ paracrine secretions are hijacked.20 The fibroblast secretome is vast. Here we focus on secreted factors in the context of colon cancer. In addition, we discuss the role of CAFs in the mechanical forces surrounding a tumor and in the metabolic machinery that sustains tumor growth.
Transforming growth factor-β is expressed in most cell types, and its subtypes are potent regulators of growth, differentiation, and migration.21 Its role in cancer development is multifaceted and can appear paradoxical. For example, at low levels, TGF-β inhibits stem cell proliferation, but at increased levels, TGF-β can promote immune modulation and angiogenesis (Fig. 3).22 The TGF-β signaling pathway exerts its large variety of effects by activating SMAD, a transcription factor, which in turn changes the expression of many genes.23 Abnormalities in the TGF-β pathway can result in tumorigenesis, and cancer can result from defective TGF-β signaling in both epithelial cells and stromal fibroblasts. During tissue injury and inflammation, TGF-β is released from epithelial cells which promotes proliferation of stromal fibroblasts to participate in tissue repair.24 Stromal fibroblasts, on the other hand, produce TGF-β to keep excessive epithelial growth in check.25 However, in colon cancer, TGF-β type II receptor (TGFβRII) mutations occur frequently, resulting in uncontrolled proliferation of epithelia.26–28 The insensitivity of the TGF-β receptor, in turn, triggers a feedback loop in which stromal cells produce excess TGF-β. At high levels, TGF-β enhances immune modulation and angiogenesis, which may further promote cancer growth.

Cancer-associated fibroblasts secrete a large number of growth factors and cytokines, including EGF, FGF, IL6, and IL8. This figure illustrates the feedback upregulation of TGF-β due to receptor insensitivity on cancer epithelia. This upregulation is also associated with an upregulation of HGF. Whereas high levels of TGF-β promote a supportive tumor microenvironment, HGF acts directly on tumor epithelia to promote tumor growth and metastasis
Oncological processes usually involve multiple signaling pathways. There is tremendous crosstalk or cross-activation among signaling pathways involving a number of cell types in the microenvironment. The interplay between the TGF-β and HGF/c-Met pathways illustrates this well (Fig. 3). In one study, a mouse model with a conditional knockout of TGFβRII showed de novo carcinogenesis in prostate and forestomach cells. In this work, HGF was upregulated, and c-Met was activated in tissue carrying the conditional knockout of TGFβRII.20 HGF and MSP (both are ligands of c-Met) are primarily expressed by stromal fibroblasts, and the HGF/c-Met pathway controls cell proliferation, survival, and metastasis via multiple signaling pathways. Hepatocyte growth factor also modulates the Wnt signaling by affecting β-catenin activity, resulting in restoration of the CSC phenotype and acquisition of an invasive phenotype.29 In another study using a TGFβRII-knockout breast cancer model, the same authors again report elevated HGF/c-Met activity. Furthermore, they demonstrate that the inhibition of HGF and MSP activity reduced proliferation of epithelial cells.30 Pharmacological inhibitors targeting components of both the TGF-β and the HGF/c-Met pathways have been developed and many have shown effectiveness in the clinical setting.
Epidermal growth factor and FGF foster the growth of the tumor and its stroma, and both factors are secreted by CAFs. Most epithelial cancers express EGF receptors (EGFR), and inhibitors of EGFR have been used successfully to treat EGFR+ cancer, including cetuximab for colon cancer.
In addition to growth factors, CAFs also secrete inflammatory cytokines/chemokines such as interleukin-6 (IL6) and interleukin-8 (IL8)/CXCL8. Interleukin-6 is abundantly expressed in colon cancer, and its level directly correlates with cancer stage and mortality. One of the pathways activated by IL8 is the JAK/STAT3 pathway, which is shown to promote cancer initiation and progression.31 Interleukin-8 is also a neutrophil chemotactic factor and like IL6, IL8 is found in large quantities in the tumor microenvironment. IL8 has roles in tumor initiation, progression, metastasis, and angiogenesis. It acts transcriptionally through a large number of signaling pathways, affecting cell survival, proliferation, and metabolism for both cancer epithelia and vascular endothelia.32 The high levels of IL6 and IL8 expressed by cancer epithelia drive a powerful positive feedback loop that promotes tumor growth and proliferation.33,34 This makes a compelling argument for the development of inhibitors and antibodies against cytokines or their receptors.35,36 For example, reparixin (formerly known as repertaxin) is an inhibitor of the IL8 receptor and is currently being studied for cancer therapy.37
Consistent with the many parallels between cancer and inflammation, CAFs can gradually gain myofibroblast function over time, mirroring the behavior of fibroblasts in tissue injury. They change their organizational structure and secretome to accommodate tumor growth. Myofibroblasts characteristically express α smooth muscle actin (α-SMA), and have smooth muscle properties.38,39 This may explain the fibrotic feature of many solid tumors, but more importantly, it offers insight into the role of mechanical forces in tumor growth and invasion.40 One could envision an intervention that disrupts the mechanical forces in the tumor microenvironment as a potential way to treat cancer. Also, the quantity of myofibroblasts, using α-SMA as a marker, correlates to colon cancer recurrence after resection.41 This could be used as a prognostic biomarker, which could help in stratifying patients with stage II colon cancer.
Cancer associated fibroblasts have adapted to support the metabolism of cancer epithelia. Colon cancer epithelia show characteristics of anaerobic metabolism. For example, they express high levels of lactate dehydrogenase-5 (LDH5) and hypoxia-inducible factor 1α (HIF1α). They also exhibit efficient glucose absorption and lactate extrusion. Opposite that of epithelia, CAFs express proteins involved in lactate absorption, lactate oxidation, and low glucose absorption.42 Further understanding of these adaptive changes may allow us to eliminate cancer epithelia by starvation of the CAFs in the microenvironment.
In summary, CAFs are transformed stromal fibroblasts that are capable of initiating cancer and supporting its growth and metastasis. In colon cancer, many of the chemical and mechanical properties of CAFs discussed here can be exploited for treatment, diagnosis, and monitoring.
Immune Cells
The association between inflammation and cancer is well established. Virchow first documented this link in 1863 when he noted leukocytes in neoplastic tissues. A number of malignancies involving gastrointestinal, genitourinary, and respiratory tracts have known associations with inflammation, including colon cancer.43 As our understanding of the immune system evolves, its implication in cancer biology becomes increasingly evident. A normal inflammatory response serves the dual purpose of immunity against infectious pathogens and repair of injured tissue. It begins with recruitment of immune cells from the blood stream. These immune cells arrive at their target following cytokine signals and exit the vasculature with the assistance of adhesion molecules expressed by endothelial cells. Proteinases from the stroma break down the extracellular matrix (ECM), allowing immune cells to infiltrate. Neutrophils are typically first to arrive, followed by macrophages and lymphocytes. Chronic inflammation not only initiates genomic defects, but also sustains these mutations as cancer cells proliferate, much like a “wound that does not heal.”44 Some of the ongoing effects of the immune system are pro-tumor, while others are anti-tumor (Fig. 4). The obvious strategy for devising cancer therapies would be to amplify the anti-tumor affects and suppress the pro-tumor affects.

Cancer changes immune cell phenotype to support tumor growth. It induces proliferation of immature myeloid cells, which produce pro-inflammatory factors that further potentiate tumor growth. At the same time, as a pathologic response, immature myeloid cells also produce factors that mediate suppression of innate and adaptive immunity, allowing unabated cancer proliferation and metastasis
Granulocytes drive the initial inflammatory response, and in cancer, they have been shown to promote angiogenesis and tumor growth.45,46 However, macrophages are the workhorses in an inflammatory environment. Macrophages present antigens and secrete pro-inflammatory cytokines (IL6, IL12, IL23, TNF-α) and anti-inflammatory cytokines (IL4, IL10, IL13, TGF-β) that coordinate balanced immune responses. Furthermore, macrophages participate in cytotoxic killing. In tumors, these tumor-associated macrophages (TAM) can be both pro-tumorigenic and anti-tumorigenic. Their polarity is dependent on the type of cancer. In most cancers, they correlate with worse prognosis. In colon cancer, however, they may have the opposite effect. This discrepancy may be the result of skewing toward one of two polar types of TAMs present in a tumor. Anti-tumor TAMs are labeled as M1. They have more antigen presenting molecules and co-stimulatory receptors for T-cells, make a number of pro-inflammatory cytokines, and have killing capabilities for pathogens and cancer cells. Pro-tumor TAMs are labeled as M2. They are more adept at tissue remodeling, driving cell proliferation and angiogenesis, and producing immunosuppressive cytokines.47–49 This model provides the premise for a treatment paradigm that can shift TAMs from the M2 to the M1 phenotype. For example cyclooxygenase-2 (COX-2) has been found to be upregulated in many cancers, and COX-2 inhibitors have been found to reduce the number and size of adenomas in patients with familial adenomatous polyposis (FAP) syndrome. A recent study on mouse polyps shows that the anti-tumor effect of COX-2 inhibitors may stem from its ability to shift TAMs from M2 to M1 phenotypes.50
Dendritic cells also come from the myeloid lineage. In a healthy organism, they reside in peripheral tissues in immature states. Upon antigen activation, they express co-stimulatory receptors, migrate to lymphoid organs, and drive T-cell activation and proliferation. In cancer, they decrease in number and are functionally altered.51 In cancer, dendritic cells become less capable of developing co-stimulatory signals and driving effector T-cell activity.52 Also, these dendritic cells express increased levels of IL10, indoleamine-2,3-dioxygenase (IDO), TGF-β, and COX-2, further potentiating their immunosuppressive effects. Furthermore, they express increased levels of vascular endothelial growth factor (VEGF) and pro-inflammatory cytokines like IL6 and IL8, which are all potent mediators of cancer growth and metastasis.53
Immature myeloid precursor cells are of intense interest as they are implicated in cancer immune suppression as well as immune suppressive states, such as that in sepsis. These cells are aptly named myeloid derived suppressor cells (MDSC). They are broadly defined as LIN−, CD11b+, HLA-DR−, and CD33+ cells in humans, and they encompass immature neutrophils, dendritic cells, and monocytes, as well as early myeloid progenitors.54 In cancer, they are released in response to CSF secreted by the cancer stroma. They allow tumor survival and growth by suppressing natural killer (NK) cell function, inhibiting cytotoxic T-cell expansion, as well as expanding regulatory T-cells that are immunosuppresive.55,56 Additionally, they can promote angiogenesis by the secretion of VEGFA, basic FGF (bFGF), and TGF-β.57 The development of cancer MDSCs may be tightly integrated with the development of individual pathological myeloid cell types and many functions of MDSCs collectively reflect those of individual pathologic granulocytes, macrophages, and dendritic cells. Targeting MDSCs for cancer therapies can be accomplished using one of three strategies:
-
1.
Facilitate their maturation into effective immune cells that can assist with cancer killing.
-
2.
Inhibit their immune suppressive effect on cytotoxic immune cell proliferation and function.
-
3.
Prohibit their production and release from bone marrow.
Many cancer drugs currently use one of these strategies, and a number of them are used to treat colorectal cancer.54
The T-lymphocyte is the main effector cell in the immune system and major checkpoints exist in the immune system that down-regulate the T-cell response. For example, cytotoxic lymphocytic antigen-4 (CTLA-4) dampens T-cell directed killing. Antagonistic antibodies for this receptor therefore liberate T-cells from such inhibition and enhance the destruction of cancer. In fact, this class of drugs, approved by the Food and Drug Administration in 2010 is the first to demonstrate a survival benefit against melanoma. Programmed death 1 (PD1) is another checkpoint affected by CTLA-4.58 Uncovering additional checkpoints and finding ways to block them will be a major component in future cancer therapy.
The CSC is a unique pro-inflammatory instrument. Instead of being recognized by the body as foreign like most pathogens or autoantigens, it can evade immune destruction and send signals that promote tumor survival and metastasis. The idea of reprogramming our own body’s immune system to combat tumors is an attractive one. A better understanding of all components of the immune system and their roles within the tumor environment is necessary to achieve this goal.
Vasculature
Although, as mentioned above, CAFs are adapted to support the anaerobic metabolism that cancer epithelia often exhibit, angiogenesis is still required to support a tumor’s high metabolic demand.42 A positive correlation exists between the degree of angiogenesis and the severity of cancer.59 For this reason, the search for angiogenesis inhibitors has received much attention in recent years. However, the exhaustive pool of knowledge gained from angiogenesis research has not, as many had hoped, yielded a cure for cancer. This is partly due to the complexity of the tumor microenvironment and partly due to a dilemma involving the problem of drug delivery. In order to deliver drugs, robust blood supply is needed, but as the tumor vasculature contracts, the effects of drugs necessarily decrease. This reduces the effectiveness of not only anti-angiogenesis drugs, but also other chemotherapeutic agents. In the same vein, hypoxic regions of tumors may harbor stem cells that have low metabolic requirements, which further contribute to chemoresistance.60 Evidence from recent glioblastoma research reveals that CSCs can undergo mesenchymal differentiation, giving rise to pericytes that support vascular tissue growth and function.61
The current generation of anti-angiogenesis therapies has been developed following the principle outlined by Judah Folkman.62 He stated that it is “helpful to think the switch to the angiogenic phenotype as a net balance of positive and negative regulators of blood vessel growth. The extent to which the negative regulators are decreased during this switch may dictate whether a primary tumor grows rapidly or slowly and whether metastases grow at all.” He further observed that positive regulators available in the tumor microenvironment, such as aFGF, bFGF, VEGF, angiogenin, by themselves do not result in faster angiogenesis required by the tumor. There must be concomitant downregulation of the negative regulators such as thrombospondin and angiostatin. In many cancers, thrombospondin is decreased with p53 mutation, and angiostatin is decreased by critical tumor mass. A number of anti-angiogenesis drugs have been developed and approved for clinical use. They target a variety of mechanisms, including those involving growth factors like VEGF, EGF and platelet-derived growth factor (PDGF), receptor tyrosine kinases (RTK), transcription factors like HIF, and molecules involved in MAPK and PI3K signaling.63 Avastin (bevacizumab), a VEGF inhibitor was the first of such drugs, and it was approved in 2004 for use in metastatic colorectal cancer. It slowed tumor growth and improved patient survival.64 The commonality among all of these drugs is that they are angiogenesis-inhibiting agents (AIA), and that they are used at late stages of cancer to retard tumor growth and prolong patient survival.
There is another burgeoning area of anti-angiogenesis research that aims to rapidly destroy early tumor vasculature either by disrupting endothelial cell cytoskeleton or inducing their apoptosis. These drugs are termed tumor vascular disrupting agents (VDA). Tumor-VDAs are given during early tumor development to initiate thrombotic events in the neovasculature and cause extensive tumor necrosis.65 These agents are in early clinical trials, and may be used alone or with other treatment modalities, such as radiation, to enhance cancer killing.
In summary, angiogenesis is a key feature in tumorigenesis. Many cellular and intercellular processes in the tumor microenvironment play a role in this activity. We have developed numerous drugs exploiting a number of important factors. While many show beneficial effects in the clinical setting, they improve the length and quality of life by only a modest margin. The next generation of tumor-VDAs has the potential to be a game changer, pending results of the clinical trials.
Conclusion
The study of CSCs has transformed over time and necessarily incorporates the tumor microenvironment. The appreciation for the complex interaction among all components of the microenvironment increases with the growth of knowledge in this area. This appreciation will allow us to create mature and comprehensive cancer treatment regimens. This review outlines a schematic of how the cancer microenvironment may behave, using a system of functional compartments including the prototypical epithelial cancer cell (the CCSC), the stem cell niche, the stroma, the immune system, and the tumor vasculature. There are many details that still need to be elucidated and integrated into this scheme. It may be helpful to think of tumors as organs, as they are composed of multiple tissue types derived from more than one embryonic origin. Future treatments of tumors, including ones in the lower gastrointestinal tract, must usurp all components of this complex organ.
Moreover, we must not neglect the fourth dimension of tumors, which is time. While CSCs evolve over time, other components of the microenvironment co-evolve. For these reasons, there may never be an all-encompassing cancer cure. In fact, an optimal outcome may be that the patient survives with a low level of existing disease, with minimal impact on their quality of life. The revolution in genetic and epigenetic research may allow us to peer into the individuality of a tumor at a particular time in space, keep pace with its progression, and orchestrate a systematic attack tailored to a single patient involving multiple modalities and therapeutic regimens.
References
Cancer facts & Figures 2013, 2013, American Cancer Society: Atlanta.
Reya T and Clevers H. Wnt signalling in stem cells and cancer. Nature 2005;434:843-850.
Clevers H. The cancer stem cell: Premises, promises and challenges. Nature medicine 2011;17:313-319.
Kelly P, Dakic A, Adams J, Nutt S, and Strasser A. Tumor growth need not be driven by rare cancer stem cells. Science (New York, N.Y.) 2007;317:337.
Gerlinger M, Rowan A, Horswell S, Larkin J, Endesfelder D, Gronroos E, Martinez P, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald N, Butler A, Jones D, Raine K, Latimer C, Santos C, Nohadani M, Eklund A, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal P, and Swanton C. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. The New England journal of medicine 2012;366:883-892.
Paget S. The distribution of secondary growths in cancer of the breast. The Lancet 1889;133:571-573.
Fidler I. The pathogenesis of cancer metastasis: The 'seed and soil' hypothesis revisited. Nature reviews. Cancer 2003;3:453-458.
Egeblad M, Nakasone E, and Werb Z. Tumors as organs: Complex tissues that interface with the entire organism. Developmental cell 2010;18:884-901.
Sato T, van Es J, Snippert H, Stange D, Vries R, van den Born M, Barker N, Shroyer N, van de Wetering M, and Clevers H. Paneth cells constitute the niche for lgr5 stem cells in intestinal crypts. Nature 2011;469:415-418.
Clevers H and Bevins C. Paneth cells: Maestros of the small intestinal crypts. Annual review of physiology 2013;75:289-311.
Krishnamurthy S, Dong Z, Vodopyanov D, Imai A, Helman J, Prince M, Wicha M, and Nör J. Endothelial cell-initiated signaling promotes the survival and self-renewal of cancer stem cells. Cancer research 2010;70:9969-9978.
Jia L, Xiangcang Y, Fan F, Ling X, Rajat B, Seth B, Federico T, Eric S, Yunfei Z, Isamu T, Dipen MM, David HH, Janusz R, Sendurai AM, Patrick Z-M, and Lee ME. Endothelial cells promote the colorectal cancer stem cell phenotype through a soluble form of jagged-1. Cancer cell 2013;23:171-185.
Rønnov-Jessen L, Petersen O, and Bissell M. Cellular changes involved in conversion of normal to malignant breast: Importance of the stromal reaction. Physiological reviews 1996;76:69-125.
Tlsty T and Hein P. Know thy neighbor: Stromal cells can contribute oncogenic signals. Current opinion in genetics & development 2001;11:54-59.
Jotzu C, Alt E, Welte G, Li J, Hennessy B, Devarajan E, Krishnappa S, Pinilla S, Droll L, and Song Y-H. Adipose tissue-derived stem cells differentiate into carcinoma-associated fibroblast-like cells under the influence of tumor-derived factors. Analytical cellular pathology (Amsterdam) 2010;33:61-79.
Mink S, Vashistha S, Zhang W, Hodge A, Agus D, and Jain A. Cancer-associated fibroblasts derived from egfr-tki-resistant tumors reverse egfr pathway inhibition by egfr-tkis. Molecular cancer research : MCR 2010;8:809-820.
Zeisberg E, Potenta S, Xie L, Zeisberg M, and Kalluri R. Discovery of endothelial to mesenchymal transition as a source for carcinoma-associated fibroblasts. Cancer research 2007;67:10123-10128.
Bhowmick N, Chytil A, Plieth D, Gorska A, Dumont N, Shappell S, Washington M, Neilson E, and Moses H. Tgf-beta signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science (New York, N.Y.) 2004;303:848-851
Kuperwasser C, Chavarria T, Wu M, Magrane G, Gray J, Carey L, Richardson A, and Weinberg R. Reconstruction of functionally normal and malignant human breast tissues in mice. Proceedings of the National Academy of Sciences of the United States of America 2004;101:4966-4971.
Bhowmick N, Neilson E, and Moses H. Stromal fibroblasts in cancer initiation and progression. Nature 2004;432:332-337.
Bierie B and Moses H. Tumour microenvironment: Tgfbeta: The molecular jekyll and hyde of cancer. Nature reviews. Cancer 2006;6:506-520.
Mishra L, Derynck R, and Mishra B. Transforming growth factor-beta signaling in stem cells and cancer. Science (New York, N.Y.) 2005;310:68-71
Siegel P and Massagué J. Cytostatic and apoptotic actions of tgf-beta in homeostasis and cancer. Nature reviews. Cancer 2003;3:807-821.
Zeisberg M, Strutz F, and Müller G. Role of fibroblast activation in inducing interstitial fibrosis. Journal of nephrology 2000;13 Suppl 3:S111-120.
Becker C, Fantini M, Schramm C, Lehr H, Wirtz S, Nikolaev A, Burg J, Strand S, Kiesslich R, Huber S, Ito H, Nishimoto N, Yoshizaki K, Kishimoto T, Galle P, Blessing M, Rose-John S, and Neurath M. Tgf-beta suppresses tumor progression in colon cancer by inhibition of il-6 trans-signaling. Immunity 2004;21:491-501.
Markowitz S, Wang J, Myeroff L, Parsons R, Sun L, Lutterbaugh J, Fan R, Zborowska E, Kinzler K, and Vogelstein B. Inactivation of the type ii tgf-beta receptor in colon cancer cells with microsatellite instability. Science (New York, N.Y.) 1995;268:1336-1338
Biswas S, Trobridge P, Romero-Gallo J, Billheimer D, Myeroff L, Willson J, Markowitz S, and Grady W. Mutational inactivation of tgfbr2 in microsatellite unstable colon cancer arises from the cooperation of genomic instability and the clonal outgrowth of transforming growth factor beta resistant cells. Genes, chromosomes & cancer 2008;47:95-106.
Biswas S, Chytil A, Washington K, Romero-Gallo J, Gorska A, Wirth P, Gautam S, Moses H, and Grady W. Transforming growth factor beta receptor type ii inactivation promotes the establishment and progression of colon cancer. Cancer research 2004;64:4687-4692.
Vermeulen L, De Sousa E Melo F, van der Heijden M, Cameron K, de Jong J, Borovski T, Tuynman J, Todaro M, Merz C, Rodermond H, Sprick M, Kemper K, Richel D, Stassi G, and Medema J. Wnt activity defines colon cancer stem cells and is regulated by the microenvironment. Nature cell biology 2010;12:468-476.
Cheng N, Bhowmick N, Chytil A, Gorksa A, Brown K, Muraoka R, Arteaga C, Neilson E, Hayward S, and Moses H. Loss of tgf-beta type ii receptor in fibroblasts promotes mammary carcinoma growth and invasion through upregulation of tgf-alpha-, msp- and hgf-mediated signaling networks. Oncogene 2005;24:5053-5068.
Waldner M, Foersch S, and Neurath M. Interleukin-6--a key regulator of colorectal cancer development. International journal of biological sciences 2012;8:1248-1253.
Waugh DJ and Wilson C. The interleukin-8 pathway in cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2008;14:6735-6741.
Korkaya H, Liu S, and Wicha M. Regulation of cancer stem cells by cytokine networks: Attacking cancer's inflammatory roots. Clinical cancer research : an official journal of the American Association for Cancer Research 2011;17:6125-6129.
Carpentino J, Hynes M, Appelman H, Zheng T, Steindler D, Scott E, and Huang E. Aldehyde dehydrogenase-expressing colon stem cells contribute to tumorigenesis in the transition from colitis to cancer. Cancer research 2009;69:8208-8215.
Guo Y, Xu F, Lu T, Duan Z, and Zhang Z. Interleukin-6 signaling pathway in targeted therapy for cancer. Cancer treatment reviews 2012;38:904-910.
Varney M, Singh S, Li A, Mayer-Ezell R, Bond R, and Singh R. Small molecule antagonists for cxcr2 and cxcr1 inhibit human colon cancer liver metastases. Cancer letters 2011;300:180-188.
Ginestier C, Liu S, Diebel M, Korkaya H, Luo M, Brown M, Wicinski J, Cabaud O, Charafe-Jauffret E, Birnbaum D, Guan J-L, Dontu G, and Wicha M. Cxcr1 blockade selectively targets human breast cancer stem cells in vitro and in xenografts. The Journal of clinical investigation 2010;120:485-497.
Otranto M, Sarrazy V, Bonté F, Hinz B, Gabbiani G, and Desmoulière A. The role of the myofibroblast in tumor stroma remodeling. Cell adhesion & migration 2012;6:203-219.
De Wever O, Demetter P, Mareel M, and Bracke M. Stromal myofibroblasts are drivers of invasive cancer growth. International journal of cancer. Journal international du cancer 2008;123:2229-2238.
Tse J, Cheng G, Tyrrell J, Wilcox-Adelman S, Boucher Y, Jain R, and Munn L. Mechanical compression drives cancer cells toward invasive phenotype. Proceedings of the National Academy of Sciences of the United States of America 2012;109:911-916.
Tsujino T, Seshimo I, Yamamoto H, Ngan C, Ezumi K, Takemasa I, Ikeda M, Sekimoto M, Matsuura N, and Monden M. Stromal myofibroblasts predict disease recurrence for colorectal cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2007;13:2082-2090.
Koukourakis M, Giatromanolaki A, Harris A, and Sivridis E. Comparison of metabolic pathways between cancer cells and stromal cells in colorectal carcinomas: A metabolic survival role for tumor-associated stroma. Cancer research 2006;66:632-637.
Balkwill F and Mantovani A. Inflammation and cancer: Back to virchow? Lancet 2001;357:539-545.
Coussens L and Werb Z. Inflammation and cancer. Nature 2002;420:860-867.
Houghton A, Rzymkiewicz D, Ji H, Gregory A, Egea E, Metz H, Stolz D, Land S, Marconcini L, Kliment C, Jenkins K, Beaulieu K, Mouded M, Frank S, Wong K, and Shapiro S. Neutrophil elastase-mediated degradation of irs-1 accelerates lung tumor growth. Nature medicine 2010;16:219-223.
Nozawa H, Chiu C, and Hanahan D. Infiltrating neutrophils mediate the initial angiogenic switch in a mouse model of multistage carcinogenesis. Proceedings of the National Academy of Sciences of the United States of America 2006;103:12493-12498.
Ong S-M, Tan Y-C, Beretta O, Jiang D, Yeap W-H, Tai J, Wong W-C, Yang H, Schwarz H, Lim K-H, Koh P-K, Ling K-L, and Wong S-C. Macrophages in human colorectal cancer are pro-inflammatory and prime t cells towards an anti-tumour type-1 inflammatory response. European journal of immunology 2012;42:89-100.
Edin S, Wikberg M, Rutegård J, and Oldenborg… P. Phenotypic skewing of macrophages in vitro by secreted factors from colorectal cancer cells. PloS one 2013;8:e74982
Erreni M, Mantovani A, and Allavena P. Tumor-associated macrophages (tam) and inflammation in colorectal cancer. Cancer microenvironment : official journal of the International Cancer Microenvironment Society 2011;4:141-154.
Nakanishi Y, Nakatsuji M, Seno H, Ishizu S, Akitake-Kawano R, Kanda K, Ueo T, Komekado H, Kawada M, Minami M, and Chiba T. Cox-2 inhibition alters the phenotype of tumor-associated macrophages from m2 to m1 in apcmin/+ mouse polyps. Carcinogenesis 2011;32:1333-1339.
Almand B, Resser JR, Lindman B, Nadaf S, Clark JI, Kwon ED, Carbone DP, and Gabrilovich DI. Clinical significance of defective dendritic cell differentiation in cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 2000;6:1755-1766.
Gabrilovich D. Mechanisms and functional significance of tumour-induced dendritic-cell defects. Nature reviews. Immunology 2004;4:941-952.
Novitskiy SV, Ryzhov S, Zaynagetdinov R, Goldstein AE, Huang Y, Tikhomirov OY, Blackburn MR, Biaggioni I, Carbone DP, Feoktistov I, and Dikov MM. Adenosine receptors in regulation of dendritic cell differentiation and function. Blood 2008;112:1822-1831.
Gabrilovich D, Ostrand-Rosenberg S, and Bronte V. Coordinated regulation of myeloid cells by tumours. Nature reviews. Immunology 2012;12:253-268.
Shojaei F, Wu X, Qu X, Kowanetz M, Yu L, Tan M, Meng Y, and Ferrara N. G-csf-initiated myeloid cell mobilization and angiogenesis mediate tumor refractoriness to anti-vegf therapy in mouse models. Proceedings of the National Academy of Sciences of the United States of America 2009;106:6742-6747.
Pylayeva-Gupta Y, Lee K, Hajdu C, Miller G, and Bar-Sagi D. Oncogenic kras-induced gm-csf production promotes the development of pancreatic neoplasia. Cancer cell 2012;21:836-847.
Motz G and Coukos G. The parallel lives of angiogenesis and immunosuppression: Cancer and other tales. Nature reviews. Immunology 2011;11:702-711.
Pardoll D. The blockade of immune checkpoints in cancer immunotherapy. Nature reviews. Cancer 2012;12:252-264.
Di Renzo M, Olivero M, Giacomini A, Porte H, Chastre E, Mirossay L, Nordlinger B, Bretti S, Bottardi S, and Giordano S. Overexpression and amplification of the met/hgf receptor gene during the progression of colorectal cancer. Clinical cancer research : an official journal of the American Association for Cancer Research 1995;1:147-154.
Mao Q, Zhang Y, Fu X, Xue J, Guo W, Meng M, Zhou Z, Mo X, and Lu Y. A tumor hypoxic niche protects human colon cancer stem cells from chemotherapy. Journal of cancer research and clinical oncology 2013;139:211-222.
Cheng L, Huang Z, Zhou W, Wu Q, Donnola S, Liu JK, Fang X, Sloan AE, Mao Y, Lathia JD, Min W, McLendon RE, Rich JN, and Bao S. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell 2013;153:139-152.
Folkman J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nature medicine 1995;1:27-31.
Cook K and Figg W. Angiogenesis inhibitors: Current strategies and future prospects. CA: a cancer journal for clinicians 2010;60:222-243.
Shih T and Lindley C. Bevacizumab: An angiogenesis inhibitor for the treatment of solid malignancies. Clinical therapeutics 2006;28:1779-1802.
Siemann D. The unique characteristics of tumor vasculature and preclinical evidence for its selective disruption by tumor-vascular disrupting agents. Cancer treatment reviews 2011;37:63-74.
Acknowledgments
We gratefully acknowledge Kristen Huang, PhD, for careful reading of the text.
Author information
Authors and Affiliations
Corresponding author
Additional information
Grant Support: Sugong Chen receives support from NIH T32 CA106493, awarded to Kevin E Behrns. Emina H. Huang is the principal investigator of NIH R01 CA142808 and R01 CA157663.
Rights and permissions
About this article
Cite this article
Chen, S., Huang, E.H. The Colon Cancer Stem Cell Microenvironment Holds Keys to Future Cancer Therapy. J Gastrointest Surg 18, 1040–1048 (2014). https://doi.org/10.1007/s11605-014-2497-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11605-014-2497-1