
Abstract
Distance geometry is a class of problems where the position of points in space is to be identified by using information about some relative distances between these points. Although continuous approaches are usually employed, problems belonging to this class can be discretized when some particular assumptions are satisfied. These assumptions strongly depend on the order in which the points to be positioned are considered. We discuss new discretization assumptions that are weaker than previously proposed ones, and present a greedy algorithm for an automatic identification of discretization orders. The use of these weaker assumptions motivates the development of a new method for computing point coordinates. Computational experiments show the effectiveness and efficiency of the proposed approaches when applied to protein instances.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
Given a positive integer \(K\) and a simple weighted undirected graph \(G=(V,E,d)\), where \(d\) maps edges \((u,v) \in E\) to positive interval weights \([\underline{d}(u,v),\bar{d}(u,v)]\), the distance geometry problem (DGP) [13] is the one of finding an embedding of the graph \(G\) in a \(K\)-dimensional space, that is a function \(\mathrm{x}: V \longrightarrow \mathbb {R}^K\) satisfying the following set of constraints:
where \(||\cdot ||\) denotes the Euclidean norm. An important subclass of DGPs is the molecular DGP (MDGP) [7], where the graph \(G\) gives information to be used for the identification of molecular conformations. In such a case, each vertex \(v \in V\) of the graph represents an atom of the molecule, while \(E\) contains edges \((u,v)\) between vertices, whose weight represents the known distances between the atoms \(u\) and \(v\). When dealing with experimentally obtained data, the distance information \(d\) is generally given by a lower and an upper bound on the distances, i.e. by the interval \([\underline{d}(u,v),\bar{d}(u,v)]\). Distances obtained from experiments of nuclear magnetic resonance (NMR) [15] are, for example, generally represented by intervals. Distances between bonded atoms, instead, can be deduced from already known X-ray obtained molecular conformations, and they can be considered as precise [14]. Naturally, an exact distance can be represented as a degenerate interval, where \(\underline{d}(u,v) = \bar{d}(u,v)\), so that the constraint (1) becomes an equality constraint.
There is a wide literature on DGPs (see for example the recent survey [13]). They can be formulated as global optimization problems in a continuous space. The objective function, to be minimized, is generally a penalty function that measures the violation of the constraints (1). One example is the widely-used largest distance error (LDE):
where \(X = (\mathrm{x}_1,\mathrm{x}_2,\dots ,\mathrm{x}_n)\) and \(n = |V|\), and where the symbol \(|\cdot |\) denotes the set cardinality. One previously proposed continuous approach is, for example, the one in [16], where the objective function is approximated by smoother functions converging to the original one. Based on the continuous approach is also the work in [9], where the concept of funnel bottom is exploited, and where the optimization process is inspired by the shape of energy landscapes. Moreover, semidefinite programming approaches have been more recently proposed in [1, 3]. The interested reader can find various interesting approaches to the DGP in a recently edited book [19].
When particular assumptions are satisfied, the search space of the DGP, which is generally a continuous space, can be discretized (see for example [11, 18]). The basic idea behind the discretization is as follows. Consider we are trying to place in the space the vertex \(u \in V\). If the distances between \(u\) and other vertices \(v_i \in V\) (with known coordinates) are available, then the possible positions for \(u\) belong to the Euclidean object given by the intersection of the spherical shells centered in the positions for the vertices \(v_i\), and having minimum radii \(\underline{d}(v_i,u)\) and maximum radii \(\bar{d}(v_i,u)\), respectively. Therefore, the discretization is possible when, for every vertex \(u \in V\), this intersection provides a discrete object, or an object that can be discretized without losing so much information.
The easiest case is the one treated for the first time in [11], where all distances are supposed to be exact (\(\underline{d}(v_i,u) = \bar{d}(v_i,u)\)), and, in dimension \(K\), there are at least \(K\) reference vertices \(v_i \in V\), for every \(u > K\). In such a case, indeed, the spherical shells degenerate to spheres, and the intersection of \(K\) spheres gives at most two points in the \(K\)-dimensional space (when an additional assumption is also satisfied, see Sect. 2). The intersection object is therefore discrete and finite. If fewer distances are available, or if not all of them are exact, then the intersection object is generally not discrete.
DGPs with interval data can however still be discretized by employing the strategy that was preliminary proposed in [12]. The idea is to allow only one of the \(K\) distances to be a nondegenerate interval, so that the intersection object has dimension 1, and we can choose a discrete subset of positions from it. This is a realistic assumption when working with biological molecules, as it is shown in Sect. 5.
This paper deals with two important facts related to the discretization. First, given a graph \(G\) representing an instance of the DGP, how to identify an order for the vertices in \(V\) so that the intersection object is (or can be considered as) discrete? Since the vertices \(v_i\) will be references for finding the position of the vertex \(u\), they must be placed earlier in the order. It is clear therefore how the concept of “order” is important for the discretization. We will formalize this problem in the case the distance information can be imprecise, and we will present an efficient greedy algorithm for its solution. Differently from previous publications, we will consider orders that satisfy weaker assumptions.
The second part of the paper gives an answer to the following question: given \(K\) reference vertices \(v_i \in V\), how to compute the Cartesian coordinates of a vertex \(u\)? Vertex positions can be identified by performing intersections of Euclidean objects. When these objects are spheres, the intersection points can be found by solving a system of quadratic equations [5]. Its solution, however, can lead to numerical instabilities [18]. Another method instead, widely used in the past, is not sensitive to errors, but requires discretization orders satisfying stronger assumptions [11]. We will propose a more efficient and reliable procedure, that will allow us to consider instances satisfying discretization orders based on the new weaker assumptions.
The rest of the paper is organized as follows. In Sect. 2, we will discuss the conditions for a DGP to be discretizable and we will briefly report the interval Branch & Prune (\(i\hbox {BP}\)) algorithm [12] for the solution of discretizable DGPs. In Sect. 3, the problem of finding discretization orders for vertices of graphs \(G\) will be discussed, and a solution method will be detailed. In Sect. 4, we will propose a new procedure for an efficient computation of Cartesian coordinates of the vertices, and we will compare to other previously employed procedures. Computational experiments, on discretizable MDGP instances, will be presented in Sect. 5. Finally, concluding remarks will be given in Sect. 6.
2 Discretizable DGPs and \(i\)BP algorithm
Let \(G=(V,E,d)\) be a simple weighted undirected graph representing an instance of the DGP. Let \(E'\) be the subset of \(E\) for which the weight associated to the edge is an exact distance (degenerate interval).
When defining a discretizable instance of the DGP, it is important to verify some properties related to the order relationship on the vertices of \(V\).
Definition 1
An order for \(V\) is a sequence \(r:\mathbb {N}\rightarrow V \cup \{ 0 \}\) (with length \(|r| \ge |V|\)) such that, for each \(v \in V\), there is an index \(i \in \mathbb {N}\) for which \(r_i = v\) [17].
We remark that, in previous publications,the repetition of vertices in the order was allowed. This artifice was used to find orders that satisfied stronger assumptions with respect to the ones we will consider in this paper [6, 12]. In this work, orders will not contain repeated vertices.
Once an order \(r\) has been assigned to \(V\), it is possible to verify, for each vertex of the graph \(G\), its predecessors and successors [17]. Let
be the subset of adjacent predecessors for the vertex \(r_i\) in the order \(r\). Let
be the subset of adjacent successors of \(r_i\) in the order \(r\). We will denote the cardinality of these sets by:
In Definition 2, we will also consider the cardinality of a subset of \(\Lambda _\alpha (r_i)\), related to exact distances:
As it is easy to remark, the considered set of vertices is similar to \(\Lambda _\alpha (r_i)\): the only difference stands in the fact that not all edges are considered, but only the ones in \(E'\).
We refer to the vertices in \(\Lambda _\alpha (r_i)\) as possible reference vertices for \(r_i\), because the distances between \(r_i\) and any other vertex in \(\Lambda _\alpha (r_i)\) are known, and all these vertices precede \(r_i\) in the order. The reference vertices can be used for finding the candidate positions for the vertex \(r_i\). We refer to the corresponding distances as the reference distances. Instead, vertices in \(\Lambda _\beta (r_i)\) have the vertex \(r_i\) as a possible reference. Throughout this paper, we refer to a vertex using the usual letters \(u\) and \(v\), but also by the symbol \(r_i\), when a certain order \(r\) on \(V\) is supposed to be defined. Also, we may refer to the vertex with the symbol \(i\), by giving in this way more emphasis to its rank in the given order.
Among the vertices in \(\Lambda _{\alpha }(r_i)\), we will need to choose \(K\) of them as references for the vertex \(r_i\). We will denote the set of selected references as \(\hat{\Lambda }_{\alpha }(r_i)\), while \(S(\hat{\Lambda }_\alpha (r_i))\) will denote the convex hull of the vertices in \(\hat{\Lambda }_\alpha (r_i)\).
We give the following formal definition of a class of DGPs, containing interval data, that can be discretized.
Definition 2
The interval Discretizable DGP (\(i\hbox {DDGP}\)).
Given a simple weighted undirected graph \(G=(V,E,d)\) and a positive integer \(K\), we say that \(G\) represents an instance of the \(i\)DDGP if and only if there exists an order \(r\) on the vertices of \(V\) verifying the following conditions:
- (a):
-
\(G_C = (V_C,E_C) \equiv G[\{r_1,r_2,\dots ,r_K\}]\) is a clique and \(E_C \subset E'\);
- (b):
-
\(\forall i \in \{ K+1,\dots ,|r| \},\, \alpha (r_i) \ge K\) and \(\alpha _{ex} (r_i) \ge K - 1\);
- (c):
-
\(\forall i \in \{ K+1,\dots ,|r| \},\, \exists \hat{\Lambda }_\alpha (r_i)\) such that \(S(\hat{\Lambda }_\alpha (r_i))\) has positive volume.
Assumption (a) requires that the graph \(G_C\), induced from the first \(K\) vertices in the order \(r\), is a \(K\)-clique. This allows us to place the first \(K\) vertices into a unique position. By assumption (b), at least \(K\) reference vertices are available for all other vertices \(r_i\), and at least \(K-1\) out of the \(K\) vertices are related to an exact distance. For each exact distance, a sphere can be defined; for each interval distance, a spherical shell can be instead defined. By assumption (c), the convex hull of the \(K\) selected reference vertices \(S(\hat{\Lambda }_{\alpha }(r_i))\) has positive volume. Under these assumptions, the intersection of \(K\) (hyper)spheres in the \(K\)-dimensional space gives at most 2 points (see Lemma 2 in [10]), while the intersection of \(K-1\) spheres and 1 spherical shell (see assumption (b)) produces two (usually disjoint) curves in dimension \(K\). In the former case, we have two possible positions for the vertex, while, in the latter, a predefined number of points can be selected from the two curves. Thus, we always have a discrete (and finite) set of possible positions for the vertex \(r_i\).

The \(i\)BP algorithm can be employed for the solution of \(i\)DDGP instances. Algorithm 1 is a sketch of such an algorithm. In the algorithm call, \(j\) is the current vertex for which we are looking for candidate positions, \(r\) is the discretization order, \(d\) represents the set of weights on the edges of \(G\) (available distances), and \(D\) is the (predefined) number of points that are taken from intervals for allowing the discretization, when the intersection gives a continuous 1-dimensional object. A preliminary version of the \(i\)BP algorithm was published in [12]. This version was tailored to dimension \(K=3\) and to special orders \(r\) with repetitions where only the reference distance \((r_{i-3},r_i)\) was allowed to be represented by an interval.
The \(i\hbox {BP}\) algorithm recursively calls itself for exploring the whole discrete search domain (that is a tree, to which we refer as \(i\)BP tree). During each call, a certain number of candidate positions are computed for the current vertex, and the feasibility of each computed position is verified. Throughout this paper, the coordinates of the vertices in the embedding can be referred to by using the symbols \(\mathrm{x}_u,\, \mathrm{x}_{r_i}\) or \(\mathrm{x}_i\), depending on the context. When referring to the coordinates, we use the italic style. For example, in dimension 3, we write \(\mathrm{x}_i = (x_i,y_i,z_i)\). The same applies for the distances: we may use the symbols \(d(u,v)\), \(d_{u,v},\,d(r_i,r_j)\) or \(d_{ij}\), depending on the context.
Every time possible positions for a vertex are computed, their feasibility can be verified by applying the so-called pruning devices (see lines 10 and 13 of algorithm 1). When we verify the feasibility with respect to the constraints (1), we can employ the pruning device named direct distance feasibility (DDF) [11]. Let \(r_j \in V\) be the current vertex for which we have a new candidate position \(\mathrm{x}_j\). DDF consists in verifying whether the inequality
with \(\varepsilon > 0\), is satisfied for every distance between \(r_j\) and its predecessors \(r_i\) that are not used in the discretization. DDF is the simplest and probably most efficient pruning device to be integrated with the \(i\hbox {BP}\) algorithm. We remark that other pruning devices can be added to \(i\hbox {BP}\): the interested reader can refer, for example, to [8] for a discussion on energy-based pruning devices.
In order to apply the \(i\hbox {BP}\) algorithm, a discretization order for the vertices in \(V\) is needed. When such an order is not available, finding a discretization order becomes a fundamental preprocessing step for solving a DGP instance. Section 3 will be devoted to this problem, and we will present an efficient greedy algorithm for its solution.
Moreover, a procedure for the computation of the candidate vertex positions is necessary for the implementation of this algorithm (see line 8 of algorithm 1). As discussed above, these positions can be identified by intersecting (hyper)spheres in the \(K\)-dimensional space. However, as we will discuss in Sect. 4, the solution of the quadratic systems derived from the sphere intersections is not the most efficient way to compute the candidate vertex positions. A more numerically stable method will be instead presented.
3 Finding discretization orders
The problem of finding a discretization order \(r\) for \(G\) such that the assumptions in definition 2 are satisfied is formalized as follows. This problem appears in the literature with two different names: the “discretization vertex order problem” (DVOP) in [10, 17] and the “trilateration ordering problem” (TOP, see [4]). In the rest of the text, we will refer to this problem as the reordering problem.
Definition 3
Given a simple weighted undirected graph \(G=(V,E,d)\) and a positive integer \(K\), establish whether there exists an order \(r\) such that assumptions (a) and (b) of the \(i\)DDGP are satisfied. Orders satisfying (a) and (b) are referred to as “discretization orders” [17].
As it can be seen from the definition, we do not explicitly consider assumption (c) of the \(i\)DDGP. In fact, the subset of possibilities for which \(S(\hat{\Lambda }_\alpha (r_i))\) does not have a positive volume has Lebesgue measure equal to zero [10].
A necessary condition for finding a discretization order for the vertices in \(V\) is that each \(v \in V\) has at least \(K\) adjacent vertices. Additional condition, to be verified for the order \(r\) to be a discretization one, is that at least \(K\) adjacents of \(v\) precede \(v\) in the order \(r\) (so that they actually are reference vertices).
In [6, 12], some handcrafted orders have been proposed for the protein backbone and some side chains. These orders required the reference vertices to be consecutive. To make this possible, some vertices needed to be repeated along the order. However, the absence of the consecutivity assumption makes the conditions for these orders weaker, so that a wider class of DGPs can be discretized and solved by our discrete approach.
When all available distances are exact, the reordering problem (see definition 3) can be solved in polynomial time by employing the greedy algorithm proposed in [10]. For any initial \(K\)-clique \(C\) that can be selected in \(V\), the algorithm proceeds by choosing the next vertex \(r_i\) for which the value of \(\alpha (r_i)\) is maximized. If this maximum possible value for \(\alpha (r_i)\) is smaller than \(K\), then the initial clique \(C\) cannot lead to a discretization order. On the other side, if such an order exists (for a given choice of the initial clique), then the algorithm is able to identify it.

Algorithm 2 is a sketch of the greedy algorithm extended for interval data [17]. There are two main differences with respect to the version for exact distances only. The first one is on line 3 of the algorithm, where it is required that all distances in the initial clique \(C\) are exact. This way, it is ensured that the initial \(K\) vertices can be placed uniquely (modulo rotation and translation). The second difference is on line 8. Instead of considering all possible vertices \(u\) for which the value of \(\alpha (r_i)\) is maximized, only the ones for which \(\alpha _{ex} (r_i)\) is at least equal to \(K-1\) are considered. This filter allows us to generate (if they exist) orders where only one of the reference distances for the vertices can be represented by an interval, as required by assumption (b) (see definition 2).
We remark that, for \(K\) fixed, the complexity of algorithm 2 is given by the complexity for finding a \(K\)-clique (polynomial), plus the complexity of its greedy part, which is still polynomial [10].
4 Computing cartesian coordinates
In order to simplify the discussion (and since we are mostly interested in MDGPs), in this section our attention will be focused on the case \(K=3\). In dimension 3, the subproblem that needs to be solved at each iteration of the \(i\hbox {BP}\) algorithm is the one of finding the intersection of three Euclidean objects (see Sect. 2). When the three reference distances are all exact, the three Euclidean objects are three spheres, whose intersection gives at most 2 points. When one of the three distances is represented by an interval (see definition 2), the third sphere becomes a spherical shell, and the intersection generally provides two disjoint curves. In order to discretize, a certain subset of points can be chosen from the two curves. From a computational point of view, this can be implemented by choosing a certain subset of distances from the available interval, and by intersecting the three spheres several times, where only the third one changes its radius in the given interval. At each recursive call of \(i\hbox {BP}\) (see algorithm 1), therefore, the main subproblem is the one of finding the points given by one or several sphere intersections.
Suppose that we need to place the vertex \(i\). Let \(i',\, i''\) and \(i'''\) be the three reference vertices for \(i\). From the equations of the spheres in the three-dimensional space, we can deduce that the intersection points can be obtained by solving the following system of quadratic equations:
A method for finding solutions to the system (4) can be found in [5], where the solutions to the quadratic system are found by solving two linear systems. This method was implemented in conjunction with a branch and prune framework for problems with exact distances in [18], but it was remarked that it can lead to strong numerical instabilities. A strategy for controlling such errors was actually integrated with the method, where different sets of references vertices were chosen and the one leading to the smallest increase in the LDE value was considered (see Eq. 2). However, the considered method, integrated with this strategy, made the overall computational cost higher.
When the discretization order on \(V\) satisfies a stronger assumption, for which the reference vertices, for every vertex \(i\), are the ones that immediately precede \(i\) in the order (consecutivity assumption), then a simpler and more reliable procedure can be employed [11]. The idea is to replace the problem of intersecting the three spheres with the problem of finding the possible torsion angles of a “backbone” of vertices that are placed in sequence accordingly to the ordering on \(V\). When all reference distances are exact (intersection of 3 spheres), it can be proved that only two torsion angles can be defined for each quadruplet of consecutive vertices. Two torsion angles correspond to two possible positions for the last vertex (in the order \(r\)) of the quadruplet.
In the following, we will use the symbol \(\theta _i\) for referring to the angle formed by the two segments \((i,i')\) and \((i',i'')\). When dealing with molecules, this angle is also referred to as bond angle. Moreover, we will use the symbol \(\omega _i\) for referring to the angle formed by the two planes defined by the two triplets \((i''',i'',i')\) and \((i'',i',i)\). Similarly, when the focus is on molecules, this angle is generally named torsion angle. We warn the reader that, even if we will use the terms “bond angle” and “torsion angle” in the following, as in molecular biology, the angles that are here considered may not be equivalent to the ones generally defined when studying biological molecules.
In order to avoid considering solutions that can be obtained by translations and rotations, we can fix the three vertices that belong to the initial clique. For instance, the first vertex can be placed in the origin of the Cartesian system of coordinates, the second one can lie on the negative \(x\)-axis, and the third vertex can be placed on the \(1\)st (or \(2\)nd) quadrant of the \(xy\)-plane [20].
If we consider consecutive reference vertices, so that \(i''' = 1\), \(i'' = 2\) and \(i' = 3\), in order to position the \(4\)th vertex, we need to compute the torsion angle \(\omega _{4}\) defined by the first four vertices. As described in [11], the cosine of \(\omega _{4}\) can be computed from the distances between pairs of vertices in the quadruplet, by employing the cosine law. The cosine of the torsion angle, together with distances and angles between segments, can be used for identifying the two positions given by the sphere intersections. Vertex positions can be computed by multiplying \(4 \times 4\) matrices defined by
whose elements depend on distances, and bond and torsion angles related to the vertices \(i, i-1, i-2, i-3\). If we consider the consecutivity assumption [11], then the Cartesian coordinates of the candidate positions, for each \(i\), can be obtained by:
This procedure is very efficient and works well in the practice [11, 12]. It requires, however, the consecutivity assumption for the reference vertices. In previous publications, therefore, orders on \(V\) satisfying this assumption were necessary. Instead, orders that do not satisfy this assumption can be obtained automatically in polynomial time (see Sect. 3): we will therefore propose a generalization of the procedure defined by Eqs. (5) and (6) that allows us to generate accurate Cartesian coordinates, while orders satisfying weaker assumptions can be employed.
Our proposition is to avoid to accumulate matrices every time we step from a vertex to another (see Eq. 6), but rather to define only one matrix, that we will call \(U_{i'}\), which is able to convert directly vertex positions from the coordinate system defined in \(i'\) to the canonical system. This conversion is possible independently from the fact the reference vertices are consecutive or not. The idea is to construct the matrix \(U_{i'}\) by using the unit vectors defining the three axes of a coordinate system defined in \(i'\).
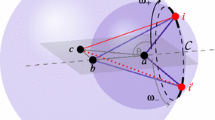
Recall that, during the execution of the \(i\hbox {BP}\) algorithm, the Cartesian coordinates for the reference vertices \(i',\, i''\) and \(i'''\) are already available along the current branch of the \(i\hbox {BP}\) tree. Let \(v_1\) be the vector from \(i''\) to \(i'\). Equivalently, let \(v_2\) be the vector from \(i''\) to \(i'''\). In order to define the matrix \(U_{i'}\), we consider the following coordinate system. The \(x\)-axis can be defined by \(v_1\): we will denote the unit vector defining this axis with the symbol \(\hat{x}\). Moreover, the vectorial product \(v_1 \times v_2\) gives as a result another vector that defines the \(z\)-axis: let us denote the corresponding unit vector with \(\hat{z}\). Finally, the vectorial product \(\hat{x} \times \hat{z}\) provides the vector that defines the \(y\)-axis (let the unit vector be \(\hat{y}\)). Figure 1 shows the coordinate system defined by the three unit vectors \((\hat{x},\hat{y},\hat{z})\). The three obtained vectors \(\hat{x}\), \(\hat{y}\) and \(\hat{z}\) can be used for defining the columns of the matrix \(U_{i'}\), which is a unitary rotation matrix. Once \(U_{i'}\) has been computed, the Cartesian coordinates for a candidate position for the vertex \(i\) can be obtained by:
Notice that Eq. (7) is equivalent to Eq. (6), in the sense that they both depend on bond angles and torsion angles, and they both provide the same result. However, there is no need to accumulate products of matrices in Eq. (7), and the consecutivity assumption is not necessary. The angles \(\theta _{i}\) and \(\omega _{i}\) can be computed using the cosine law and exploiting the available distances \(d_{i',i},\, d_{i'',i}\) and \(d_{i''',i}\), as well as the positions of the previous vertices \(i',\, i''\) and \(i'''\) (see Fig. 1).

The reference vertices \(i''',\, i''\) and \(i'\) induce a system of coordinates. The angles \(\theta _{i}\) and \(\omega _{i}\) are the spherical coordinates of \(i\) for the system centered in \(i'\).
There is an axial symmetry around the \(x\)-axis which defines a circle of possible positions for the vertex \(i\). If we suppose that the distance \(d_{i''',i}\) is exact, only two possible positions (\(i_{+}\) and \(i_{-}\) in the figure), corresponding to the torsion angles \(\omega _{+}\) and \(\omega _{-}\), can be computed for the vertex \(i\). These positions are symmetric with respect to the plane defined by \(i''',\, i''\) and \(i'\). If \(d_{i''',i}\) is instead an interval, then two symmetric arcs can be identified on the circle in Fig. 1, where a predefined number of sample points can be selected (see algorithm 1 in Sect. 2).
5 Computational experiments
This section presents some computational experiments where the considered instances are based on the discretization orders found by algorithm 2, and the \(i\hbox {BP}\) algorithm (see algorithm 1) is integrated with the procedure detailed in Sect. 4 for the computation of the Cartesian coordinates of the vertices. All codes were written in C programming language and all the experiments were carried out on an Intel Core 2 Duo \(@\) 2.4 GHz with 2GB RAM, running Mac OS X. The codes have been compiled by the GNU C compiler v.4.0.1 with the -O3 flag.
The purpose of our first set of experiments is to compare the accuracy of the vertex coordinates obtained by applying the procedure in Sect. 4 to the one that was previously employed, which is based on the solution of \(4 \times 4\) quadratic systems (see [18]). In this set of experiments, we consider instances generated from proteins having known conformation, which can be downloaded from the protein data bank (PDB) [2]. Each PDB record consists in a set of atomic coordinates for a given protein: we generate instances by computing the distances between all the possible pairs of hydrogens in the molecule, and by keeping only the ones smaller than a predefined threshold \(\delta \). In this way, we simulate data obtained from NMR experiments, because all available distances are generally between hydrogens, and only short-range distances can be detected. The threshold \(\delta \) usually ranges between 5Å and 6Å: we set \(\delta = 5.5\)Å, because, with this value, the discretization assumptions (see definition 2) do not hold if the hydrogen atoms are ordered as in the PDB files. In this case, reordering the atoms of the molecule becomes a necessary preprocessing for applying the \(i\hbox {BP}\) algorithm. The same set of instances was considered in the experiments presented in [10]. We point out that all distances are here considered as exact. This allows us to analyze the accuracy of the two compared procedures.
Table 1 shows some computational experiments. The name of the instances correspond to the label of the protein conformation on the PDB. Moreover, the size \(n\) of the instance, in terms of number of vertices (hydrogens), and the cardinality \(|E|\) of the edge set (i.e. the total number of available distances) are given. Experiments are performed by using the \(i\hbox {BP}\) algorithm integrated with the new procedure detailed in Sect. 4 (see “New procedure” in the table), as well as with \(i\hbox {BP}\) integrated with the procedure presented in [18] and based on the solution of quadratic systems (recall that, because of the error propagation, a strategy is considered for keeping such errors as low as possible). In both cases, we evaluate the experiments through the following three indices: #S is the total number of solutions found by \(i\hbox {BP}\), LDE evaluates the quality of the best obtained solution \(X\) (see Eq. (2), where the lower bounds \(\underline{d}\) and the upper bounds \(\bar{d}\) coincide in this case), and finally we monitor the CPU time, in seconds. In all these experiments, the tolerance \(\varepsilon \) in the DDF pruning device (see Eq. 3) is set to \(10^{-3}\).
As Table 1 shows, the \(i\hbox {BP}\) algorithm, integrated with the new presented procedure, outperforms the older version on all instances. The LDE values of the best found solutions are better (smaller) or compatible to the ones found when solving the quadratic systems, while the executions are always faster when using the new procedure.
In our second set of experiments, we consider fragments of proteins consisting of short sequences of amino acids, where their atoms are automatically sorted by algorithm 2. Differently from the previous experiments, all the atom kinds are here considered, and interval data are included in the instance. We use the following procedure for generating the instances. From some PDB files that were also considered in Table 1, we extract a random protein fragment consisting of a predefined number of consecutive amino acids. For each fragment, we compute all the distances between pairs of atoms. In order to simulate realistic instances, we consider as exact all distances smaller than a certain threshold \(\ell _b\): we suppose that all such distances are related to chemical bonds. Moreover, all distances between two atoms that are detected as bonded to a common atom are also considered as exact (they form the so-called bond angles). Finally, distances between atoms separated by 2 bonded atoms, as well as distances between pairs of hydrogen atoms (which fall below the threshold \(\delta \)), are considered as intervals. From the computed exact distance, an interval is generated by applying a random perturbation (having maximal amplitude 2.5Å) to the exact distance. The subset of considered exact and interval distances form an instance of the DGP with interval data. Even if we are aware that this procedure could fail in identifying atomic bonds correctly (because it only makes use of a distance threshold), we found its use convenient for an easy generation of instances that are relatively close to real NMR instances. None of these generated instances satisfies the discretization assumptions (see definition 2) when the order given to the atoms is the one we found in the PDB files. Therefore, the greedy algorithm sketched in algorithm 2 is used for reordering the atoms of the instances in a way that they can be discretized. This preprocessing step took <0.10 s for each instance.
Table 2 shows some computational experiments with the second set of instances. When generating such instances, we set \(\ell _b = 2.5\)Å and \(\delta = 5.0\)Å. From every protein, we select a certain number \(n_{aa}\) of consecutive amino acids (the fragment) and we apply the procedure above for the generation of the instance. Additionally to the information reported in Table 1, we also report the number \(D\) of discretization points to be taken from the curves (see Sect. 2) in order to allow the discretization in presence of interval data. The reported \(D\) value is actually the smallest one that allows us to find at least one solution for the instance. The \(i\hbox {BP}\) algorithm is employed for finding only one solution, which is the leftmost solution in the \(i\hbox {BP}\) tree. The LDE value (see Eq. 2) in this solution is used for evaluating the performances of the algorithm. The tolerance \(\varepsilon \) in (3) is here set to \(10^{-1}\) in all experiments. Each experiment took <1 CPU time second.
As the table shows, the \(i\hbox {BP}\) algorithm is able to solve instances formed by up to about 500 atoms. For every instance, the obtained LDE value for the first found solution is always about \(10^{-4}\), which implies that all distance constraints are satisfied with, at most, a relatively small violation.
The considered instances consist of backbone atoms, as well as side chains atoms, of a protein [15]. Differently from previous works (see for example [12]), we did not define any a priori order for the backbone and the side chains, but we rather automatically found a discretization order for the whole instance. This implies a wider applicability of this approach to any kind of instance. Moreover, we can also claim that our orders are more adapted to the instances at hand, because a different ad-hoc order can be identified for each instance, even when considering instances belonging to the same class (such as the MDGP).
Finally, we remark that, after having computed the first solution, the \(i\hbox {BP}\) algorithm could continue searching the tree, and find other solutions. However, this possibility was not considered in this work, because some issues related to the way we currently discretize the interval distances must be better investigated.
6 Conclusions
We considered two important aspects for the discretization of DGPs. First, instances of the DGP can be discretized only if its vertices are sorted in a way for which the discretization assumptions are satisfied. Second, the core of the \(i\hbox {BP}\) algorithm, which we employ for the solution of discretizable DGP instances, is the computation of candidate positions for the vertices of the graph. This paper discusses an algorithm for the automatic reordering of DGP instances so that they can be discretized, and proposes a new procedure for the computation of vertex candidate positions. Presented computational experiments show the effectiveness and efficiency of the new proposed approaches.
This paper represents another little but yet important step for the solution of real-life DGPs by a discrete approach. As mentioned in the Introduction, one of the most studied classes of DGPs is the MDGP, where experimental data about molecules are obtained by NMR experiments, and the possible conformations for such molecules are searched. For the first time, we are able to handle protein instances containing side chains where an order for their atoms is not predefined, but it is rather automatically identified and adapted to the instance at hand.
The final aim is however solving, by employing the \(i\hbox {BP}\) algorithm, MDGP instances where real NMR data are contained. One big obstacle to an efficient solution of such instances is the discretization in presence of interval data. The current strategy consists in choosing a discrete subset of representative distances from each interval used in the discretization process. When the entire interval is composed by embeddable distances, each representative distance can lead to a feasible solution, which may imply the identification of many “too similar” solutions. On the other hand, when only a small part of the interval contains embeddable distances, the number of chosen samples is crucial, because at least one of them should lead to an embedding.
References
Alipanahi, B., Krislock, N., Ghodsi, A., Wolkowicz, H., Donaldson, L., Li, M.: Determining protein structures from noesy distance constraints by semidefinite programming. J. Comput. Biol. 20(4), 296–310 (2013)
Berman, H., Westbrook, J., Feng, Z., Gilliland, G., Bhat, T., Weissig, H., Shindyalov, I., Bourne, P.: The protein data bank. Nucleic Acids Res. 28, 235–242 (2000)
Biswas, P., Lian, T., Wang, T., Ye, Y.: Semidefinite programming based algorithms for sensor network localization. ACM Trans. Sen. Netw. 2, 188–220 (2006)
Cassioli, A., Günlük, O., Lavor, C., Liberti, L.: Discretization vertex orders in distance geometry, working paper (2014)
Coope, I.D.: Reliable computation of the points of intersection of \(n\) spheres in \(n\)-space. ANZIAM J. 42, 461–477 (2000)
Costa, V., Mucherino, A., Lavor, C., Carvalho, L.M., Maculan, N.: On suitable orders for discretizing molecular distance geometry problems related to protein side chains. In: IEEE Conference Proceedings, pp. 397–402. Workshop on Computational Optimization (WCO12), FedCSIS12, Wroclaw, Poland (2012)
Crippen, G.M., Havel, T.F.: Distance Geometry and Molecular Conformation. John Wiley & Sons, New York (1988)
Gonçalves, D., Mucherino, A., Lavor, C.: Energy-based pruning devices for the bp algorithm for distance geometry. In: IEEE Conference Proceedings, pp. 335–340. Workshop on Computational Optimization (WCO13), FedCSIS13, Krakow, Poland (2013)
Grosso, A., Locatelli, M., Schoen, F.: Solving molecular distance geometry problems by global optimization algorithms. Comput. Optim. Appl. 43(1), 23–37 (2009)
Lavor, C., Lee, J., John, A.L.S., Liberti, L., Mucherino, A., Sviridenko, M.: Discretization orders for distance geometry problems. Optim. Lett. 6(4), 783–796 (2012)
Lavor, C., Liberti, L., Maculan, N., Mucherino, A.: The discretizable molecular distance geometry problem. Comput. Optim. Appl. 52, 115–146 (2012)
Lavor, C., Liberti, L., Mucherino, A.: The interval branch-and-prune algorithm for the discretizable molecular distance geometry problem with inexact distances. J. Glob. Optim. 56(3), 855–871 (2013)
Liberti, L., Lavor, C., Maculan, N., Mucherino, A.: Euclidean distance geometry and applications. SIAM Review 56(1), to appear (2014)
Linge, J.P., Nilges, M.: Influence of non-bonded parameters on the quality of nmr structures: a new force field for nmr structure calculation. J. Biomol. NMR 13(1), 51–59 (1999)
Malliavin, T.E., Mucherino, A., Nilges, M.: Distance geometry in structural biology: new perspectives. In [19] pp. 329–350 (2013)
Moré, J.J., Wu, Z.: Distance geometry optimization for protein structures. J. Glob. Optim. 15, 219–223 (1999)
Mucherino, A.: On the identification of discretization orders for distance geometry with intervals. In: Proceedings of Geometric Science of Information (GSI13). Lecture Notes in Computer Science 8085, pp. 231–238. France, Paris (2013)
Mucherino, A., Lavor, C., Liberti, L.: The discretizable distance geometry problem. Optim. Lett. 6(8), 1671–1686 (2012)
Mucherino, A., Lavor, C., Liberti, L., Maculan, N. (eds.): Distance Geometry: Theory, Methods and Applications, p 410. Springer, New York (2013)
Thompson, H.B.: Calculation of cartesian coordinates and their derivatives from internal molecular coordinates. J. Chem. Phys. 47, 3407–3410 (1967)
Acknowledgments
We wish to thank Carlile Lavor and Leo Liberti for the fruitful comments to this paper. We are also thankful to Brittany Region (France) for financial support.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Gonçalves, D.S., Mucherino, A. Discretization orders and efficient computation of cartesian coordinates for distance geometry. Optim Lett 8, 2111–2125 (2014). https://doi.org/10.1007/s11590-014-0724-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11590-014-0724-z