
Abstract
The Eh-pH diagrams for Ni-Co-Mn-CO3-H2O system at various temperatures and ion concentrations are simulated via OLI studio based on the fundamentals of thermodynamic equilibrium. A co-existence area for NiCO3, CoCO3, and MnCO3 is observed visually from the Eh-pH diagrams, which thermodynamically proves the stability of these species in aqueous solutions, and the possibility of co-precipitating polymetallic carbonate. The simulation results also demonstrate that a higher temperature and/or a more dilute solution are not in favor of the co-precipitation. With the predicted pH ranges from the Eh-pH diagrams, a confirmative experiment was conducted to synthesize Ni0.13Co0.13Mn0.54(CO3)0.8, the precursor for preparing Li1.2Ni0.13Co0.13Mn0.54O2, which is a promising cathode material for next-generation LIBs. The physical properties of both materials are characterized in detail, and the electrochemical performance for the final cathode material was tested. The results show that Ni2+, Co2+, and Mn2+ ions in solution are homogeneously co-precipitated in the form of polymetallic carbonate. The Li1.2Ni0.13Co0.13Mn0.54O2 material obtained from the carbonate precursor has a typical structure of Li- and Mn-rich cathodes and yields an initial discharge capacity of 296.0 mAh g−1 at 0.1 C and 188.1 mAh g−1 after 100 cycles at 1 C rate. It was verified that the OLI-assisted Eh-pH simulation is consistent with the experimental measurements.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Rechargeable lithium-ion batteries (LIBs) have been considered as one of the most reliable paths for energy storage for a long time under today’s severe global energy and environmental concerns for greenhouse gases [1,2,3]. In the past few years, LIBs have seen a leap-frog development in both research and applications [4,5,6], as one of the promising energy storage devices in electric and hybrid electric vehicles (EVs and HEVs). These developments have also promoted the utilization of high-value metals such as Ni, Co, and Mn. Even so, lowering the cost of production and increasing the energy density of LIBs remain the most important metrics of success [7, 8]. Among the popular cathode materials (LiCoO2, LiMn2O4, LiFePO4, and LiNixCoyMn1-x-yO2), the lithium-rich and manganese-based layered compounds seem to provide an ideal solution because of the relatively lower production cost and high discharge capacity (> 250 mAh g−1) [9, 10]. This category of materials is often described by the chemical formula of xLi2MnO3·(1–x)LiMeO2 (0 < x < 1, Me = Ni, Co, Mn, etc.), where the composite oxide Li1.2Ni0.13Co0.13Mn0.54O2 has been largely studied and proved to possess excellent comprehensive electrochemical performance [11,12,13].
Among the various synthesis methods for Li- and Mn-rich cathode materials, such as the Pechini [14], the Sol-gel [15, 16], the solvothermal/hydrothermal method [17, 18], and the sacrificial template [19], co-precipitation coupled with a follow-up calcination with a Li source is the most widely used synthesis method to prepare these materials [20, 21]. Co-precipitating three or more metal ions uniformly is a challenging process. Conditions, including but not limited to, temperature, pH, ion concentration, etc., have a significant impact on the micro-morphology, crystallinity, and structure of the resulting precursor material, which in turn affects the physical, chemical, and electrochemical properties of the final cathode materials [22, 23]. Until now, many researchers have discussed several key parameters during the precursor synthesis, such as ligand selection [20], nucleation and growth mechanisms [22], and feeding ways [24]. For example, Xiang et al. [9] performed a log[M]-pH calculation based on equilibrium reactions to predict the residual transition metal concentrations in ammonium bicarbonate solutions. Nevertheless, there still exists a certain discrepancy because the equilibrium constants are all for 25 °C but the actual reactions are taking place at 55 °C. Our previous work [23] reported on a thermodynamic analysis for the polymetallic hydroxide co-precipitation process which provided guidance for the synthesis of LiNi0.5Co0.2Mn0.3O2 (NCM 523) hydroxide precursor, where the thermodynamic data at elevated temperatures were computed. However, many calculations, especially those of the heat capacity and activity coefficient, had to be simplified due to their complexity in manual calculation. Therefore, although progress has been achieved on the thermodynamics of co-precipitation processes for LIBs [25,26,27], there is still room for improvements and a more comprehensive analysis of the electrochemical equilibria of the process, which is different from the hydroxide system, and could have important significance in material synthesis.
In this work, polymetallic carbonate precipitation was studied both theoretically and experimentally. A series of Eh-pH diagrams related to the co-precipitation of Ni-Co-Mn carbonate were constructed based on fundamental reaction equilibria and simulated by the OLI AQ framework. Different temperatures and ion concentrations were tested in the simulation to identify compositional variation tendencies in the co-precipitating process. An experimental verification was also attempted by synthesizing a Ni0.13Co0.13Mn0.54(CO3)0.8 precursor and in turn Li1.2Ni0.13Co0.13Mn0.54O2 cathode materials. The accuracy and the reliability of the constructed Eh-pH diagrams were also assessed.
Theories and experiments
Thermodynamic calculation
All reduction half-reactions occurring in the Me-CO3-H2O (Me = Ni, Co, Mn) system is typically expressed by Eq. (1):
The Gibbs free energy change (ΔGT) for the above reaction at temperature T is given by Eq. (2):
where \( {\varDelta}_r{G}_T^{\varTheta } \) is the standard Gibbs free energy of reaction at temperature T, in kJ·mol−1. R is the universal gas constant, J·mol−1·K−1; T is the absolute temperature, K; and α is the activity of species in Eq. (1).
At elevated temperatures, the standard Gibbs free energy of a reaction is given by the following equation [23]:
where \( {\varDelta}_r{G}_{298}^{\varTheta } \) and \( {\varDelta}_r{S}_{298}^{\varTheta } \) are the standard Gibbs free energy and standard entropy of reaction at 298 K; \( \varDelta {C}_p^{\varTheta } \) is the average heat capacity change of Eq. (1) from 298 to T K, in J·K−1. In Eq. (3), the heat capacity change is not taken as temperature dependent, instead an average value over the temperature ranging from 298 to T K is used.
Construction of Eh-pH diagrams was performed using the aqueous (AQ) model in OLI Studio Stream Analyzer v.9.6.3 (OLI Systems, Inc., USA). The AQ framework is using an electrolyte activity coefficient model that predicts the properties of electrolytes, in which the mean activity coefficient, 훾±, is computed using the Bromley-Zemaitis activity model [28, 29]. This expression is a combination of the Debye-Hückel long-range electrostatic interactions and a semi-empirical expression for short-range interactions between cations and anions [30]. The generalized form of the implemented activity coefficient is
where A is the Debye-Hückel coefficient; z is the ion charge number; I is the ionic strength; B, C, and D are empirical, temperature-dependent cation-anion interaction parameters.
At elevated temperatures, the revised Helgeson Kirkham Flowers method (HKF model) [31, 32] is applied to essentially calculate a more accurate \( \varDelta {C}_p^{\varTheta } \) value from statistical thermodynamics.
Materials synthesis and characterization
The carbonate precursor Ni0.13Co0.13Mn0.54(CO3)0.8 (denoted here as NCM-P) was synthesized by a co-precipitation approach. Firstly, a certain amount (Ni:Co:Mn = 1:1:4.15 in molar ratio) of NiSO4·6H2O, MnSO4·H2O, and CoSO4·7H2O (Xilong Scientific Co., Ltd., China) was dissolved in de-ionized water to form a 1 mol L−1 mixed solution and pumped into a batch reactor with vigorous stirring at 600 rpm. Meanwhile, a 1 mol L−1 Na2CO3 (Sinopharm Chemical Reagent Co., Ltd., China) solution used as the precipitator was also fed into the reactor. The reactor was heated in a water bath, and the temperature was maintained at 55 °C. The co-precipitation process was operated under atmospheric pressure with no need of inert-gas protection. The pH was monitored and adjusted at 7.2 ± 0.2 using the 1 mol L−1 Na2CO3 solution. After 10 h of aging, spherical carbonate precipitates were filtered, washed with de-ionized water, and dried in an air-dry oven overnight at 105 °C. The Li1.2Ni0.13Co0.13Mn0.54O2 cathode material (abbreviated as LNCMO) was prepared by the high-temperature solid-state method [33]. The obtained Ni0.13Co0.13Mn0.54(CO3)0.8 precursor was weighed and mixed with powder Li2CO3 (Ganfeng Lithium Co., Ltd., China) at a Li/Me (Me = Ni + Co + Mn) molar ratio of 1.5. The raw materials were mixed in a material mixer (M10, Beijing Grinder Instrument Co., Ltd., China) at the speed of 60 rpm for 1 h. After that, the mixture was calcined in a muffle furnace at 500 °C for 6 h and then at 850 °C for 12 h in air.
X-ray diffraction (XRD) analysis was performed to identify the crystal structures of the synthesized materials via a Rigaku TTR-III diffractometer using a Cu Kα radiation. The XRD data were collected in the 2θ range of 10–80° with a scanning rate of 2° min−1. And the Rietveld refinement was conducted via the GSAS/EXPGUI program to obtain the cell parameters. The particle morphology was observed, and the surface elemental distribution was measured by scanning electron microscopy equipped with energy dispersive spectroscopy (SEM/EDS; JSM-6360LV, JOEL). Elemental analysis of the as-prepared material was determined by inductively coupled plasma atomic emission spectrophotometry (ICP-AES; ICAP-7400, Thermo Fisher Scientific). Solutions to be measured were prepared by dissolving 0.1 g of the precursor or as-prepared material in 5 mL of 1 mol L−1 HNO3 under stirring for 30 min at room temperature, and then diluting 1000 times with DI-water. Regarding the electrochemical testing, the prepared Li1.2Ni0.13Co0.13Mn0.54O2 cathode material was fabricated to CR2016 type coin cells, whose detailed assembly procedures were similar with our previous reports [34, 35]. After being activated at 0.1, 0.2, and 0.5 C (1 C = 250 mA g−1) for one cycle each, the cycling test was conducted on a LAND test system (CT2001A, Wuhan LAND Electronics Co., Ltd.) over 2.0–4.6 V (vs. Li/Li+) at 1 C rate and at room temperature.
Results and discussions
Eh-pH diagrams for the Ni-Co-Mn-CO3-H2O system
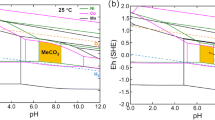
To identify each stable phase, separate Eh-pH diagrams for Ni-CO3-H2O, Co-CO3-H2O, and Mn-CO3-H2O systems were prepared by OLI as shown in Fig. 1. The temperature was set to 25 °C and the molalities of Ni, Co, and Mn to 0.1 mol kg−1, respectively. The pH in the simulation was adjusted by using H2SO4 and Na2CO3 as titrants. The divalent Me2+ ions transform into different species at different pH and Eh (vs. standard hydrogen electrode, SHE) values (Fig. 1). The predominant areas of carbonates for all Ni, Co, and Mn were identified, indicating that it is possible for the three metal ions to be co-precipitated as a complex carbonate at a certain overlapping pH and Eh ranges. According to these diagrams, Ni2+ ions start to precipitate as NiCO3 at pH 6.48 and end at 8.17; the corresponding Eh range is from − 0.376 to 0.600 V. For Co2+, the pH and Eh windows for CoCO3 precipitation are 5.22–10.52 and from − 0.526 to 0.581 V, respectively. Similarly, MnCO3 is stable between pH from 5.12 to 12.00 and Eh from − 1.445 to 0.692 V.

Eh-pH diagrams for a Ni-CO3-H2O, b Co-CO3-H2O, and c Mn-CO3-H2O systems at 25 °C and [Me2+] = 0.1 mol kg−1
In order to verify the specific region for co-precipitating Ni2+, Co2+, and Mn2+ ions as a complex carbonate, the Eh-pH diagram for the Ni-Co-Mn-CO3-H2O system is presented in Fig. 2a by overlapping the three single Me-CO3-H2O systems. It is clear that the co-precipitation area is mainly controlled by the formation of NiCO3 since its stable region is relatively smaller than those of Co and Mn. Therefore, the most suitable pH and Eh ranges for co-precipitating all three metal ions are from 6.48 to 8.17 and from − 0.376 to 0.503 V, respectively. When the temperature rises to 55 °C and the molalities of metal ions remain the same, the Eh-pH diagram converts to Fig. 2b, where the stable area for MeCO3 significantly shrinks to pH from 6.24 to 7.10 and Eh from − 0.341 to 0.411 V. On the other hand, the region of Me(OH)2 stability is enlarged by the temperature increase. These observations demonstrate that in theory Ni2+, Co2+, and Mn2+ ions can be co-precipitated by adjusting the pH and Eh values within appropriate ranges. Furthermore, the co-precipitation process of MeCO3 is negatively affected at higher temperatures.

Eh-pH diagrams for Ni-Co-Mn-CO3-H2O system at a 25 °C and [Me2+] = 0.1 mol kg−1; b 55 °C and [Me2+] = 0.1 mol kg−1
Simulation results for lower ion concentrations are presented in Fig. 3. The influence of concentration on the predominance area of MeCO3 was evaluated by setting the molality for each of Ni, Co, and Mn to 0.01 mol kg−1. The effect of ion concentration decrease by a factor of 10 shows that the MeCO3 region in Fig. 3a is dramatically reduced when compared with that in Fig. 2a under the same temperature. At elevated temperatures, as shown in Fig. 3b, the predominance areas of Ni(OH)2, Co(OH)2, and Mn(OH)2 further expand due to the chemical equilibrium shift and the stable region of NiCO3 ceases to exist. As expected in precipitation processes, the results illustrate that lowering the ion concentration is also unfavorable to the co-precipitation process of MeCO3.

Eh-pH diagrams for Ni-Co-Mn-CO3-H2O system at a 25 °C and [Me2+] = 0.01 mol kg−1; b 55 °C and [Me2+] = 0.01 mol kg−1
On the basis of the above discussion, the ion molalities of Ni2+, Co2+, and Mn2+ in the simulation were set as 0.13, 0.13, and 0.54 mol kg−1, respectively, in order to be consistent with the stoichiometric ratio of Ni0.13Co0.13Mn0.54(CO3)0.8, which is the precursor for Li1.2Ni0.13Co0.13Mn0.54O2 cathode material. The calculated Eh-pH diagrams are depicted in Fig. 4, where similar outcomes are observed. These results were then used for an experimental verification described below in the next section.

Eh-pH simulations for the synthesis of Ni0.13Co0.13Mn0.54(CO3)0.8 at a 25 °C and b 55 °C (where [Ni2+] = 0.13 mol kg−1, [Co2+] = 0.13 mol kg−1, and [Mn2+] = 0.54 mol kg−1)
Experimental verification
Given that the stable region of MeCO3 is contracted as temperature goes up, whereas elevated temperature are beneficial for the kinetics of the precipitation process and the crystallography of the final precursor, and that the pH range of 6.26–7.48 shown in Fig. 4b is large enough for experimental control and monitor, it was selected that the trial experimental conditions of temperature and pH were 55 °C and 7.2 ± 0.2, respectively. Consequently, NCM-P and LNCMO were synthesized following the experimental procedures described previously in the experimental section. X-ray diffraction (XRD) was carried out to confirm the crystal structures for both NCM-P and LNCMO. As depicted in Fig. 5a, the as-synthesized precursor (NCM-P) conforms well to the hexagonal structure of the MnCO3 phase with the R-3c space group (JCPDS #86-0173) [22]. The sharp peaks also indicate that the precursors are mainly crystalline, which is in agreement with the SEM images shown in Fig. 6. In addition, there are no impurities observed in the XRD pattern, demonstrating that all the transition metal ions are co-precipitated into the mixed carbonate phase, without forming oxides or hydroxides. Moreover, the molar ratio of Ni:Co:Mn in the precursor was found to be 0.130:0.132:0.539 via ICP-AES, which is very close to the desired values of 0.13:0.13:0.54. The above further confirms that the transition metal ions in the solution were stoichiometrically co-precipitated. These evidences reveal that the simulated Eh-pH diagrams are consistent with our experiments. Figure 5b shows the XRD patterns for the final Li1.2Ni0.13Co0.13Mn0.54O2 powder (LNCMO) after calcination with Li2CO3. All main diffraction patterns are indexed to the hexagonal α-NaFeO2 structure with R-3m space group (JCPDS #74-0919), except for a series of small and weak peaks around 21–23°, which belong to the Li2MnO3-like phase (C2/m space group, JCPDS #73-0152) [15, 36]. Meanwhile, the clear splitting of both (006)/(102) and (108)/(110) doublets also indicates the highly ordered layered structure for LNCMO [34, 37]. The detailed lattice parameters from Rietveld refinement were listed in Table S1, where the relatively low R factor confirmed the reliability of the refinement results. Moreover, the molar ratio of Li:Ni:Co:Mn in the final cathode material was examined to be 1.364:0.146:0.148:0.617 by ICP-AES, which is close to the theoretical values of 1.2:0.13:0.13:0.54.

XRD patterns for the synthesized a Ni0.13Co0.13Mn0.54(CO3)0.8 precursor and b Li1.2Ni0.13Co0.13Mn0.54O2 cathode material

a SEM images and b EDS mapping for the synthesized Ni0.13Co0.13Mn0.54(CO3)0.8 precursor
Figure 6a shows the SEM images of the prepared Ni0.13Co0.13Mn0.54(CO3)0.8 precursor, which has a quasi-spherical shape with a diameter of 8 to 12 μm. At larger magnification, a porous and rough morphology can be seen at the surface of the precursor. EDS mapping of the obtained precursor is also shown in Fig. 6b, where the distributions of Ni, Co, and Mn on the particle surface are seen as homogeneous. This clearly demonstrates that the Ni2+, Co2+, and Mn2+ ions in the mixed solution were uniformly co-precipitated into the carbonate precursor.
To further investigate the electrochemical performances of the synthesized LNCMO sample, a cyclic test was performed from 2.0 to 4.6 V at 1 C (current density of 250 mA g−1). Before that, the cells were activated by charging and discharging for one cycle at 0.1, 0.3, and 0.5 C, respectively, over the same voltage range. The initial charge/discharge curves for LNCMO are portrayed in Fig. 7a, where the charge curve consists of a positive slope region below 4.5 V and a relatively long plateau at ~ 4.5 V. The positive slope region is assigned to the extraction of Li+ ions from the layered structure due to the Ni2+/Ni4+ and Co3+/Co4+ reaction [38]. The long plateau is attributed to the removal of Li2O from the Li2MnO3 structure [22, 39, 40]. This shows typical electrochemical behavior of a Li- and Mn-rich composite material [12, 15]. As shown in Fig. 7a, the LNCMO sample delivers a first-discharge capacity of 296.0 mAh g−1 at 0.1 C, which is reasonable for being less than the calculated theoretical discharge capacity (314 mAh g−1) of Li1.2Ni0.13Co0.13Mn0.54O2 [41]. Such a high capacity at 4.6 V could be attributed to the reversible oxygen redox chemistry (O2−/O−) or/and the charge compensation of high-valent ions, according to the reports [42,43,44,45]. Regarding the cyclic performance (Fig. 7b), the LNCMO sample yields 188.1 mAh g−1 after 100 cycles at 1 C rate, corresponding to a retention rate of 84.5%. Moreover, the average voltage changes as a function of cycle numbers was illustrated in Fig. S2, where the average voltage fades from 3.465 to 3.092 V after 100 cycles. It further demonstrates that the Li1.2Ni0.13Co0.13Mn0.54O2 cathode material was successfully synthesized with the guidance of Eh-pH diagrams.

a Initial charge/discharge profile at 0.1 C between 2.0 and 4.6 V; b cyclic performance of the prepared Li1.2Ni0.13Co0.13Mn0.54O2 cathode material at 1 C between 2.0 and 4.6 V
Conclusions
Eh-pH diagrams for the Ni-Co-Mn-CO3-H2O system at two temperatures (25 and 55 °C) and molalities (0.01 and 0.1 mol kg−1) were constructed using OLI software. A co-precipitation predominance area for the metal carbonates was identified in the diagrams. The calculated pH and Eh ranges under different conditions constitute a good reference for guiding the carbonate precursor synthesis process for LIBs cathode materials. Moreover, increasing the temperature and/or decreasing the ion concentration leads to shrinkage of the MeCO3 predominance area, which is detrimental to the co-precipitation process. Guided by the Eh-pH diagrams, the Ni0.13Co0.13Mn0.54(CO3)0.8 carbonate precursor was successfully synthesized with no embedded impurities, where the Ni, Co, and Mn were homogeneously distributed and close to the desired stoichiometric ratio. Upon calcination with Li2CO3, a final Li1.2Ni0.13Co0.13Mn0.54O2 cathode material was found to present a typical crystal structure and electrochemical characteristics for Li- and Mn-rich materials. It delivers an initial discharge capacity of 296.0 mAh g−1 at 0.1 C and a competitive capacity of 188.1 mAh g−1 after 100 cycles at 1 C rate. In summary, the OLI-assisted Eh-pH diagram simulation was verified to be reliable in theoretically predicting the co-precipitation process of the precursor. The latter is expected to be helpful in other related applications as well, including material synthesis via wet-chemical methods, element doping for material modification by carbonate precipitation, wet recovering of spent LIBs, and so on.
References
Shang G, Tang Y, Lai Y et al (2019) Enhancing structural stability unto 4.5 V of Ni-rich cathodes by tungsten-doping for lithium storage. J Power Sources 423:246–254. https://doi.org/10.1016/j.jpowsour.2019.03.072
Chen D, Mahmoud MA, Wang J-H, Waller GH, Zhao B, Qu C, el-Sayed MA, Liu M (2019) Operando investigation into dynamic evolution of cathode–electrolyte interfaces in a Li-ion battery. Nano Lett 19:2037–2043. https://doi.org/10.1021/acs.nanolett.9b00179
Schipper F, Nayak P, Erickson E et al (2017) Study of cathode materials for lithium-ion batteries: recent progress and new challenges. Inorganics 5:32. https://doi.org/10.3390/inorganics5020032
Wu Y (2015) Lithium-ion batteries: fundamentals and applications. CRC Press
Zhao L, Wu Q, Wu J (2018) Improving rate performance of cathode material Li1.2Mn0.54Co0.13Ni0.13O2 via niobium doping. J Solid State Electrochem 22:2141–2148. https://doi.org/10.1007/s10008-018-3912-4
Deng S, Li Y, Dai Q et al (2019) Structure and primary particle double-tuning by trace nano-TiO2 for a high-performance LiNiO2 cathode material. Sustain Energy Fuels 3:3234–3243. https://doi.org/10.1039/C9SE00487D
Xiao B, Sun X (2018) Surface and subsurface reactions of lithium transition metal oxide cathode materials: an overview of the fundamental origins and remedying approaches. Adv Energy Mater 8:1802057. https://doi.org/10.1002/aenm.201802057
Chen JJ, Li ZD, Xiang HF et al (2015) Enhanced electrochemical performance and thermal stability of a CePO4-coated Li1.2Ni0.13Co0.13Mn0.54O2 cathode material for lithium-ion batteries. RSC Adv 5:3031–3038. https://doi.org/10.1039/C4RA11370E
Xiang Y, Yin Z, Li X (2014) Synthesis and characterization of manganese-, nickel-, and cobalt-containing carbonate precursors for high capacity Li-ion battery cathodes. J Solid State Electrochem 18:2123–2129. https://doi.org/10.1007/s10008-014-2461-8
Zhang H, Yang T, Han Y et al (2017) Enhanced electrochemical performance of Li1.2Ni0.13Co0.13Mn0.54O2 by surface modification with the fast lithium-ion conductor Li-La-Ti-O. J Power Sources 364:272–279. https://doi.org/10.1016/j.jpowsour.2017.08.050
Li L, Zhang X, Chen R et al (2014) Synthesis and electrochemical performance of cathode material Li1.2Co0.13Ni0.13Mn0.54O2 from spent lithium-ion batteries. J Power Sources 249:28–34. https://doi.org/10.1016/j.jpowsour.2013.10.092
Du Z, Peng W, Wang Z et al (2018) Improving the electrochemical performance of Li-rich Li1.2Ni0.13Co0.13Mn0.54O2 cathode material by LiF coating. Ionics (Kiel) 24:3717–3724. https://doi.org/10.1007/s11581-018-2556-9
Liu H, Chen C, Du C et al (2015) Lithium-rich Li1.2Ni0.13Co0.13Mn0.54O2 oxide coated by Li3PO4 and carbon nanocomposite layers as high performance cathode materials for lithium ion batteries. J Mater Chem A 3:2634–2641. https://doi.org/10.1039/C4TA04823G
Pechen LS, Makhonina EV, Rumyantsev AM, Koshtyal YM, Pervov VS, Eremenko IL (2018) Effect of the synthesis method on the functional properties of lithium-rich complex oxides Li1.2Mn0.54Ni0.13Co0.13O2. Russ J Inorg Chem 63:1534–1540. https://doi.org/10.1134/S0036023618120173
He W, Liu J, Sun W et al (2018) Coprecipitation-gel synthesis and degradation mechanism of octahedral Li1.2Mn0.54Ni0.13Co0.13O2 as high-performance cathode materials for lithium-ion batteries. ACS Appl Mater Interfaces 10:23018–23028. https://doi.org/10.1021/acsami.8b04023
Jin X, Xu Q, Liu X, Yuan X, Liu H (2016) Improvement in rate capability of lithium-rich cathode material Li[Li0.2Ni0.13Co0.13Mn0.54]O2 by Mo substitution. Ionics (Kiel) 22:1369–1376. https://doi.org/10.1007/s11581-016-1675-4
Qiu S, Fang T, Zhu Y et al (2019) Li1.2Mn0.6Ni0.2O2 with 3D porous rod-like hierarchical micro/nanostructure for high-performance cathode material. J Alloys Compd 790:863–870. https://doi.org/10.1016/j.jallcom.2019.03.282
Han J, Zheng H, Hu Z et al (2019) Facile synthesis of Li-rich layered oxides with spinel-structure decoration as high-rate cathode for lithium-ion batteries. Electrochim Acta 299:844–852. https://doi.org/10.1016/j.electacta.2019.01.078
Zhao C, Wang X, Liu R et al (2014) β-MnO2 sacrificial template synthesis of Li1.2Ni0.13Co0.13Mn0.54O2 for lithium ion battery cathodes. RSC Adv 4:7154. https://doi.org/10.1039/c3ra45428b
Zhao T, Chen S, Li L et al (2013) Synthesis, characterization, and electrochemistry of cathode material Li[Li0.2Co0.13Ni0.13Mn0.54]O2 using organic chelating agents for lithium-ion batteries. J Power Sources 228:206–213. https://doi.org/10.1016/j.jpowsour.2012.11.099
Ju J-H, Ryu K-S (2011) Synthesis and electrochemical performance of Li(Ni0.8Co0.15Al0.05)0.8(Ni0.5Mn0.5)0.2O2 with core–shell structure as cathode material for Li-ion batteries. J Alloys Compd. https://doi.org/10.1016/j.jallcom.2011.05.060
Xiang Y, Li J, Liao Q, Wu X (2019) Morphology and particle growth of Mn-based carbonate precursor in the presence of ethylene glycol for high-capacity Li-rich cathode materials. Ionics (Kiel) 25:81–87. https://doi.org/10.1007/s11581-018-2569-4
Li L, Li Y, Li L, Chen N, Han Q, Zhang X, Xu H (2017) Thermodynamic analysis on the coprecipitation of Ni-Co-Mn hydroxide. Metall Mater Trans B Process Metall Mater Process Sci 48:2743–2750. https://doi.org/10.1007/s11663-017-0985-x
Xiang Y, Yin Z, Li X (2014) An improved carbonate precipitation method for the preparation of Li1.2Ni0.12Co0.12Mn0.56O2 cathode material. Ionics (Kiel) 20:163–168. https://doi.org/10.1007/s11581-013-0981-3
Wang D, Belharouak I, Zhou G, Amine K (2013) Synthesis of lithium and manganese-rich cathode materials via an oxalate co-precipitation method. J Electrochem Soc 160:A3108–A3112. https://doi.org/10.1149/2.016305jes
Xie H, Du K, Hu G et al (2015) Synthesis of LiNi0.8Co0.15Al0.05O2 with 5-sulfosalicylic acid as a chelating agent and its electrochemical properties. J Mater Chem A 3:20236–20243. https://doi.org/10.1039/C5TA05266A
Zhou F, Xu L, Kong J (2018) Co-precipitation synthesis of precursor with lactic acid acting as chelating agent and the electrochemical properties of LiNi0.5Co0.2Mn0.3O2 cathode materials for lithium-ion battery. J Solid State Electrochem 22:943–952. https://doi.org/10.1007/s10008-017-3837-3
Bromley LA (1973) Thermodynamic properties of strong electrolytes in aqueous solutions. AICHE J 19:313–320. https://doi.org/10.1002/aic.690190216
Zemaitis JF (1980) Predicting vapor-liquid-solid equilibria in multicomponent aqueous solutions of electrolytes. ACS Symposium Series:227–246
Lencka MM, Riman RE (1993) Thermodynamic modeling of hydrothermal synthesis of ceramic powders. Chem Mater 5:61–70. https://doi.org/10.1021/cm00025a014
Beverskog B, Puigdomenech I (1996) Revised pourbaix diagrams for iron at 25–300 °C. Corros Sci 38:2121–2135. https://doi.org/10.1016/S0010-938X(96)00067-4
Plyasunov AV, Shock EL (2001) Correlation strategy for determining the parameters of the revised Helgeson-Kirkham-Flowers model for aqueous nonelectrolytes. Geochim Cosmochim Acta 65:3879–3900. https://doi.org/10.1016/S0016-7037(01)00678-0
Zou W, Xia F-J, Song J-P et al (2019) Probing and suppressing voltage fade of Li-rich Li1.2Ni0.13Co0.13Mn0.54O2 cathode material for lithium-ion battery. Electrochim Acta 318:875–882. https://doi.org/10.1016/j.electacta.2019.06.119
Li Y, Deng S, Chen Y et al (2019) Dual functions of residue Li-reactive coating with C4H6CoO4 on high-performance LiNiO2 cathode material. Electrochim Acta 300:26–35. https://doi.org/10.1016/j.electacta.2019.01.083
Chen Y, Li Y, Tang S et al (2018) Enhanced electrochemical properties of the Cd-modified LiNi0.6Co0.2Mn0.2O2 cathode materials at high cut-off voltage. J Power Sources 395:403–413. https://doi.org/10.1016/j.jpowsour.2018.05.088
Ming L, Zhang B, Cao Y et al (2018) Effect of Nb and F co-doping on Li1.2Mn0.54Ni0.13Co0.13O2 cathode material for high-performance lithium-ion batteries. Front Chem 6:1–12. https://doi.org/10.3389/fchem.2018.00076
Gao J, Huang Z, Li J, He X, Jiang C (2014) Preparation and characterization of Li1.2Ni0.13Co0.13Mn0.54O2 cathode materials for lithium-ion battery. Ionics (Kiel) 20:301–307. https://doi.org/10.1007/s11581-013-0991-1
Croy JR, Kim D, Balasubramanian M et al (2012) Countering the voltage decay in high capacity xLi2MnO3•(1–x)LiMO2 electrodes (M=Mn, Ni, co) for Li+-ion batteries. J Electrochem Soc 159:A781–A790. https://doi.org/10.1149/2.080206jes
Cabana J, Johnson CS, Yang X-Q et al (2010) Structural complexity of layered-spinel composite electrodes for Li-ion batteries. J Mater Res 25:1601–1616. https://doi.org/10.1557/JMR.2010.0206
Gao Y, Patel RL, Shen K-Y, Wang X, Axelbaum RL, Liang X (2018) Boosting the electrochemical performance of Li1.2Mn0.54Ni0.13Co0.13O2 by atomic layer-deposited CeO2 coating. ACS Omega 3:906–916. https://doi.org/10.1021/acsomega.7b01922
Yabuuchi N, Yoshii K, Myung S-T et al (2011) Detailed studies of a high-capacity electrode material for rechargeable batteries, Li2MnO3−LiCo1/3Ni1/3Mn1/3O2. J Am Chem Soc 133:4404–4419. https://doi.org/10.1021/ja108588y
Assat G, Tarascon J-M (2018) Fundamental understanding and practical challenges of anionic redox activity in Li-ion batteries. Nat Energy 3:373–386. https://doi.org/10.1038/s41560-018-0097-0
Li X, Qiao Y, Guo S et al (2018) Direct visualization of the reversible O2−/O− redox process in Li-rich cathode materials. Adv Mater 30:1705197. https://doi.org/10.1002/adma.201705197
Radin MD, Vinckeviciute J, Seshadri R, Van der Ven A (2019) Manganese oxidation as the origin of the anomalous capacity of Mn-containing Li-excess cathode materials. Nat Energy 4:639–646. https://doi.org/10.1038/s41560-019-0439-6
Han S, Xia Y, Wei Z et al (2015) A comparative study on the oxidation state of lattice oxygen among Li1.14Ni0.136Co0.136Mn0.544O2 , Li2MnO3, LiNi0.5Co0.2Mn0.3O2 and LiCoO2 for the initial charge–discharge. J Mater Chem A 3:11930–11939. https://doi.org/10.1039/C5TA02161H
Acknowledgments
Mr. S. Deng is thankful for the support from Central South University for the CSU Special Scholarship for Study Abroad at the University of Toronto. OLI Systems Inc. is also acknowledged for providing access to the OLI software at the UofT.
Funding
This study received financial support from the Government of Chongzuo, Guangxi Zhuang Autonomous Region, China (GC Joint Special Fund No. FA2019015) and Science and Technology Department of Guangxi Zhuang Autonomous Region, China (Guangxi Special Fund for Scientific Center and Talent Resources, No. AD18281073).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOC 192 kb)
Rights and permissions
About this article

Cite this article
Deng, S., Chen, Y., Kolliopoulos, G. et al. Thermodynamic and experimental analysis of Ni-Co-Mn carbonate precursor synthesis for Li-rich cathode materials. Ionics 26, 2747–2755 (2020). https://doi.org/10.1007/s11581-020-03439-2
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-020-03439-2