
Abstract
As a representative of transition metal sulfides, MoS2 has a graphene-like structure and has recently been widely used as an anode in lithium ion batteries. However, the disadvantages of poor electrical conductivity and cycle performance limit its application. Here, hollow MoS2/reduced graphene oxide (rGO) composites were successfully synthesized by a hydrothermal method using polystyrene microspheres as hard templates. The hollow MoS2 microspheres with a size of about 180 nm are distributed on the surface of graphene sheets. The hollow MoS2/rGO composites exhibit superior electrochemical properties as anode materials for lithium-ion batteries compared with the hollow MoS2 microspheres. They show a high reversible capacity of 752.4 mAh g−1 at 500 mA g−1 after 100 cycles with negligible decay as well as a high-rate performance (620.8 mA h g−1, 1 A g−1). The excellent lithium storage performance of the hollow MoS2/rGO composites can be attributed to the synergistic effect of the hollow MoS2 microspheres and graphene sheets. Graphene effectively suppresses the agglomeration of the MoS2 and maintain the structural stability of the material.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Lithium-ion battery has the advantages of high-energy density and long cycle life and has been widely used as the power source of portable electronic devices [1, 2]. Graphite is considered as the main anode in the current commercial lithium-ion battery. However, the low theoretical capacity of graphite makes it difficult for lithium-ion batteries to meet the application requirements in some fields such as high-energy density and high-power density [3,4,5,6]. Particularly, the development of new-energy electric vehicles puts a higher demand on the new generation of lithium-ion batteries. Therefore, the development of the anode material with a high specific capacity, an excellent cycle performance, and a high-rate performance for the next generation of high-performance lithium-ion battery is of great significance.
As a kind of layered material with two-dimensional structure, MoS2 has the advantages of a high theoretical specific capacity, a simple preparation process, and low cost. It has aroused great attention of researchers and is regarded as a very promising anode material of lithium-ion battery [7,8,9,10]. However, like most two-dimensional layered materials, MoS2 has the problems of poor conductivity and easy agglomeration during cycling, resulting in poor cycle stability [11,12,13,14,15,16,17].
To overcome the shortcomings of poor cycling performance of MoS2 nanosheets, three-dimensional MoS2 materials, such as MoS2 nanospheres [18,19,20], hollow MoS2 spheres [21,22,23], and core-shell MoS2 spheres [24, 25], have been prepared using a self-assembly or a hard template method. Constructing three-dimensional structure of MoS2, to some extent, can solve the problem of agglomeration of MoS2. However, its cycle stability is still not ideal at high current densities for a long period of cycling [26].
Recently, the application of graphene in lithium-ion batteries has been extensively studied. Graphene has a high conductivity, large surface area, good flexibility, and superior lithium deintercalation performance compared with other carbon materials [27, 28].
In addition, graphene can also act as a buffer layer to prevent the aggregation of nanoparticles in the cycle [29,30,31]. The composites of graphene and MoS2 with different morphologies have been reported. Guo et al. reported the synthesis of MoS2 nanowall/graphene composites by a solvothermal method [32]. The specific capacity of the MoS2 nanowall/graphene composites can stay 700 mAh g−1 after 100 cycles at a current of 500 mA g−1, which is much higher than that of bulk MoS2. Hou et al. reported the synthesis of C3N4/N-doped graphene/MoS2 hybrid nanosheets [33]. The specific capacity of the composites can remain at 855 mAh g−1 at 100 mA g−1 after 100 cycles. The results indicate that graphene can significantly enhance the MoS2 cycle performance. Therefore, the combination of graphene and MoS2 can not only promote the transfer of electrons during the electrochemical reaction but also prevent the accumulation and pulverization of MoS2. However, there is still the problem of volume expansion during charging and discharging for MoS2 of above composites.
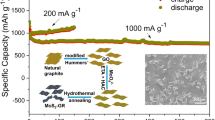
In this work, we report a novel hollow MoS2/rGO composites synthesized by the combination of a hard template method and a hydrothermal method. The MoS2 microspheres with a hollow structure can effectively alleviate the problem of volume expansion during charging and discharging. The addition of rGO can improve the conductivity of composite materials. At the same time, the sheet structure of rGO can also inhibit the volume expansion of MoS2 and improve its cycling performance. Therefore, hollow MoS2/rGO composites exhibit a high reversible capacity up to 763.4 mAh g−1 at a current density of 500 mA g−1 as well as excellent cycling stability and rate capability.
Experimental
Synthesis of sulfonated PS microspheres
The mini emulsion polymerization procedure of cross-linked sulfonated polystyrene (PS) microspheres was based on a previously report of styrene polymerization with some modification [34]. Firstly, 0.024 g surfactant (SDBS) was dissolved in 24 g deionized water. Meanwhile, 6 g monomers mixture (mass ratio of St:DVB = 20:1) was mixed with 0.12 g HD and stirred for 20 min. Secondly, two phases continued stirring for 1 h at 300 r min−1 for pre-emulsification. After sonicated for 10 min, the resultant solution was turned into a white milk-like mini emulsion. The as-obtained mini emulsion was transferred into a 250-ml flask and purged with a nitrogen stream for 30 min. After adding 0.12 g KPS, the polymerization was initiated and reacted at 70 °C for 4 h. Finally, the product was separated on filter paper and washed with ethanol and deionized water repeatedly. The white powder was obtained after dried at 60 °C under vacuum.
Three grams of PS was dispersed in 30 ml of concentrated sulfuric acid, stirring at 40 °C for 4 h. After the reaction, the yellow product was centrifuged and washed with ethanol. The final product was freeze-dried for 24 h.
Synthesis of hollow MoS2/rGO composites
Graphene oxide was synthesized from natural flake graphite powder (crystalline, 300 mesh) by a modified Hummers method [35]. Hollow MoS2/rGO was prepared using a hydrothermal method. Typically, 0.075 g PS spheres were dispersed in 30 ml deionized water. Sodium molybdate (0.2 g) and 0.4 g thiourea were added and stirred to dissolve at room temperature. Then, 1 ml hydrochloric acid and 100 mg graphene oxide were added into the solution and stirred for 30 min. The solution was transferred to a 100-ml Teflon-line stainless steel autoclave and annealed at 200 °C for 24 h. The reacted product was washed alternately with water and ethanol and centrifuged. After drying, the hollow MoS2/rGO composites were ground into powder and calcined in a furnace at 800 °C in argon atmosphere for 3 h. The black hollow MoS2/rGO composites were obtained after annealing.
For comparison, the hollow MoS2 microspheres were also prepared at similar conditions without the introduction of graphene oxide.
Characterization
The crystal structure of the products was confirmed by X-ray powder diffraction (XRD) on a Rigaku Ultima IV powder diffraction system using Cu Kα radiation with a scanning rate of 10° min−1 between 5° ≤ 2θ ≤ 80°. Field emission scanning electron microscopic (SEM) images were recorded with a Hitachi S-4800 microscope set at 15 kV. Transmission electron micrographs (TEM) and selected area electron diffraction (SAED) pattern were performed with a JEM-2010 microscope operated at 150 kV. X-ray photoelectron spectroscopy (XPS) analysis was used to evaluate the chemical states of samples on a PHI 5300 ESCA commercial instrument. Raman spectra were collected using a Renishaw inVia-Reflex Raman Microscope with a 532 nm excitation laser.
Electrochemical measurements
The electrodes were prepared by dispersing 70 wt% hollow MoS2/rGO composites, 20 wt% Super P, and 10 wt% polyvinylidene fluoride binder in N-methyl-2-pyrrolidone solvent to produce a slurry. The resultant slurry was pasted on Cu foil and dried in a vacuum oven to completely remove the solvent and water. The mass loading of the active material on the electrode was about 1.0–1.5 mg cm−2. CR2025 coin cells were fabricated for electrochemical measurements. Lithium foil was used as the counter electrode, and Cellgard 2400 polypropylene microporous film acted as separators. The electrolyte solution was 1 m LiPF6 dissolved in a 1:1:1 volume mixture of ethylene carbonate (EC), dimethyl carbonate (DMC), and diethyl carbonate (DEC). The cells were assembled in a glove box filled with high purity Ar. The CV curves were carried out on a CHI 660D electrochemical workstation at a scan rate of 0.2 mV S−1 from a range of 0.01 to 3.0 V versus Li/Li+. The galvanostatic discharge–charge tests were conducted on a Land CT2001A battery system at different current densities within between 0.005 and 3.0 V versus Li/Li+. The electrochemical impedance spectroscopy (EIS) of the electrode was carried out in a frequency range of 100 kHz to 0.01 Hz.
Results and discussion
Hollow MoS2/rGO composites were fabricated directly using a hydrothermal method in the presence of hard templates. The general procedure is demonstrated in Fig. 1. First, PS microspheres were prepared by a mini emulsion polymerization procedure. The PS microspheres were sulfurized with sulfuric acid to obtain sulfonated PS microspheres. Second, the sulfonated PS microspheres were mixed with GO suspension, sodium molybdate, and thiourea to get the homogeneous MoS2–GO–PS suspension. The suspension was subsequently treated hydrothermally. With the progress of hydrothermal reaction, MoS2 nanosheets were continuously deposited on the surface of PS microspheres due to the presence of polystyrene sulfonic acid on the surface of the PS microsphere.

Schematic illustration of the fabrication process and the structure of the resulting hollow MoS2/rGO composites
After cooling naturally, the black precipitates were collected by centrifugation, washed with water and ethanol. The PS/MoS2/GO composites were obtained after drying in a vacuum oven. Finally, the as-obtained PS/MoS2/GO composites were annealed in a conventional tube furnace. Hollow MoS2/rGO composites were obtained due to the decomposition of PS spheres and the reduction of GO during calcination.
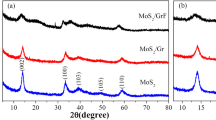
Figure 2a, b shows the XRD patterns of hollow MoS2 microspheres and hollow MoS2/rGO composites. From Fig. 2a, sharp diffraction peaks appear at 13.1°, 32.3°, 38.5°, 49.0°, and 58.5°, respectively, which indicate the presence of MoS2 with good crystallinity. The corresponding diffraction peaks are (002), (100), (103), (105), and (110) which are coincided well with the standard card of MoS2 (JCPDS no. 37-1492). XRD pattern of hollow MoS2/rGO composites is shown in Fig. 2b. It can be observed that hollow MoS2/rGO composites also have the characteristic diffraction peaks of MoS2, but the intensity of the peaks is a litter lower than hollow MoS2 microspheres due to the existence of graphene.

XRD patterns of (a) hollow MoS2 and (b) hollow MoS2/rGO composites
Figure 3a shows the TEM image of sulfonated PS microspheres. It can be seen that the size of the sulfonated PS microspheres is about 80 nm. Figure 3b, c shows the SEM and TEM images of hollow MoS2 microspheres. As is seen from Fig. 3b, the size distribution of the MoS2 microspheres is estimated to be in the range from 120 to 180 nm. The hollow structure of MoS2 microspheres can be clearly observed from Fig. 3c, and the dimension of hollow section of MoS2 microspheres is consistent with the size of the sulfonated PS microspheres. Meanwhile, from Fig. 3c, we also found that the hollow MoS2 microspheres are assembled by MoS2 nanosheets. The hollow architectures not only facilitate electron and mass transfer but also accommodate the volume expansion which will benefit to promote electrochemical performance. Figure 3d–f shows the SEM and TEM images of hollow MoS2/rGO composites. Some aggregated MoS2 microspheres which distributed on the graphene sheet are observed in Fig. 3d. From Fig. 3e, the diameter of hollow MoS2/rGO composites is approximately 180 nm and the hollow structure is also confirmed by TEM image in Fig. 3e. As shown in Fig. 2f, the clear lattice fringes of 0.62 nm were observed shown by arrows which were attributed to the (002) plane of the layered MoS2 [27,28,29].

a TEM image of sulfonated PS microspheres. b SEM image of hollow MoS2 microspheres. c TEM image of hollow MoS2 microspheres. d SEM image of hollow MoS2/rGO composites. e, f TEM images of hollow MoS2/rGO composites
Raman spectra of hollow MoS2/rGO composites are shown in Fig. 4a. The two dominant peaks of hollow MoS2/rGO can be clearly observed at 383 and 404 cm−1 which correspond to E2g and A1g modes of the hexagonal MoS2, respectively. The E2g mode correlates to the in-layer displacement of Mo and S atoms, whereas the A1g mode relates to the out-of-layer symmetric displacements of S atoms along the C axis [36, 37]. The existence of MoS2 is also exactly confirmed by A1g peak at 404 cm−1. Two wide peaks at ~ 1360 and 1600 cm−1 are attributed to D and G bands of carbon materials. To determine the weight content of graphene in the composites, TG measurements were carried out from room temperature to 600 °C in flowing air at a ramp rate of 10 °C min−1. As shown in Fig. S2, the weight loss of pure MoS2 is 8.4% due to the conversion of MoS2 to MoO3 in air. A more significant weight loss for hollow MoS2/rGO is observed because of the co-effect of carbon combustion and conversion of MoS2 to MoO3 between 250 and 480 °C. The weight percentage of MoS2 in hollow MoS2/rGO is calculated to be 81.0%. Thus, the total content of carbon in hollow MoS2/rGO is 19%.

a Raman spectra of hollow MoS2/rGO composites. b–d XPS spectra of hollow MoS2/rGO: high-resolution spectra of Mo 3d (b), S 2p (c), and C 1s (d)
X-ray photoelectron spectroscopy (XPS) analysis was used to evaluate the oxidation state and composition of hollow MoS2/rGO in Fig. 4b–d. As shown in Mo 3d spectrum of Fig. 4b, two strong peaks appeared at around 229.3 and 232.4 eV can be ascribed to Mo3d 5/2 and Mo3d 3/2 binding energies of MoS2, respectively, whereas the peak at 226.3 eV can be indexed as S 2s [38, 39]. From Fig. 4c, two peaks are shown at 162.0 and 163.1 eV which are corresponded to the S 2p3/2 and S 2p1/2 binding energies of MoS2, respectively. Especially, the C 1s peak (Fig. 4d) can be resolved into two peaks at 284.3 and 285.7 which are indexed to sp2 carbon, sp3 carbon, and carbon in C–O single bond, respectively [40]. The oxygen content of hollow MoS2/rGO composites decreases rapidly as compared with graphene oxide (Fig. S3), suggesting a remarkable reduction of GO into graphene in the furnace at 800 °C.
The electrochemical properties of the hollow MoS2/rGO composite for reversible Li+ storage were investigated by eletrochemical measurements. The results are shown in Fig. 5. Figure 5a is a cyclic voltammogram and a charge/discharge curve of a hollow MoS2/rGO composite. As seen from Fig. 5a, the hollow MoS2/rGO composite shows two reduction peaks around 0.2 V and 0.75 V in the first lap scan. The reduction peak near 0.75 V belongs to Li + embedded in MoS2 to form LixMoS2. The reduction peak near 0.2 V corresponds to the reaction LixMoS2 → Mo + 2Li2S [41, 42]. After the second cycle, the two reduction peaks shift. Two peaks appeared at 1.0 V and 1.7 V represent the reaction of Li and Mo to form LixMoS2 and S to Li2S. The strong oxidation peaks at about 2.4 V in the three discharge processes correspond to the formation of MoS2. In Fig. 5b, the discharge capacity gradually decreases as the discharge progresses. After the first charge–discharge cycle, the charge–discharge curve within the voltage range of 0.01 to 2.0 V corresponds to the storage of lithium ions and electrons in the pores of the interface between Li2S and metal Mo nanoparticles [41, 43].

Electrochemical performances of the hollow MoS2/rGO for Li-ion batteries. a CV curves from 3.0 to 0.01 V at a scanning rate of 0.2 mV s−1. b Galvanostatic charge–discharge profiles at 500 mA g−1. c Cycling performance of hollow MoS2 microspheres and hollow MoS2/rGO at a current density of 500 mA g−1. d Rate capability performance of hollow MoS2/rGO
Figure 5c shows the cyclic performance of hollow MoS2/rGO composites and hollow MoS2 microspheres. It can be seen that the hollow MoS2/rGO composites have better cyclic stability compared with hollow MoS2 microspheres. After 100 cycles, the specific capacity of hollow MoS2/rGO composites remains at 752.4 mAh g−1, corresponding to a coulombic efficiency of 99.15%. However, the hollow MoS2 microspheres show a significant capacity decrease. This improved electrochemical performance may attribute to synergistic effect of the hollow MoS2 microspheres and graphene sheets.
The rate capability performance of hollow MoS2/rGO composites is shown in Fig. 5d. When the current density increases, the discharge capacity decreases. The discharge capacity of hollow MoS/rGO composites remains at 620.8 mAh g−1 and a current rate of 1 A g−1. When the current density returns to 100 mA g−1 again, the discharge capacity was restored to 715.4 mAh g−1, indicating hollow MoS2/rGO composites have excellent current rate capability performance.
Meanwhile, the morphology of the hollow MoS2/rGO electrode after 100 cycles is shown in Fig. S4. From Fig. S4, the microsphere morphology in the hollow MoS2/rGO can still retain after 100 cycles which indicates that the graphene in the hollow MoS2/rGO composites could effectively buffer large volume changes and inhibit the aggregation of MoS2 microspheres.
The Nyquist plots of the hollow MoS2/rGO electrode and hollow MoS2 microspheres are shown in Fig. 6. The Nyquist plots are fitted by an equivalent electrical circuit (inset of Fig. 6), and the fitting impedance data is displayed in Table S1. The Nyquist plots of two electrodes are constituted by the internal resistance (Rs), the impedance (Rf) of the electrode/electrolyte interface and the charge–transfer impedance (Rct). From Fig. 6 and Table S1, both of the electrodes show similar Nyquist profiles with a semicircle in the high-frequency region and a straight line in the low-frequency region. The two electrodes have similar Rs and Rf values, while the hollow MoS2/rGO electrode reveals a much smaller charge–transfer resistance (Rct = 52.9 Ω) associated with the semicircle than the hollow MoS2 microspheres (Rct = 82.1 Ω), indicating hollow MoS2/rGO electrode has better rate performance due to the rapid charge transfer reaction for Li+ insertion and extraction.

Nyquist plots of the hollow MoS2/rGO and hollow MoS2 microsphere electrodes and the equivalent circuit for describing the EIS behavior (inset)
Conclusions
In conclusion, we employ sulfonated PS microsphere as a hard template and hydrothermal method to prepare hollow MoS2/rGO composites. The hollow MoS2 microspheres have a diameter of about 180 nm and uniformly disperse on the graphene sheets. Compared to hollow MoS2, hollow MoS2/rGO composites exhibit outstanding cycle performance at high currents. At a current of 500 mA g−1, the specific capacity of hollow MoS2/rGO composites still reaches 752.4 mAh g−1 after 100 cycles. The strategy of introducing graphene effectively enhances the electrical conductivity of the composites. Meanwhile, it can also suppress the volume expansion of MoS2, which greatly improves the cycle performance of the composites.
References
Arico AS, Bruce P, Scrosati B, Tarascon J-M, Van Schalkwijk W (2005) Nanostructured materials for advanced energy conversion and storage devices. Nat Mater 4:366–377
Marom R, Amalraj SF, Leifer N, Jacob D, Aurbach D (2011) A review of advanced and practical lithium battery materials. J Mater Chem 21:9938–9954
Winter M, Brodd RJ (2004) What are batteries, fuel cells, and supercapacitors? Chem Rev 104:4245–4269
Simon P, Gogotsi Y (2008) Materials for electrochemical capacitors. Nat Mater 7:845–854
Yan J, Wang Q, Wei T, Fan Z (2014) Recent advances in design and fabrication of electrochemical supercapacitors with high energy densities. Adv Energy Mater 4:1300816
Larcher D, Tarascon J-M (2015) Towards greener and more sustainable batteries for electrical energy storage. Nat Chem 7:19–29
Hu Z, Wang L, Zhang K, Wang J, Cheng F, Tao Z, Chen J (2014) MoS2 nanoflowers with expanded interlayers as high-performance anodes for sodium-ion batteries. Angew Chem Int Ed 53:12794–12798
David L, Bhandavat R, Singh G (2014) MoS2/graphene composite paper for sodium-ion battery electrodes. ACS Nano 8:1759–1770
Zhu C, Mu X, van Aken PA, Yu Y, Maier J (2014) Single-layered ultrasmall nanoplates of MoS2 embedded in carbon nanofibers with excellent electrochemical performance for lithium and sodium storage. Angew Chem 53:2152–2156
Cook JB, Kim HS, Yan Y, Ko JS, Robbennolt S, Dunn B, Tolbert SH (2016) Mesoporous MoS2 as a transition metal dichalcogenide exhibiting pseudocapacitive Li and Na-ion charge storage. Adv Energy Mater 6:1501937
Hu X, Zhang W, Liu X, Mei Y, Huang Y (2015) Nanostructured Mo-based electrode materials for electrochemical energy storage. Chem Soc Rev 44:2376–2404
Zhang L, Wu HB, Yan Y, Wang X, Lou XWD (2014) Hierarchical MoS2 microboxes constructed by nanosheets with enhanced electrochemical properties for lithium storage and water splitting. Energy Environ Sci 7:3302–3306
Yang L, Wang S, Mao J, Deng J, Gao Q, Tang Y, Schmidt OG (2013) Hierarchical MoS2/polyaniline nanowires with excellent electrochemical performance for lithium-ion batteries. Adv Mater 25:1180–1184
Cao X, Shi Y, Shi W, Rui X, Yan Q, Kong J, Zhang H (2013) Preparation of MoS2-coated three-dimensional graphene networks for high-performance anode material in lithium-ion batteries. Small 9:3433–3438
Sen UK, Johari P, Basu S, Nayak C, Mitra S (2014) An experimental and computational study to understand the lithium storage mechanism in molybdenum disulfide. Nanoscale 6:10243–10254
Jeong J-M, Lee KG, Chang S-J, Kim JW, Han Y-K, Lee SJ, Choi BG (2015) Ultrathin sandwich-like MoS2@N-doped carbon nanosheets for anodes of lithium ion batteries. Nanoscale 7:324–329
Fang X, Guo X, Mao Y, Hua C, Shen L, Hu Y, Wang Z, Wu F, Chen L (2012) Mechanism of lithium storage in MoS2 and the feasibility of using Li2S/Mo nanocomposites as cathode materials for lithium–sulfur batteries. Chem-Asian J 7:1013–1017
Bai Z, Zhang Y, Zhang Y, Guo C, Tang B (2015) Hierarchical MoS2@carbon microspheres as advanced anodes for Li-ion batteries. Chem-Eur J 21:18187–18191
Ding S, Zhang D, Chen JS, Lou XWD (2012) Facile synthesis of hierarchical MoS2 microspheres composed of few-layered nanosheets and their lithium storage properties. Nanoscale 4:95–98
Lu Y, Zhao Q, Zhang N, Lei K, Li F, Chen J (2016) Facile spraying synthesis and high-performance sodium storage of mesoporous MoS2/C microspheres. Adv Funct Mater 26:911–918
Cai Y, Yang H, Zhou J, Luo Z, Fang G, Liu S, Pan A, Liang S (2017) Nitrogen doped hollow MoS2/C nanospheres as anode for long-life sodium-ion batteries. Chem Eng J 327:522–529
Wang Y, Yu L, Lou XW (2016) Synthesis of highly uniform molybdenum-glycerate spheres and their conversion into hierarchical MoS2 hollow nanospheres for lithium-ion batteries. Angew Chem Int Ed 55:7423–7426
Wang L, Ma Y, Yang M, Qi Y (2015) Hierarchical hollow MoS2 nanospheres with enhanced electrochemical properties used as an electrode in supercapacitor. Electrochim Acta 186:391–396
Wang Y, Kang W, Cao D, Zhang M, Kang Z, Xiao Z, Wang R, Sun D (2018) A yolk-shelled Co9S8/MoS2-CN nanocomposite derived from a metal-organic framework as a high performance anode for sodium ion batteries. J Mater Chem A 6:4776–4782
Zhang Y, Sun W, Rui X, Li B, Tan HT, Guo G, Madhavi S, Zong Y, Yan Q (2015) One-pot synthesis of tunable crystalline Ni3S4@amorphous MoS2 core/shell nanospheres for high-performance supercapacitors. Small 11:3694–3702
Wu Z-S, Ren W, Wen L, Gao L, Zhao J, Chen Z, Zhou G, Li F, Cheng H-M (2010) Graphene anchored with Co3O4 nanoparticles as anode of lithium ion batteries with enhanced reversible capacity and cyclic performance. ACS Nano 4:3187–3194
Novoselov KS, Geim AK, Morozov SV, Jiang D, Zhang Y, Dubonos SV, Grigorieva IV, Firsov AA (2004) Electric field effect in atomically thin carbon films. Science 306:666–669
Huang X, Yin Z, Wu S, Qi X, He Q, Zhang Q, Yan Q, Boey F, Zhang H (2011) Graphene-based materials: synthesis, characterization, properties, and applications. Small 7:1876–1902
Zhou G, Wang D-W, Li F, Zhang L, Li N, Wu Z-S, Wen L, Lu GQ, Cheng H-M (2010) Graphene-wrapped Fe3O4 anode material with improved reversible capacity and cyclic stability for lithium ion batteries. Chem Mater 22:5306–5313
Luo J, Zhao X, Wu J, Jang HD, Kung HH, Huang J (2012) Crumpled graphene-encapsulated Si nanoparticles for lithium ion battery anodes. J Phys Chem Lett 3(13):1824–1829
Vinayan B, Ramaprabhu S (2013) Facile synthesis of SnO2 nanoparticles dispersed nitrogen doped graphene anode material for ultrahigh capacity lithium ion battery applications. J Mater Chem A 1(12):3865–3871
Guo J, Chen X, Jin S, Zhang M, Liang C (2015) Synthesis of graphene-like MoS2 nanowall/graphene nanosheet hybrid materials with high lithium storage performance. Catal Today 246:165–171
Hou Y, Li J, Wen Z, Cui S, Yuan C, Chen J (2014) N-doped graphene/porous g-C3N4 nanosheets supported layered-MoS2 hybrid as robust anode materials for lithium-ion batteries. Nano Energy 8:157–164
Landfester K, Bechthold N, Tiarks F, Antonietti M (1999) Miniemulsion polymerization with cationic and nonionic surfactants: a very efficient use of surfactants for heterophase polymerization. Macromolecules 32:2679–2683
Hirata M, Gotou T, Horiuchi S, Fujiwara M, Ohba M (2004) Thin-film particles of graphite oxide 1: high-yield synthesis and flexibility of the particles. Carbon 42:2929–2937
Wang Z, Chen T, Chen W, Chang K, Ma L, Huang G, Chen D, Lee JY (2013) CTAB-assisted synthesis of single-layer MoS2-graphene composites as anode materials of Li-ion batteries. J Mater Chem A 1:2202–2210
Jin Z, Shin S, Kwon DH, Han S-J, Min Y-S (2014) Novel chemical route for atomic layer deposition of MoS2 thin film on SiO2/Si substrate. Nanoscale 6:14453–14458
Ko YN, Kang Y, Park S (2014) Superior electrochemical properties of MoS2 powders with a MoS2@void@MoS2 configuration. Nanoscale 6:4508–4512
Becerril HA, Mao J, Liu Z, Stoltenberg RM, Bao Z, Chen Y (2008) Evaluation of solution-processed reduced graphene oxide films as transparent conductors. ACS Nano 2:463–470
Stankovich S, Dikin DA, Piner RD, Kohlhaas KA, Kleinhammes A, Jia Y, Wu Y, Nguyen ST, Ruoff RS (2007) Synthesis of graphene-based nanosheets via chemical reduction of exfoliated graphite oxide. Carbon 45:1558–1565
Miki Y, Nakazato D, Ikuta H, Uchida T, Wakihara M (1995) Amorphous MoS2 as the cathode of lithium secondary batteries. J Power Sources 54:508–510
Chang K, Chen W (2011) L-cysteine-assisted synthesis of layered MoS2/graphene composites with excellent electrochemical performances for lithium ion batteries. ACS Nano 5:4720–4728
Wang Q, Li J (2007) Facilitated lithium storage in MoS2 overlayers supported on coaxial carbon nanotubes. J Phys Chem C 111:1675–1682
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOC 1396 kb)
Rights and permissions
About this article

Cite this article
Xue, H., Jiao, Q., Du, J. et al. Hollow MoS2/rGO composites as high-performance anode materials for lithium-ion batteries. Ionics 25, 4659–4666 (2019). https://doi.org/10.1007/s11581-019-03041-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-019-03041-1