
Abstract
The present paper deals with the studies concerning electrochemical properties of the expanded graphite/iron oxide/carbon (EG/Fe2O3/C) composite produced by deep cathodic reduction. The electrochemical activity of EG/Fe2O3/C composite was investigated in alkaline solution by cyclic voltammetry technique. Electrochemical features of EG/Fe2O3/C were found to be strongly influenced by the conditions under which the measurements were performed. The interpretation of electrochemical measurements was supported by the scanning electron microscope (SEM) observation revealing the appearance of carbon fibers of a different diameter as the main component of the carbon layer within the examined EG/Fe2O3/C composite. The X-ray diffraction (XRD) and energy dispersive spectrometry (EDS) analysis have confirmed the presence of Fe2O3 layer within the EG/Fe2O3/C composite. Additionally, the latter analysis has provided information on changes within the chemical composition of EG/Fe2O3/C caused by electrochemical treatment.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Expanded graphite (EG), being a product of exfoliation of graphite intercalation compounds (GIC), exhibits interesting properties such a developed surface area, high adsorption capacity, enhanced concentration of surface defects, high surface activity, low bulk density, and high thermal resistivity [1, 2]. Owing to these features, EG has found an application as heavy oil sorbent [3, 4], material for the generation and storage of energy in alkaline batteries [5], fuel cells [6], and supercapacitors [7] as an electrode material for electrooxidation of various types of organic compounds [8–10]. EG can be also used in preparation of various composite materials, including carbon-polymer [11, 12], carbon-metal [13], and carbon-carbon [14]. On the other hand, low density of EG associated with its strong ability to be compressed, followed by the reduction of its sorption capacity, may be a disadvantage hindering composite formation. Due to these properties, chemical vapor deposition (CVD) may be used as an alternative way for synthesis of composites based on the EG matrix [14, 15]. In the CVD method, carbon obtained by carbonization of hydrocarbons is deposited onto the surface of metal or metal oxide catalyst thus forming metal-C or metal oxide-C composites. The efficiency of carbon formation by carbonization is strongly influenced by the type of hydrocarbon used as a carbon source and the catalytic properties of metal or metal oxide in the reaction of hydrocarbon carbonization [14–16]. A number of theories have been proposed describing the mechanism of carbon growth over metal or metal oxide catalysts. One of such theories, elucidating the synthesis of carbon fibers, postulates that carbon atoms or clusters formed on the surface of the catalyst diffuse through its structure and finally precipitate on the opposite side of the synthesized fiber. In this case, the total duration of the synthesis is limited by the catalyst activity [16]. The type of the formed products is also influenced by the conditions of carbonization, namely temperature, reaction mixture composition, and reaction time [14–16]. Among the potential products of the CVD process, carbon nanotubes (CNTs), carbon nanofibers (CNFs), and carbon filaments are currently considered. Note that the synthesis of such products is accompanied by the formation of amorphous carbon, usually an undesired byproduct in the CVD synthesis.
From the view point of electrode capacity of Fe-air batteries, it is important to find an anode material of increased porosity enabling better access of electrolyte to its active surface [17]. As another factors determining the high capacity of iron anodes in Fe-air batteries can be considered increased resistivity against passivation as well as increased conductivity and low self-discharge. Owing to the increased conductivity, the Fe/C composite can be applied as anode material instead of porous Fe. One of the method commonly used to solve the problem of anode passivation as well as to reduce the self-discharge processes is based on the modification of electrolyte by the use of sulfide admixture (K2S, Na2S, PbS) [18, 19]. However, the particular role of sulfides is not yet well known, but some theories postulate that the interaction of sulfides with electrode surface occurring within the electrode/electrolyte interface should be recognized as a crucial factor determining the electrochemical activity of Fe/C anodes in Fe-air batteries [18, 19].
Our previous work on ozone modification of EG/Fe/C composite have revealed that strong oxidation of composite results in considerable modification of its textural and chemical properties [20]. The intensive reaction between ozone and C layer of EG/Fe/C composite removes amorphous carbon uncovering carbon fibers of a different diameter. Simultaneously, the oxidation of Fe to Fe2O3 occurs. The changes in physicochemical properties of ozonated EG/Fe/C composite induced valuable changes within its electrochemical behavior demonstrated in alkaline solution.
The describing work [20] does not explain exhaustively detailed role of factor employed to modification of EG/Fe/C composite as well as does not provides enough information for comprehensive electrochemical characteristics in the region of cathodic reduction.
The purpose of this work was to obtain and to study the properties of EG/Fe2O3/C composite produced by deep cathodic reduction. Electrochemical measurements were carried out in alkaline solutions in the potential range of rapid hydrogen evolution using cyclic voltammetry technique. In order to improve the understanding of the examined processes, the electrochemical properties of EG/Fe2O3/C composite were discussed in conjunction with morphological (SEM) and structural (XRD analyses) properties as well as its chemical composition (EDS).
Experimental
Preparation of EG/Fe2O3/C
The EG/Fe2O3/C composite was prepared according to a four-step synthesis. Expanded graphite (EG) being a matrix of target composite was prepared by thermal exfoliation of a stage-5 graphite intercalation compound with sulfuric acid (H2SO4-GIC). Process of exfoliation was conducted in a muffle furnace at the temperature of 800 °C for 4 min in air atmosphere. In order to obtain the EG/Fe composite, the electrodeposition of Fe from the bath composed of FeSO4 × 7H2O, FeCl2 × 4H2O, (NH4)2SO4, and (NH4)2C2O4 was performed. Galvanostatic deposition of Fe was carried out by passing through the electrode made of EG current density equal to 1 A dm-2. The such prepared EG/Fe composite worked as a catalyst for acetylene carbonization realized by chemical vapor deposition (CVD) method, yielding the EG/Fe/C composite. The mentioned carbonization was carried out at 650 °C for 30 min. in a tube furnace coupled to the flowmeter system ensuring the control of the amounts of C2H2 and N2 gases passing through the quartz reactor. In order to remove the amorphous carbon formed onto EG/Fe surface due to acetylene carbonization, the EG/Fe/C composite was heat treated at 500 °C for 1 h in air atmosphere. Since under such conditions, the layer of Fe was oxidized to Fe2O3, the final composite will be referred to as EG/Fe2O3/C [21].
Physicochemical characterization
The morphology of EG/Fe2O3/C, especially its outer layer was studied by scanning electron microscopy (SEM) (S-3400 N, Hitachi microscope) using a voltage acceleration of electron beam equal to 15 keV. The information on EG/Fe2O3/C composition before and after its thermal treatment at 500 °C for 1 h as well as after cathodic reduction were acquired from the energy dispersive spectrometry (EDS) analysis.
In order to characterize the structure of the EG/Fe2O3/C composite, the X-ray diffraction (XRD) measurement was performed using CuKα radiation (PW-1710, Philips).
Electrochemical treatments
The electrochemical properties of EG/Fe2O3/C composite were examined in three electrode cell by cyclic voltammetry method. The electrochemical measurements were performed in water solution of 0.5 M KOH. The working electrode EG/Fe2O3/C being in a powder consistency was placed in a pocket made of porous polymer material, in which graphite rod (3 mm in diameter and 50 mm in length), playing a role of current collector, was beforehand inserted. In each experiment, the mass of the working electrode was equal to 20 mg. Hg/HgO/0.5 M KOH (+0.117 V vs. the normal hydrogen electrode) was a reference electrode whereas graphite rod of a similar parameters to current collector was used as a counter electrode. The particular description of electrode preparation procedure can be found in previous works [9, 22]. The electrochemical researches were carried out with three different scan rates 0.1, 1.0, and 10 mV s−1 within the two potential ranges −1.25 ↔ 0.0 and −1.5 ↔ 0.0 V. Prior to each measurement, the electrode was kept at an open circuit in the investigated electrolyte for 30 min. After starting the measurements, the potential was swept from the rest potential of electrode (E R) in the negative direction. Electrochemical measurements were carried out using AUTOLAB potentiostat-galvanostat (model PGSTAT 30).
Results and discussion
The structure, morphology, and chemical composition of EG/Fe2O3/C composite
Due to thermal treatment of EG/Fe/C composite carried out at 500 °C in air, amorphous carbon, being an undesired byproduct in the CVD synthesis, was removed from the outer part of composite. Simultaneously, the Fe component of EG/Fe/C composite underwent oxidation to Fe2O3 [21]. The abovementioned hypothesis has been partially confirmed by the EDS analysis. As can be seen from the data included in Table 1, no signal arising from the oxygen has been found for the untreated composite (sample EG/Fe/C). The oxidative treatment of the investigated composite (sample EG/Fe2O3/C) results in appearance of oxygen (1.39 at.%), most likely bounded with Fe as a Fe2O3. Owing to heat treatment, the amorphous carbon being the side product of acetylene carbonization has been removed from the carbon layer of the investigated composite resulting in uncovering the surface of composite.
The similar effect has been previously observed for ozone treated EG/Fe/C [20]. On the other hand, the increased amount of oxygen within the EG/Fe2O3/C composite gathered after its heat treatment can be also explained by the formation of oxygen functionalities on the surface of carbon layer. It is known that thermal treatment of carbon materials realized in air atmosphere favors generation of surface oxygen groups.
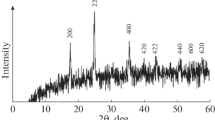
The occurrence of Fe2O3 within the structure of investigated composite is also confirmed by XRD analysis. XRD pattern displayed in Fig. 1 involves diffraction peaks of 0.267, 0.250, 0.183, 0.169, 0.148, and 0.143 nm referred to the diffraction line (104), (110), (204), (116), (214), and (300), respectively. According to the literature data, the abovementioned signals can be largely ascribed to α-Fe2O3 [23, 24]. The regarded pattern also contains signals arising from the carbon phase (denoted as C). Among them, peaks of 0.338 and 0.167 nm indexed as (002) and (004), respectively, seems to be of highest importance because these peaks most likely correspond to nanofibers existing within the outer part of the examined composite [25].

XRD pattern for EG/Fe2O3/C
To acquire an additional information on carbon layer within the EG/Fe2O3/C composite, the SEM analysis was performed. SEM images recorded for EG/Fe2O3/C composite are shown in Fig. 2a–c. From these images, it is clear that the acetylene carbonization over the EG/Fe composite followed by heat treatment at 500 °C in air atmosphere brought about the formation of carbon layer in which carbon fibers are the main component. The SEM images show a characteristic tangle of carbon fibers varying in their length and diameter. Among the carbon fibers shown in Fig. 2b–c, one can also find the fibers of diameter expressed in nanoscale. White points seen at the end of fibers structure likely correspond to the existence of iron particles playing a role of catalyst during the acetylene carbonization. The occurrence of catalyst particles within the outer part of synthesized fibers suggests that the process of fiber growth proceeds through its cracking, followed by its diffusion, and finally precipitation at the opposite part of catalyst [16]. This may be an explanation why the remaining Fe catalyst occupies the outer regions of synthesized carbon fibers.

SEM micrographs for EG/Fe2O3/C
Study of electrochemical properties of EG/Fe2O3/C composite
Electrochemical properties of EG/Fe2O3/C were studied by cyclic voltammetry method using 0.5 M solution of KOH as an electrolyte. Cyclic voltammograms (CVs) recorded in the potential range −1.25 ↔ 0.0 V with scan rate 0.1 mV s−1 are plotted Fig. 3. The CVs clearly indicate that the Fe2O3 component of EG/Fe2O3/C composite is accessible to the electrolyte thereby enabling redox reactions with its participation. During the anodic polarization one can observe current peak a 1 at −0.76 V in response to the previously performed cathodic reduction illustrated by the cathodic peak (c 1 *) appearing at the potential of −1.15 V. The details of peaks positions as well as current charges are given in Table 2. The considered pair of peaks can be ascribed to the redox reactions which proceed according to Eq. 1[19, 24, 26].

Voltammetric curves for EG/Fe2O3/C electrode recorded in 0.5 M KOH within the potential range −1.25 ↔ 0.0 V and with scan rate 0.1 mV s−1
The asymmetric shape of the anodic peak allows us to infer that it most likely has contributions from the oxidation of Fe(II) to Fe(III) and also from oxidation of metallic Fe to Fe(OH)2 (Eq. 2). The obtained results therefore well agree with those reported by Hang et al. [19].
This electrochemical feature well agrees with the results reported by Hang et.al. [19]. A small anodic peak (peak a 2 ) seen in Fig. 3 at −0.94 V for the second and third cycle probably also corresponds to oxidation of metallic Fe to Fe(OH)2. According to some authors, the considered transformation proceeds through the intermediate step in which the generation of intermediate product Fe(OH)a takes place (Eq. 3). Within the subsequent step, Fe(OH)a undergoes oxidation to Fe(OH)2 (peak a 1 ) according to the Eq. 4 [19, 24]
Note that electrochemical behavior of examined composite during cathodic polarization is considerably changed after the first cycle. Cathodic peak (c 1 *) being the only cathodic peak on CV recorded for the first cycle, upon cycling splits into two separated effects c 1 and c 2 (Fig. 3) (Table 2). It cannot be excluded that during the first cycle, the signal associated with the reduction of Fe(III) → Fe(II) overlaps with that corresponded to Fe(II) → Fe transformation. In consequence, on CV recorded for the first cycle appears a broad cathodic peak c 1 *. On cycling, the peaks arising from above mentioned reactions become separated. The reduction of Fe(OH)2 to Fe is depicted as a cathodic wave (c 2 ) (much more visible for the third cycle), whereas the Fe(III) → Fe(II) transformation is represented by the well-defined peak denoted as c 1 . The significant changes in electrochemical features of EG/Fe2O3/C observed after the first cycle can be tentatively explained by the differences in Fe forms which are taking part in the electrochemical processes. The abovementioned hypothesis is partially confirmed by the investigations on electrochemical behavior of graphite intercalation compounds with Fe ions reported by Skowroński and Urbaniak [24].
It is worth to note that the intensity of cathodic peak c 1 gradually decreases on cycling, whereas the intensity of anodic peak a 1 increases (Table 2). Such a feature may indicates that the amount of Fe(OH)2 undergoing electrode reactions significantly enhances on cycling. On the other hand, the increment in intensity of peak a 1 may be also explained by the enhanced efficiency of Fe formation during the preceded cathodic reduction of Fe(OH)2 to metallic Fe (peak c 2 ). We assume that in the first polarization cycle the amount of metallic Fe is lower in comparison to Fe formed during the cathodic polarization of second and third cycles. Owing to this behavior, the higher amount of metallic Fe may undergoes oxidation to Fe(OH)2, which is illustrated by the increase in intensity of peak a 1 . The abovementioned explanations are in consistency with the assumption that peak a1 corresponds to both oxidation of Fe(II) → Fe(III) and Fe → Fe(II) [19].
Much more valuable information on the electrochemical behavior of EG/Fe2O3/C can be gained from the electrochemical investigations, in which the potential range was extended towards the more negative potentials ensuring deep cathodic reduction of investigated composite electrode.
Cyclic voltammograms for EG/Fe2O3/C composite recorded in 0.5 M KOH within the potential range −1.5↔0.0 V with scan rate 0.1 mV s−1 are plotted in Fig. 4. As can be seen, the curves significantly differ compared to CVs recorded within the narrow potential range (Fig. 3). The most pronounced difference is associated with the appearance of coupled redox peaks arising from the redox transitions of Fe ↔ Fe(II). For the first cycle, a broad cathodic peak attributed to Fe generation (c 2 *) is observed at the potential of −1.28 V (see Table 3). After the reversal of polarization, anodic peak (a 2 ) of Fe oxidation to Fe(OH)2 can be seen at around −0.91 V. For the first cycle, the CV also involves pair of redox peaks being attributed to Fe(II) ↔ Fe(III) transformation (c 1 *and a 1 ). It is worth to note that an extension of potential range causes the changes in electrochemical behavior of EG/Fe2O3/C exhibited during the successive scanning. Cathodic peak of Fe formation (c 2 ) has a much more defined shape and is of a 0.13 V shifted towards the less negative potentials compared to the first cycle (peak c 2 *). Simultaneously, the maximum of cathodic peak arising from the reduction of FeOOH to Fe(OH)2 (c 1 ) shifts towards the less negative potentials. However, the location of anodic peaks corresponded to oxidation reactions of Fe/Fe(II) (a 1 ) and Fe(II)/Fe(III) (a 2 ) remains almost unchanged for the second and third cycle but its intensity as well as current charges are changed (Table 3). Additionally, anodic peak associated with the Fe(OH)2 → FeOOH reaction (a 1 ) becoming more irregular and asymmetric during the second and third cycle. Such a behavior allows us to infer that it most likely has contributions from the oxidation of Fe(II) to Fe(III) and also transformation of intermediate Fe(OH)a into Fe(OH)2 (Eqs. 3 and 4) [19, 24]. This hypothesis is supported by the discrepancy in current charges of coupled redox peaks a 1 /c 1 (Table 3). For ideally reversible system, the regarded relation should be similar. For the EG/Fe2O3/C composite, the current charge registered for peak c 1 is considerably lower compared to charge for peak a 1 .

Voltammetric curves for EG/Fe2O3/C electrode recorded in 0.5 M KOH within the potential range −1.5 ↔ 0.0 V and with scan rate 0.1 mV s−1
Some valuable information on the chemical composition of the investigated composite due to its electrochemical treatment are acquired from the EDS analysis. After electrochemical investigation performed through 3 cycles in potential range −1.5 ↔ 0.0 V with scan rate 0.1 mV s−1 in 0.5 M KOH, the sample EG/Fe2O3/C was subjected to EDS analysis (see Table 1). The deep cathodic reduction of EG/Fe2O3/C performed under potentials of rapid hydrogen evolution brought about an increase in amount of Fe and O instead of C component. Due to interactions between intensively liberating hydrogen and EG/Fe2O3/C, the C layer becomes more porous, thereby enabling the penetration of electrolyte molecules to active Fe2O3. It cannot be excluded that interactions of active hydrogen with the EG/Fe2O3/C composite modify its chemical composition, most likely by incorporation of K ions. The formation of such species within the considered composite may be partially proved by EDS analysis revealing the appearance of K atoms as well as increased amount of oxygen. As an additional result of cathodic treatment of EG/Fe2O3/C, the Fe/O ratio decreased. Such a behavior may indicate the partial transformation within the iron oxide (see Table 1).
The strength and results of interactions of active hydrogen with the EG have been investigated in our previous work [27].
To understand the influence of scan rate on electrochemical features of EG/Fe2O3/C, the additional measurements under two different scan rates were undertaken. Figure 5 displays CVs for EG/Fe2O3/C composite recorded in 0.5 M KOH in potential range −1.5 ↔ 0.0 V using scan rates of 1.0 and 10 mV s−1. Taking into account the fact that during the previous measurements within the initial cycles, the investigated composite exhibited significant electrochemical instability demonstrated by some potential displacement, for the present investigations, only the third cycles were selected. To facilitate the comparison, curve for the investigated composite recorded during the third cycle with scanning equal to 0.1 mV s−1 was added to the regarded figure. As it was expected, the increased scan rate caused considerable enhancement in current charges which is accompanied by the significant shift of peaks location. The scale of these changes strongly depends on the type of regarded reaction. The most pronounced increase in intensity with simultaneous significant potential shift is noted at sweep rate of 10 mV s−1 for the pair of peaks ascribed to the redox reactions Fe(II) ↔ Fe(III) (c 1 and a 1 ). Such a behavior allows the assumption that under such conditions of voltammetric investigations, the compromise between the scan rate and diffusion of electrolyte through the carbon layer of EG/Fe2O3/C composite is reached.

Comparison of voltammetric curves for EG/Fe2O3/C recorded during the third cycles in 0.5 M KOH within the potential range −1.5 ↔ 0.0 V with scan rates 0.1, 1.0, and 10 mV s−1
The intensity as well as current charge of peaks associated with redox reactions of Fe ↔ Fe(II) (c 2 and a 2 ) for the scan rate of 1.0 mV s−1 are very closed to that noted for scanning equal to 0.1 mV s−1, simultaneously being considerable lower compared to that recorded at scan rate 10 mV/s. It indicates that during the measurements performed with scan rate of 0.1 and 1.0 mV s−1, the comparable amounts of Fe reactant are taking part in the redox reactions.
The most important feature of the EG/Fe2O3/C caused by its deep potentiodynamic reduction performed with scan rates 1.0 and 10 mV s−1 seems to be the appearance of anodic current peak a 3 . Its potential location (−1.03 and −0.95 V for the scan rate 1.0 and 10 mV s−1, respectively) justify the assumption that it likely arises from the oxidation of hydrogen. The appearance of the regarded peak suggests that during the preceded cathodic polarization, the sorption of hydrogen onto EG/Fe2O3/C takes place. The presence of a similar anodic peak is reported by Hang et al [19]. As opposed to our investigations, peak of hydrogen oxidation appears on CVs when the KOH electrolyte was modified by addition of K2S and the FeS was inserted into the electrode material (Fe/C) [19]. In our researches, no additives were used to modify both KOH electrolyte and electrode material. At this stage of investigations, it is difficult to give entire explanation why the mentioned reaction of hydrogen oxidation (peak a 3 ) can be observed only under increased scan rates. One of the possible explanations is that during the slower scanning, the adsorbed hydrogen is released from the EG/Fe2O3/C after the reversal of polarization at −1.5 V.
Conclusion
The obtained results disclosed that the electrochemical properties of EG/Fe2O3/C composite exhibits high electrochemical activity towards the redox reactions of Fe ions as evaluated by cyclic voltammetry in alkaline electrolyte. As evidenced by the SEM observations, the outer part of EG/Fe2O3/C is mainly composed of carbon fibers of different length and diameter including carbon nanofibers. The XRD and EDS analysis revealed that due to thermal treatment at 500 °C in air, the Fe component of the investigated composite exists in form of Fe2O3. Electrochemical features of the examined EG/Fe2O3/C was found to be strongly depended on the potential range and scan rate of voltammetric measurements. The dominate electrode effect of the investigations carried out in the potential range −1.25 ↔ 0.0 V appears to be an occurrence of coupled redox peaks corresponded to Fe(OH)2 ↔ FeOOH transformation. The extension of potential range to −1.5 ↔ 0.0 V brought about an appearance of additional pair of peaks assigned to redox reactions of Fe ↔ Fe(OH)2. This effect indicates that owing to deep cathodic reduction of EG/Fe2O3/C, it is possible to separate the signals arising from the redox reactions of Fe ↔ Fe(II) and Fe(II) ↔ Fe(III). The electrochemical measurements realized under increased scan rate also revealed an appearance of anodic current peak attributed to oxidation reaction of hydrogen. It is worth to note that abovementioned effect was achieved without any modification of electrolyte or electrode material.
To acquire the comprehensive information on the composition of carbon component of EG/Fe2O3/C, the structural investigations are planned. For better understanding the mechanism of electrochemical reactions especially between active hydrogen and the surface of composite, additional investigations involving detailed analysis of changes within the properties and composition of EG/Fe2O3/C created during its electrochemical investigations are necessary.
References
Chung DDL (1987) Review J Mater Sci 22:4190
Furdin G (1998) Fuel 77:479
Tryba B, Kaleńczuk RJ, Kang F, Inagaki M, Morawski AW (2000) Mol Cryst Liq Cryst 340:113
Toyoda M, Inagaki M (2000) Carbon 38:199
Vieira F, Cisneros I, Sansiviero MTC, Miranda AM, Rosa NG, Lima UB, Mohallem NDS (2006) J Phys Chem Solids 67:1208
Bhattacharya A, Hazra A, Chatterjee S, Sen P, Laha S, Basumallicket I (2004) J Power Sources 136:208
Mitra S, Lokesh KS, Sampath S (2008) J Power Sources 185:1544
Ramesh P, Sivakumar P, Sampath S (2003) Electroanalysis 15:1850
Skowroński JM, Krawczyk P (2004) J Solid State Electrochem 8:442
Bielicka-Daszkiewicz K, Krawczyk P, Nowicka K (2012) Electrochim Acta 80:22
Xiao P, Xiao M, Gong K (2001) Polymer 42:4813
Zheng W, Wong S-C (2003) Compos Sci Technol 63:225
Afanasov IM, Shornikova ON, Avdeev VV, Lebedev OI, van Tendeloo G, Matveev AT (2009) Carbon 47:513
Zhao J, Guo Q, Shi J, Liu L, Jia J, Liu Y, Wang H (2009) Carbon 47:1747
Randall L, Wal V, Hall LJ (2003) Carbon 41:659
Deck CP, Vecchio K (2005) Carbon 43:2608
Huang K-C, Chou K-S (2007) Electrochem Commun 9:1907
Souza CAC, Carlos IA, Lopes M, Finazzi GA, de Almeida MRH (2004) J Power Sources 132:288
Hang BT, Watanabe T, Egashira M, Watanabe I, Okada S, Yamaki J (2006) J Power Sources 155:461
Krawczyk P (2013) Carbon 65:374
Zhygotsky AG (2000) J Therm Anal Calorim 62:575
Krawczyk P, Skowroński JM (2010) J Appl Electrochem 40:91
Hassan MF, Rahman MM, Guo ZP, Chen ZX, Liu HK (2010) Electrochem Acta 55:5006
Urbaniak J, Skowroński JM, Olejnik B (2010) J Solid State Electrochem 14:1629
Endo M, Kim YA, Takeda T, Hong SH, Matusita T, Hayashi T, Dresselhaus MS (2001) Carbon 39:2003
Periasamy P, Ramesh Babu B, Venkatakrishna Iyer S (1996) J Power Sources 58:35
Krawczyk P, Skowroński JM (2012) Electrochim Acta 79:202
Acknowledgements
Financial support from National Science Centre of Poland (grant No. N507 479337) is acknowledged.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Krawczyk, P., Rozmanowski, T. Preparation and electrochemical properties of EG/Fe2O3/C composite. Ionics 21, 59–66 (2015). https://doi.org/10.1007/s11581-014-1154-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11581-014-1154-8