
Abstract
The phylogeny of the Psathyrellaceae has received much attention recently. Despite repeated efforts for inferring a stable phylogeny that can serve as a basis for reclassification of the Psathyrellaceae, extensive taxonomic rearrangements have been withheld by several factors; among others, inadequate taxon sampling in several clades and low support values for critical relationships. In this paper, we present a well-resolved, robust phylogeny and morphological circumscriptions for 14 clades of the Psathyrellaceae. Sequence data from a matrix of four nuclear genes (approximately 4,700 characters, including recoded indels) and various phylogenetic methods were used to infer relationships. Unexpected relationships and other morphologically informed phylogenetic hypotheses have been tested by constraint analyses. Nearly fully resolved consensus trees have been obtained, with strong support for most of the large clades and several early evolutionary events. We identified poorly sampled regions of the phylogeny and discuss potential additions and extensions of the phylogeny for future sampling. Prospects for and potential pitfalls of a future reclassification are discussed in detail.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The mushroom family Psathyrellaceae Vilgalys, Moncalvo & Redhead contains small to large, dark-spored agarics, which are generally considered difficult to identify to species. Many of the taxa are deliquescent, well known for their ability to digest themselves by means of autodigestive chitinases (Kües 2000). These, known as "inky caps" or Coprinus s.l., include species used as model organisms in various fields, spanning fungal ontogeny, breeding biology, developmental biology and genomics (Kemp 1975; Kües 2000; Stajich et al. 2010). The type genus of the family, Psathyrella (Fr.) Quél. by definition contains the non-autodigesting species, mainly decomposers of leaf-litter or wood, more rarely living on dung, or parasitizing other fungi (Kits van Waveren 1985; Singer 1986; Smith 1972). Some of the species are well-known edible mushrooms, such as Ps. aff. hymenocephala, which is used as a spice in Haiti (Nieves–Rivera 2001), or C. micaceus and C. atramentarius, which are sometimes collected for the table, although the latter is also known to be toxic when consumed with alcohol.
Traditionally, the family (formerly treated under the name Coprinaceae) included two large genera, Coprinus Pers. (Fig. 1a–f) and Psathyrella (Fig. 1g–l), although several other genera have also been included. For instance, Pseudocoprinus Kühner was introduced to accomodate partially deliquescent species such as Coprinellus disseminatus (Fig. 1e), which is now classified into Coprinellus P. Karst. (Kühner 1928). The genera Ozonium Link and Hormographiella Guarro & Gené were established to accommodate the conidial anamorphs of certain species now classified in Coprinellus. These anamorph taxa, especially Hormographiella verticillata, have been reported as opportunistic human and animal pathogens in several case reports of severe or even fatal infections (Cáceres et al. 2006; Rampazzo et al. 2009; Verweij et al. 1997). The name Coprinaceae was used for many decades (Kühner and Romagnesi 1953; Moser 1983; Singer 1986), but following early phylogenetic studies (Hopple and Vilgalys 1994, 1999) Coprinus was split into four genera: Coprinus s.s., Coprinellus (Fig. 1c–e), Coprinopsis P. Karst. (Fig. 1a, f) and Parasola Redhead, Vilgalys and Hopple (Fig. 1b), with Coprinus s.s. belonging to the Agaricaceae (Redhead et al. 2001). A consequence of the transfer of Coprinus s.s. to the Agaricaceae was that the name Coprinaceae became a synonym of Agaricaceae, necessitating the establishment of the new family name Psathyrellaceae.

Examples of different groups of Psathyrellaceae. a Coprinopsis strossmayeri, showing rich fibrillose patchy veil coverage on the cap, b Parasola leiocephala, c Coprinellus sp., a Coprinellus species lacking all types of veil, d Coprinellus micaceus, with a thin layer of granular veil remnants on the cap, e Coprinellus disseminatus, f Partially digested, mature pileus of Coprinopsis stangliana. Autodigestion starts from pileus edge and progresses concentrically in parallel with spore maturation. g Young specimens of Psathyrella gordonii with rich fibrillose veil coverage, h Psathyrella fibrillosa, i Lacrymaria vellutina, j Psathyrella gracilis, the type species of the genus, k Psathyrella ammophila, l Psathyrella panaeoloides, a species with very scanty veil, m Spores of Coprinopsis lagopus, n Cheilocystidia of Psathyrella prona, o Cheilocystidia of Parasola plicatilis, p Spores of Coprinopsis calospora (Holotype), q Cheilocystidia of Ps. lutensis, r Lageniform cystidia from the stipe surface of Coprinellus pusillulus, s Microstructure of veil elements of Coprinopsis friesii wth thick walls and diverticulate, coralloid appearance
The main morphological features used in the reclassification of coprinoid fungi were the structure of the pileipellis (cap cuticle), the presence or absence of the universal veil and its microstructure. Coprinopsis was defined as being deliquescent, having a cuticular pileipellis, always having a veil of various structures (globose, hyphal, diverticulate, etc.) and devoid of caulocystidia. This genus contains several well-known taxa, such as C. cinerea, C. lagopus or C. atramentaria. Species of Coprinellus, on the other hand, possess hymeniform (cellular) pileipellis, and veil, if present, consists of globose cells. Several Coprinellus species have short, hair-like structures, pileocystidia on the cap surface and caulocystidia-like structures on the stipes (Fig. 1r), whereas such structures are never observed in Coprinopsis. Parasola is the only genus consistently lacking all types of velar structures and having a strongly grooved, parasol-like, membranous pileus. Deliquescence in this genus is incomplete or lacking. According to Redhead et al. (2001), pleurocystidia are always present, but this view has been challenged by subsequent publications (Nagy et al. 2009; Padamsee et al. 2008). An additional species, Coprinus patuoillardii (= C. cordisporus), has not been placed in any of the above-mentioned genera, since it was grouped together with psathyrelloid species.
Although Psathyrella was also found to be paraphyletic, it has not been reclassified, awaiting more species and more loci to better understand their phylogeny and morphological evolution. Sampling of additional Psathyrella taxa revealed that the three coprinoid genera were not as uniform as formerly thought; they include non-deliquescent psathyrelloid taxa as well. Accordingly, Larsson and Örstadius (2008) placed Psathyrella conopilus in Parasola, and Psathyrella marcescibilis and P. pannucioides in Coprinopsis. Several studies attempted to decipher patterns of the phylogeny of Psathyrella taxa, but the resolving power of the analyses has been limited by gene or taxon sampling. Individual analyses uncovered single or a few clades, but a well-supported multigene phylogeny of the Psathyrellaceae phylogeny is still lacking.
In this study, we build on formerly published phylogenies (Hopple and Vilgalys 1999; Larsson and Örstadius 2008; Nagy et al. 2010, 2011; Padamsee et al. 2008; Vasutova et al. 2008; Walther et al. 2005), and extend those with additional taxa and gene sequences (beta-tubulin) to achieve a comprehensive view of Psathyrellaceae phylogeny. We conducted phylogenetic analyses based on four nuclear genes, covering most of the morphologically established groups of the family in order to recover a robust, well-supported phylogeny of the family. We analyzed 141 specimens using two different Bayesian approaches and models, as well as ML and MP bootstrapping. We also identified morphological characters that support the phylogeny, with the aim to provide morphological key characters that correspond to recovered clade structure, and discuss aspects of a future reclassification of the Psathyrellaceae.
Materials and methods
Taxon sampling
We extended previous sampling of taxa (98 specimens, Nagy et al. 2011) by several species of the Psathyrellaceae from Europe (134 ingroup taxa). Since the most severe drawback of the current classification of the family is the paraphyly of the genus Psathyrella and the ambiguous positions of psathyrelloid clades, our objective was to recover as many lineages of the Psathyrellaceae as possible. To this end, the design of our taxon sampling took advantage of formerly published phylogenies (Larsson and Örstadius 2008; Nagy et al. 2010, 2011; Padamsee et al. 2008; Vasutova et al. 2008), extensive type studies and a database of > 700 internal transcribed spacer (ITS) sequences of Psathyrellaceae. Initially, we applied rough morphological definitions for the larger clades recovered in the above mentioned studies, and applied a denser sampling in those clades. To identify hitherto unknown clades, we searched the literature for species not fitting in any of the formerly inferred larger clades, and inferred pilot phylogenies based on only one or two genes (ITS, large subunit (LSU) or both) with many hundred sequences. Taxa appearing as new clades were included in the subsequent four-loci analyses. Genbank sequences from our previous studies (Nagy et al. 2009, 2010, 2011) and of Ps. pennata, Ps. spintrigeroides, Ps. romagnesii, Ps. sphaerocystis, Ps. cotonea, Ps. stercoraria, Ps. larga, and Ps. gossypina (Accession numbers in order: DQ389710, DQ389696, DQ389716, DQ389708, AM712283, DQ389670, DQ389694, AM712294, Larsson and Örstadius 2008; Vasutova et al. 2008) were included in the study. The complete list of specimens used in this study, their origins, Accession Numbers and identifiers are listed in Supplementary Table 1.
The Bolbitiaceae have been shown to be closely related to the Psathyrellaceae (Matheny et al. 2006; Moncalvo et al. 2002), so we used representatives of this family plus Mythicomyces as outgroup in our analyses. GenBank sequences of Cystoagaricus strobilomyces have been included in the analyses, since evidence from former studies suggested that this belongs to the Psathyrellaceae (Vellinga 2004). Singer (1986) placed the genus Macrometrula in the Psathyrellaceae; therefore, we examined the type of this species, but found it to be a very badly preserved representative of a species closer to the Agaricaceae or the Pluteaceae.
DNA extraction, PCR and sequencing
Genomic DNA was extracted from 2–20 mg of dried herbarium specimens by means of the DNeasy Plant Mini kit (Qiagen), following the manufacturer’s instructions. PCR amplification targeted four loci: the nrLSU (ca. 1,500 bp), ITS (ca. 700 bp), tef1 (ca. 1,200 bp) and btub (ca. 500 bp) genes, and employed the primer combinations LROR/LR7, ITS1/ITS4, 983 F/2218R and B12R_psa/B36F_psa for the nrLSU, ITS, tef1 and btub genes, respectively (for primer sequences, see Nagy et al 2011). Amplification followed standard protocols (White et al. 1990). PCR clean-up and sequencing was performed by LGC Genomics (Berlin) using the same primers as above, except for the tef1 gene, for which an internal reverse primer (1567R) was used. Individual readings were assembled to contigs using the PreGap and Gap4 programs of the Staden package (Staden et al. 2000).
Alignments, congruence tests and modeling sequence evolution
Sequences of nrLSU, tef1 and btub were aligned by ClustalX (Thompson et al. 2002), followed by minor manual adjustments. ITS sequences were aligned by Probalign, a probabilistic alignment algorithm found to reduce the proportion of incorrectly inferred homologies in sequences with numerous indel events (Roshan and Livesay 2006). Probalign was launched with default parameters. Indels in the ITS region were recoded as a partition of binary characters by using the simple indel coding algorithm (Simmons and Ochoterena 2000), as implemented in FastGap 1.2 (Borchsenius 2007). This approach treats indels as one evolutionary event.
ITS alignments are frequently criticized for alignment errors, especially in data sets spanning significant taxonomic ranges. In order to examine the sensitivity of our conclusions to potential phylogenetic noise originating from incorrectly inferred homologies in the ITS1 and ITS2 regions, we performed phylogenetic analyses with all the aligned regions, as well as with ambiguously aligned regions excluded. To examine how different treatments of gapped regions affect the topology and support values of trees, we identified ambiguously aligned regions by GBlocks 1.0 by using a “less stringent” set of parameters, allowing up to half of the sequences to contain gaps in the final alignment (Castresana 2000). This trimmed ITS alignment was subjected to the same phylogenetic analyses as the complete one.
To check for topological congruence between single-locus alignments, we performed maximum likelihood (ML) bootstrapping in 100 replicates using the parallel version of RAxML 7.0.4 (Stamatakis 2006). Mutually exclusive, strongly supported clades (MLBS ≥ 70 %) were considered indicative of significant topological incongruence. Based on our former results obtained on a more restricted set of specimens and formal model testing (Nagy et al. 2011), the GTR + G model of sequence evolution have been used for all genes. Because gamma approximation accounts for the same phenomenon as P-invar and it has been raised that using both can reduce the identifiability of parameters in the analyses (Stamatakis 2006), we omitted the invariant sites model from the analyses. For the concatenated data set, the following partitioned model was used (Nagy et al. 2011): the ITS1, 5.8S, ITS2 and nrLSU regions were modeled by a GTR + G model, while for the two protein coding genes, two site-specific rate models were invoked by dividing the tef1 and btub genes into 1st, 2nd, and 3rd codon positions, and estimating a GTR + G model for each codon position in each gene. This resulted in ten partitions of nucleic acid supplemented with recoded indel data as the 11th partition. Model parameters were unlinked between partitions in the Bayesian analyses. The concatenated alignment, as well as the Bayesian consensus tree, are available in TreeBase (Accession Number: 11724).
Constraint analyses
Several morphologically informed phylogenetic hypotheses were tested by constraint analyses (summarized in Table 1). Constraint trees were designed either to test unexpected relationships, nodes contradicting previously published phylogenies, or the affinities of morphologically similar taxa. Mesquite 2.74 (Maddison and Maddison 2009) was used to create constraint trees, choosing the most conservative way of imposing the constraints. This often required several clades to be collapsed to polytomy, which has later been resolved according to the ML solution around the particular node. RAxML was used to compute the unconstrained trees, to find the ML solution for polytomous nodes of constrained trees and to calculate single-site likelihoods. Trees were inferred in ten replicates in all cases. Single-site likelihoods were then imported into CONSEL (Shimodaira and Hasewaga 2001) to calculate confidence intervals on the trees. CONSEL was run with default settings, and priority was given to the results of the approximately unbiased test (Shimodaira and Hasewaga 2001). P values ≤0.05 were considered significant.
MP and ML bootstrap analyses
We estimated nodal support for the trees by maximum parsimony (MP) and ML bootstrapping. Parsimony analyses have been performed using PAUP 4.0b10 (Swofford 2002). Initial, exploratory MP searches have been performed in 1,000 replicates (TBR branch swapping, MULTREES = YES), keeping only three of the shortest trees per replicate. Subsequently, more thorough branch swapping has been performed on the trees saved during the first cycle. Bootstrapping was initiated in 1,000 replicates with TBR branch swapping, stepwise sequence addition and ten replicates of branch swapping per bootstrap replicate. A 50 % bootstrap consensus tree has been computed in PAUP.
For ML bootstrapping, we used the parallel version of PhyML 3.0 (Guindon and Gascuel 2003). One thousand bootstrap replicates were analyzed under the GTR + G model of evolution. Rate heterogeneity was modeled by a gamma distribution discretized using four categories. Branch swapping was set to SPR.
Bayesian analyses
We inferred trees by using two different Bayesian approaches, under two different partitioned models found optimal for a smaller version of this data set in our former study (Nagy et al. 2011). The first, outlined above, was used in MrBayes 3.1.2 (Altekar et al. 2004), while a mixture model approach using three GTR + G matrices for each of the sites of the partitions described above was implemented by using BayesPhylogenies 1.0 (Pagel and Meade 2006). The latter was found to improve likelihood values significantly (based on Bayes Factors), and to converge to more realistic branch length estimates than MrBayes (Nagy et al. 2011). Since topological differences between the two types of analyses have not yet been examined in detail, we compare them in this paper. Sampling frequency was set to 1,000 for both programs. The indel matrix was appended to the end of the concatenated alignment in both Bayesian analyses, and a correction for constant characters not included in the matrix was imposed ("coding = variable" in MrBayes and "cb = noconst" in BayesPhylogenies). Indel data were modeled by a two-parameter Markov model. MrBayes was launched with four incrementally heated chains and two replicates, while BayesPhylogenies was run with one chain, in three replicates.
Convergence of likelihood values was checked in Tracer (Rambaut and Drummond 2008). Since topological convergence was proved difficult to achieve and preliminary Bayesian runs failed to converge on satisfyingly stable split posteriors [as deduced from the average standard deviation of split frequencies (in MrBayes) and the cumulative and compare functions of AWTY (Wilgenbusch et al. 2004)], we performed long runs, where the Markov chains were run for 100 million generations in both programs. Burn-in values were determined by inspecting likelihood and topological convergence.
Trees sampled after convergence were used to compute 50 % Majority Rule consensus trees. This was done by combining trees sampled from paired or replicated MCMC runs. Runs that failed to converge to the same posterior as the others were excluded from consensus analyses. On the basis of consensus trees, we compared Bayesian posterior probabilities and branch length estimates obtained from the MrBayes and the BayesPhylogenies runs. The notations BPPb and BPPm hereafter refer to the BayesPhylogenies and MrBayes posterior probabilities, respectively.
Results
Data sets and congruence tests
We sampled and analyzed four nuclear loci for 140 specimens of the Psathyrellaceae and outgroup taxa. Of these, Bolbitius vitellinus, Conocybe lactea, Agrocybe preacox, Ps. melanthina, Ps. pennata, Ps. spintrigeroides, Ps. romagnesii, Ps. sphaerocystis, Ps. cotonea, Ps. stercoraria, Ps. larga, Ps. gossypina and Cystoagaricus strobilomyces were represented only by the ITS and LSU regions, while for Coprinopsis friesii only LSU and btub fragments amplified successfully. All other species were represented by all four, or in the case of ten specimens, by three loci. The final concatenated matrix contained 4,717 characters in total. Of this, 3,952 were nucleic acid and 765 were binary indel characters obtained by recoding gaps in the ITS alignment. The length of the individual partitions was 730 for the ITS1, 172 for the 5.8S rRNA, 712 for the ITS2, 1,297 for the nrLSU, 664 for the tef1 and 375 for the btub gene. Of these, the number of variable positions was 528 for the ITS1, 27 for the 5.8S rRNA, 469 for the ITS2, 484 for the nrLSU, 435 for the tef1and 189 for beta-tubulin. There were two intron regions in the tef1and the btub genes each, which were deleted from the alignments, due to their excessive variability. To examine the effects of excluding highly variable regions of the ITS locus from the alignment, we identified “ambiguously aligned” regions by GBlocks, which resulted in a final alignment length of 318 bp.
We found that incongruence between the single locus alignments was caused by two specimens, Coprinellus sp. (SZMC-NL-0978) and Psathyrella aff. lutensis (SZMC-NL-4301). These were deleted from the alignments, since they appeared in different positions in the single-gene ML trees and caused major discrepancies both in combined Bayesian analyses and congruence tests. The results of the constraint analyses are summarized in Table 1.
Bayesian analysis
Likelihood values converged very rapidly to stationarity, usually within the first 2 million generations. The three replicated runs of BayesPhylogenies terminated after 19 million generations, but converged to roughly the same likelihood values (-lnL = 51431–51451). A Bayes Factor test indicated small differences between the three runs (logBF = 2.13–8.44). On the other hand, paired MrBayes runs converged to more different likelihood values (-lnL = 52011–52049, logBF = 16.59), of which run 1 produced insufficient convergence, so we excluded it from the calculation of consensus trees.
Consensus trees and support values
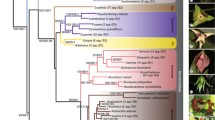
The different analyses yielded the same results with regard to the gross topology of the trees. Based on analyses of convergence, we established the burn-in as 70 million and 10 million generations in MrBayes and BayesPhylogenies, respectively. The 50 % majority rule consensus trees have a high resolution, with most larger clades receiving strong support values (Fig. 2). Out of the four different analyses, the two Bayesian trees are topologically closest to each other, conflicting only in the placement of some minor clades. The parsimony and ML trees also conflicted with the Bayesian ones in deeper internal branches. For instance, the Lacrymaria clade was placed in different positions in the analyses. In the MP consensus and the ML trees, it was placed sister to Coprinopsis (MPB: 93 %, MLB: 100 %), whereas both Bayesian analyses placed it close to Coprinellus (BPPm: 1.00, BPPb: 1.00) (see Fig. 2).


50 % Bayesian Majority Rule consensus tree. Support values above branches are given as: Maximum Parsimony bootstrap / Maximum Likelihood bootstrap / Bayesian Posterior probability from MrBayes / Bayesian Posterior probability from BayesPhylogenies. A dash (−) represents that the clade was either not present on the tree, or the support values were < 50 %/0.50. An asterisk (*) denoted 100 % or 1.00 support for bootstrap and Bayesian analyses, respectively. Support values for specimens of the same species have been removed. Coprinoid species, as defined by Nagy et al. (2011) (complete or partial deliquescence, bimorphic basidia, voluminous cystidia, pseudoparaphyses and plicate pileus), are denoted by checked circles, while non-deliquescent psathyrelloid taxa are denoted by striped circles. Outgroup taxa are identified by the Accession numbers of their ITS sequences
Trees inferred from the GBlocks-curated alignment, i.e. from which gapped regions of the ITS locus were deleted (Suppl. Fig. 1), were congruent with analyses from the complete alignment, with the exception of some incongruent nodes around Coprinellus. The candolleana clade was split to two subclades, one (containing Ps. candolleana, Ps. leucotephra, Ps. badiophylla) was nested within Coprinellus, while the remaining species, Ps. typhae, together with the calcarea clade, were inferred as a tritomy, sister to Coprinellus. Further, as a result of GBlocks-curation, the number of unresolved nodes in the clade of Coprinellus hiascens—C. congregatus increased from zero to four.
Based on our trees and former studies, we distinguish 14 major clades within the Psathyrellaceae (Fig. 2): /spadiceogrisea, /fusca, /Coprinus patouillardii, /gordonii, /cotonea, /calcarea, /Coprinellus, /candolleana, /gracilis, /Cystoagaricus, /Lacrymaria, /Coprinopsis, /pseudonivea, and /Parasola. All of these received significant support values from at least three analyses, except for the fusca (−/−/.59/.80) and Coprinellus (−/−/.91/1.00) clades. These are discussed in greater detail in the next sections.
Discussion
Relationships between early branching clades
There is persistent contradiction between different published papers and different analyses with regard to the topology at the basal nodes of the phylogeny. This most severely affects the position of the Lacrymaria clade, the genera Coprinopsis and Parasola, but also affects the gracilis clade and Coprinellus. Paradoxically, posterior probabilities can be high for alternative placements of a clade, even with extensive data sets, as seen in the Lacrymaria clade. Studies utilizing only one to two genes usually failed to find significant support for the placement of this clade, whereas multilocus analyses provided contradicting (Larsson and Örstadius 2008; Padamsee et al. 2008; Vasutova et al. 2008), often strongly supported, results (Nagy et al. 2009, 2010). Its position varied from being the sister group to the rest of the Psathyrellaceae, to the sister group of Coprinopsis or Coprinellus plus the other psathyrelloid clades. Based on three genes, we formerly sustained the latter placement, although with moderate support (Nagy et al. 2010). Sensitivity of topologies to model selection has also been examined on the basis of a more restricted taxon sampling with four genes (Nagy et al. 2011). Inference under some of the models of sequence evolution (JC69 with and without partitioning, GTR + G without partitioning, Site-Specific Rates /SSR/ model, codon-based and mixture models) placed the Lacrymaria clade sister to all other Psathyrellaceae with significant support (BPP: 1.00 in all cases), whereas the minority of tested models (JC69 + G without partitions, GTR + G partitioned) produced a relationship congruent with the tree shown in this study (BPP: 0.64–0.82). We observed no correlation between taxon sampling and the position of this clade. The prevalence of trees with the Lacrymaria clade as the most basal one prompts us to regard this relationship as more probable. Consistent with this, constraint analysis cannot reject the possibility that /Lacrymaria is sister to the rest of the Psathyrellaceae (P = 0.09, Table 1).
A counterexample of the Lacrymaria clade is the Cystoagaricus clade, which seems to have a very stable position in the tree. It has been inferred in its current placement in several former studies as well (Larsson and Örstadius 2008; Nagy et al. 2011; Padamsee et al. 2008; Vasutova et al. 2008), where it has been called Ps. larga (Larsson and Örstadius 2008; Padamsee et al. 2008), Ps. delineata, Cystoagaricus, (Padamsee et al. 2008) or Ps. gossypina + Ps. delineata (Vasutova et al. 2008) clade. In all previous studies (except for Nagy et al. 2011), the /Cystoagaricus clade was split to smaller, non-monophyletic groups.
The parasola clade has been inferred as basal in the Psathyrellaceae, although in previous studies, it was repeatedly grouped together with Coprinopsis (e.g. Hopple and Vilgalys 1999; Nagy et al. 2010, 2011; Padamsee et al. 2008). In fact, this relationship would seem probable, since both clades contain primarily deliquescent species, but constraint analyses rejected such a relationship significantly at p ≤ 0.00001.
There are two major clades in which the branching order has been somewhat uncertain: Coprinellus and the fusca—Coprinus patouillardii clades. The monophyly of Coprinellus has been questioned in a former study (Nagy et al. 2010), where the two large clades therein have been inferred as non-monophyletic, placed along a completely unbalanced backbone of the tree. However, constraint analyses showed that their monophyly is plausible on the basis of the approximately unbiased test. The other complex in which it has been difficult to resolve the internal branching order with confidence is the clade formed by the /Coprinus patouillardii, /cotonea and /fusca. In addition, finding morphological definitions for these clades is difficult, which suggests that these taxa might have experienced rapid morphological diversification that erased the historical signature of their relationships.
Phylogenetic relationships and morphological traits
In this study, we extended sampling of psathyrelloid taxa substantially, with the aim to recover all lineages of the family. This untangled several clades that formerly proved hard to define, because of sparse sampling and the lack of key species from the phylogeny. However, it is possible that certain lineages are still missing from the phylogeny, which should be examined by even more sampling of taxa from morphologically heterogeneous and/or tropical groups. In the following paragraphs, we discuss the 14 major clades and their morphological characteristics.
In the spadiceogrisea clade (MPB: 50 %, MLB: 72 %, BPPm: 1.00, BPPb: 1.00, Fig. 1k) the basidiomes are fairly large (> 3 cm), non-deliquescent with medium-sized, ellipsoid-subphaseoliform spores (7–9 μm), and fibrillose, scanty veil that is visible only on young specimens. The gill edge is lined mainly with poorly developed globose-sphaeropedunculate cells (paracystidia), whereas true, utriform cheilocystidia are very scarce. The presence of this monophyletic assemblage, corresponding to subsection Spadiceogriseae of Kits van Waveren (1985) has already been noted by Vasutová et al. (2008) and Larsson and Örstadius (2008), independently of each other.
The calcarea clade (MPB: 70 %, MLB: 67 %, BPPm: 1.00, BPPb: 1.00): the ML tree and the MP consensus tree supports the placement of this clade sister to the /candolleana, although with very weak support. Although these species are morphologically most similar to Ps. prona, so that Ps. calcarea has been treated as a variety of the latter (as Ps. prona var utriformis), trees with these three species on one clade were significantly rejected by the approximately unbiased test (Table 1). Traditional taxonomic treatments considered these species closely related to representatives of the gracilis clade (Kits van Waveren 1985; Larsson and Örstadius 2008; Smith 1972). Although not forming a monophyletic assemblage, these two clades also fall in the vicinity of each other in the Bayesian analyses. In support of their relatedness are several former studies (Larsson and Örstadius 2008; Nagy et al 2010; Padamsee et al. 2008), where the calcarea clade (≈ section Atomatae), the gracilis clade (≈ section Psathyrella) and Ps. bipellis (=Ps. odorata, section Bipelles) showed high affinity to each other or formed a monophyletic group, although support for this relationship was lacking in all cases. However, those studies sampled more species, such as Ps. potteri, Ps. prona, Ps. effibulata, Ps. orbitarum etc. Representatives of these clades/sections share a similar habit, ellipsoid spores longer than 10 μm and scanty veil. Therefore, we raise the possibility that they actually are monophyletic, but more data and denser sampling is necessary to substantiate that.
The fusca clade (MPB: -, MLB: -, BPPm: 0.59, BPPb: 0.80, Fig. 1h, l) constitutes a huge heterogeneous assemblage, regarding both morphology and traditional taxonomy. Therefore, it is difficult to provide an unequivocal morphological definition. The spores are shorter than 10 μm in most species, and the cheilocystidia are mostly abundant along the gill edge. All sampled Psathyrella species with lageniform and/or acute fusiform cystidia and spores shorter than 10 μm (except for Ps. fagetophila) are nested within this clade. These traits discriminate this clade from the spadiceogriseae clade, in which the spores are also shorter than 10 μm, but the cheilocystidia of those species are very scarce and utriform when present. Species of other clades have longer spores or they lack pleurocystidia, or the cystidia are distinctly thick-walled (> 1 μm). Species of subsection Lutenses and section Pennatae of Kits van Waveren (1985) are included in this clade, as well as Ps. sphaerocystis, which is characterized by a granular veil. Kits van Waveren (1985) placed this species in section Cystopsathyra of subgenus Psathyra. It seems that psathyrelloid species with granular veil do not form a monophyletic group, since Ps. sp. (NL-2349), an undescribed species with a granular veil, fell within the gracilis clade. Further, it is questionable whether the granular veil in psathyrelloid taxa is of the same origin as that of Coprinellus or Coprinopsis.
The Coprinus patouillardii clade (MPB: 100 %, MLB: 100 %, BPPm: 1.00, BPPb: 1.00, also known as C. cordisporus) contains small species with deliquescent basidiomes, granular veil and more or less hexagonal, lentiform spores. This group consists of two species on our trees out of a total of three (see Keirle et al. 2004). Earlier studies have also proved its affinities to the fusca clade (Hopple and Vilgalys 1999; Larsson and Örstadius 2008; Nagy et al. 2010, 2011; Padamsee et al. 2008; Vasutová et al. 2008; Walther et al. 2005), and constraint analyses reinforce its position there (Padamsee et al. 2008). However, its exact placement within/near this clade is uncertain, as shown by the results of the constraint analyses. Trees with C. patouillardii placed as a sister group of all other species of the fusca clade cannot be rejected; moreover, when forced to monophyly with Coprinellus (as a sister group of that), the likelihoods decreased only slightly (trees could not be rejected at p ≤ 0.05). This implies that—given our data—C. patouillardii may occupy several places on the tree with nearly equal probability. In support of the above finding, development of the veil in early-median phases of the ontogeny sheds more light on the relationship of this clade to /Coprinellus. The velar structures of C. patouillardii are not sharply delimited from cell layers of the pileipellis, whereas it is clearly demarcated from the underneath layers of palisade in Coprinellus (Reijnders 1979, as C. cordisporus). In this respect, C. patouillardii is similar to the C. cortinatus group, which belongs to the pseudonivea clade.
The gordonii clade (MPB: 100 %, MLB: 100 %, BPPm: 1.00, BPPb: 1.00, Fig. 1g) contains species with whitish or pale grayish brown, medium-sized basidiomes, with thick fibrillose, white veil when young, cuticular pileipellis, and almost opaque spores, which are about 10 μm. We examined whether these species can be placed in Coprinopsis, with which it shares the cuticular pileipellis and spore shape, but it seems that such a relationship is improbable (au test pvalues: 10−4 – 6 × 10−35). This, interestingly, at the same time entails multiple independent origins and/or losses of cuticular pileipellis in the Psathyrellaceae.
The cotonea clade (MPB: 95 %, MLB: 95 %, BPPm: 1.00, BPPb: 1.00) is recognized here as a monophyletic group. Both species are morphologically peculiar in that their basidiomes are large, non-deliquescent, and the veil is whitish, abundant, cottony, blackening with age. The two species here come in a rather isolated position and their placement is somewhat uncertain. On preliminary phylogenies, their position varied and ML analyses placed them within the fusca clade. However, trees where they are forced outside the fusca clade could not be rejected by the au test (0.22 < p < 0.46). Because of their superficial similarity to species of the Cystoagaricus clade, primarily the size of the basidiomes, and the darkening of veil on ageing, we tested the monophyly of these two groups. However, the p values were close to 0.01, thereby rejecting the monophyly of the cotonea and Cystoagaricus clades. Because the constraint analyses provide evidence for an unstable position for this clade on the tree, we raise the possibility that they represent an independent, enigmatic lineage of the Psathyrellaceae similar to the C. patouillardi clade.
/Coprinellus (MPB: -, MLB: -, BPPm: 0.91, BPPb: 1.00 (Fig. 1c,d,e,r)) is characterized by hymeniform pileipellis, presence or absence of a granular veil, and deliquescent or collapsing basidiomes. When the veil is lacking, the cap is covered by lageniform, hair-like pileocystidia. The two subclades therein correspond to subsection Setulosi p.p. (C. hiascens–C. congregatus) and subsections Domestici and Micacei (C. verrucispermus–C. truncorum). The former is characterized primarily by the lack of veil, while the latter generally has rich granular veil on the cap. Exceptions from this are the taxa originally placed in subsection Setulosi, such as C. disseminatus, C. verrucispermus, C. curtus etc., which are nested in the clade of veiled species. These species, however, have both veil and pilocystidia, and their spores are often mitriform, very similar to that of C. micaceus.
The candolleana clade (MPB: 100 %, MLB: 100 %, BPPm: 1.00, BPPb: 1.00) contains species with rather pale basidiomes lacking pleurocystidia, but having a fibrillose, scanty veil which is visible only on young specimens. The affinities of this clade to Coprinellus have already been noted by Hopple and Vilgalys (1999) and many subsequent studies (Walther et al. 2005; Padamsee et al. 2008; Larsson and Örstadius 2008; Vasutová et al. 2008; Nagy et al. 2010; 2011).
The gracilis clade (MPB: -, MLB: 73 %, BPPm: 1.00, BPPb: 1.00, Fig. 1j) is named after Ps. gracilis, the type species of Psathyrella. Although we did not include it in the present data set, two very closely related species, Ps. corrugis (also known as Ps. gracilis f. corrugis) and Ps. pseudogracilis are included, and there is phylogenetic evidence for these species belonging to the same clade (Larsson and Örstadius 2008; Vasutová et al. 2008). These species are characterized by slender, non-deliquescent basidiomes, often rooting in the substrate. Lageniform pleurocystidia and cheilocystidia are always present, spores on average longer than 10 μm, regular ellipsoid, and veil fibrillose, scanty and visible only on young specimens. Species of this clade have been placed in section Psathyrella of Kits van Waveren (1985) and section Psathyrella, subsection Psathyrellae of Smith (1972). On the basis of morphology, Ps. microrhiza also falls in the vicinity of Ps. gracilis and Ps. pseudogracilis, which is in contradiction with its phylogenetic position in the spadiceogrisea clade (MPB: 50 %, MLB: 72 %, BPPm, BPPb: -) on our phylogeny. Similarly, Larsson and Örstadius (2008) inferred an ambiguous position for this taxon.
The Cystoagaricus clade (MPB: 65 %, MLB: 69 %, BPPm: 1.00, BPPb: 1.00) has already appeared on the tree published by Padamsee et al. (2008), although Ps. delineata (=Ps. gossypina) was excluded. It contains large species with mostly firm stipe and cap, where the cap surface is often covered by appressed scales, which are darker than the background, sometimes vanishing somewhat and then remaining only in the center of pileus. Exceptions are Ps. gossypina and Ps. larga, which have smooth pileus surface. The spores are small (6–8 μm), and smooth or subangular (in Cy. strobilomyces). However, this clade is very heterogeneous and more research is needed to find better morphological characters to circumscribe it.
Species of the Lacrymaria clade (MPB: 100 %, MLB: 100 %, BPPm: 1.00, BPPb: 1.00) have been placed in section Spadiceae and the genus/section Lacrymaria by traditional taxonomists (Kits van Waveren 1985; Smith 1972). Two subclades corresponding to taxonomic grouping can be recognized, of which the Lacrymaria clade contains species with large, ornamented, almost opaque spores, a thick, fibrillose veil composed of pigmented hyphae, and large basidiomes (Fig. 1i). The Psathyrella spadicea clade (here missing species like Ps. cernua, Ps. conissans etc. of former studies) is characterized by thick-walled cystidia, small (6–9 μm) pale spores and lacking a veil. It seems difficult to establish a uniform morphological definition for the whole Lacrymaria clade, due to the extraordinary morphological disparity exhibited by the above mentioned subclades. However, to date, no published phylogenetic analyses question their monophyly. Lacrymaria lacrymabunda and its close allies have been treated a section of Psathyrella by certain authors (e.g. Smith 1972), but that classification does not provide any specific morphological trait for the two groups concerned here.
The Coprinopsis clade (MPB: 91 %, MLB: 98 %, BPPm: 1.00, BPPb: 1.00, Fig. 1a, f, p, s) contains both deliquescent and non-deliquescent taxa. The pileipellis is cuticular, or in a few taxa, hymenidermal (C. poliomallus), the veil is well-developed, composed of globose, filamentous or diverticulate elements. The veil on the margin of the pileus is powdery, as opposed to the pseudonivea clade (see below); when partially "beard-like", then the fruiting body is grayish and the spores are shorter than 9 μm. The psathyrelloid species, Ps. melanthina is also nested in the Coprinopsis clade, and should be recombined in this genus. These have cuticular pileipellis, like other Coprinopsis species, grayish non-deliquescent basidiomes and grayish, fibrillose scaly veil on the cap.
The pseudonivea clade (MPB: 100 %, MLB: 100 %, BPPm: 1.00, BPPb: 1.00) takes an isolated position basal to all other Coprinopsis species. Characteristic for this clade is that the veil forms a special “beard-like” structure at the cap margin of young basidiomes, composed of long white fibrils connecting cap margin to the stipe and a few globose cells in between. The basidiomes are usually pure white or whitish, sometimes brown (Ps. submicrospora). The pileipellis is cuticular when the fruiting body is deliquescent, and cuticular or hymenidermal when non-deliquescent. Psathyrelloid members of this clade have scanty veil on the cap surface, but beard-like fibrils on the margin. Separation of this clade from the rest of the Coprinopsis clade is proposed for the first time in this study. Its morphological uniformity, basal relationship and the strong support values raise it as an enigmatic lineage of Psathyrellaceae, deserving more attention in the future. The position of non-deliquescent members is noteworthy. In contrast to our previous thoughts (Nagy et al. 2010), they split into several smaller clades and are not inferred as the basal ones in Coprinopsis. This implies more shifts between fruiting body types (i.e. between non-deliquescent and deliquescent) than inferred without the inclusion of Ps. pannucioides, Ps. submicrospora, and Ps. melanthina (Nagy et al. 2010). Our study is the first to include multiple representatives of the “C. cortinatus complex” in phylogenetic studies, such as C. coniophorus, C. bellulus, or C. cortinatus. Of these tiny species, the two latter have not been classified in Coprinopsis by Redhead et al. (2001), probably due to their superficial similarity to certain Coprinellus species. Our study, however, provides unequivocal evidence for their placement close to Coprinopsis pseudonivea, with which they share the characteristic beard-like veil on the cap margin.
The Parasola clade (MPB: 100 %, MLB: 100 %, BPPm: 1.00, BPPb: 1.00, Fig. 1b, o) has been recovered in almost all phylogenetic studies of Psathyrellaceae as monophyletic (Hopple and Vilgalys 1999; Larsson and Örstadius 2008; Nagy et al. 2010, 2011; Padamsee et al. 2008; Vasutova et al. 2008; Walther et al. 2005), including a detailed study of species limits (Nagy et al. 2009). The lack of any velar structures and caulocystidia uniquely defines this genus. Interestingly, the basidiomes are collapsing (incompletely deliquescent); if not collapsing (psathyrelloid), then there are thick-walled, brown setulae on the pileus. The spores are usually rounded-triangular, flattened, which is rather peculiar for the Psathyrellaceae. Only the pseudonivea clade and the Coprinus patouillardii clade contain species with strongly flattened, often angular spores. The position of P. conopilea varied somewhat across different phylogenies, and appeared either as a sister to all other Parasola taxa, or on a weakly supported clade together with P. auricoma. Constraint analyses confirm the plausibility of P conopilea being basal to all other Parasola taxa. This basal relationship would imply gradual loss of pilocystidia, the emergence of partial deliquescence, as well as gradual flattening and expansion of the spores within the genus Parasola (Nagy et al. 2009).
Classification of Psathyrellaceae
Of the several taxonomic difficulties in fungal classification, that of the Psathyrellaceae is an exemplary case. Early phylogenetic studies and subsequent reclassification of coprinoid lineages (Coprinellus, Coprinopsis and Parasola) left the genus Psathyrella para/polyphyletic (Redhead et al. 2001). In the following years, several studies tried to decipher phylogenetic relationships within the family (Larsson and Örstadius 2008; Nagy et al. 2009, 2010, 2011; Padamsee et al. 2008; Vasutová et al. 2008; Walther et al. 2005), which added valuable information used here as a basis for designing a balanced taxon sampling. One of the major objectives during this study—which we consider an absolute prerequisite for a classification—was to uncover all major lineages within the family, and this has also strongly relied on previous studies. The available published sequence data allows us to conclude that most, if not all, major clades have been uncovered. Fortunately, there is very little phylogenetic uncertainty about the major clades within the family, and uncertainty at early splits of the phylogeny has no or little impact on a reclassification in the future.
In conjunction with former studies (Hopple and Vilgalys 1999; Larsson and Örstadius 2008; Nagy et al. 2010, 2011; Padamsee et al. 2008; Vasutová et al. 2008; Walther et al. 2005), our results reinforce the para/polyphyly of Psathyrella, and warrant extensive rearrangement of the genus into new genera. The clade containing Ps. gracilis, the type species of Psathyrella, is sister to Coprinellus and /Candolleana, and forms a clade of its own with strong support. In our opinion, the current topology and the uncertainties in the placement of some clades would make it very impractical to consider a large psathyrelloid "supergenus", i.e. treating all the clades as one genus. This would save the generic name Psathyrella for most of the species, but in that case, species of Coprinellus and the Coprinus patouillardii complex would have to be merged with the psathyrelloid species (if the Lacrymaria clade is excluded, and in that case even Coprinopsis, Parasola and the Cystoagaricus clade would have to be included). We think the most reasonable solution for the paraphyly of Psathyrella requires splitting it as currently circumscribed into several smaller genera. If recognized as a separate genus, the gracilis clade should evidently retain the name Psathyrella (Fr.) Quél., because the type species belongs to this clade. For other clades, other generic names should be sought, although their arrangement should not necessarily follow the clade structure presented here. Also, whether deliquescent taxa should be merged with non-deliquescent ones, as would be the case with Coprinus patouillardii or when merging the candolleana clade with Coprinellus, should be considered carefully, even if some non-deliquescent species have already been recombined in deliquescent genera (e.g. Coprinopsis marcescibilis or Parasola conopila; Larsson and Örstadius 2008). It seems that such mixed genera cannot be avoided in a natural classification, although this makes any future identification keys either highly unnatural or very hard to write and use.
The placement of the “Coprinus cortinatus complex” in Coprinopsis entails reconsideration of the generic definition of Coprinopsis as given by Redhead et al. (2001). The pileipellis of these species plus C. poliomallus (shown to belong to Coprinopsis by Nagy et al. 2010) is hymenidermal, whereas that of other Coprinopsis species, including non-deliquescent ones, is cuticular. However, the combination of hyphal veil cells and sphaerocysts, and the whitish colours support their inclusion in Coprinopsis.
Deeper branches of the phylogeny presented here are not robust; more data, or even phylogenomic approaches, may be necessary to resolve the earliest evolutionary events. Similarly, morphology provides no stronger evidence for any grouping of the larger clades, due to extensive convergence and difficulties in recognizing homologous traits. However, the larger clades can be defined by morphological traits unambiguously. The generality of these clade definitions should be tested by the inclusion of more taxa in the phylogeny, especially from tropical regions or the southern hemisphere. Although other traits may also prove helpful in circumscribing certain clades, the morphological definitions presented here represent a step forward in formulating taxonomic descriptions of clades when a natural classification is to be designed.
References
Altekar G, Dwarkadas S, Huelsenbeck JP, Ronquist F (2004) Parallel Metropolis coupled Markov chain Monte Carlo for Bayesian phylogenetic inference. Bioinformatics 20:407–415
Borchsenius F (2007) FastGap 1.0.8. Software distributed by the authors at (http://192.38.46.42/aubot/fb/FastGap_home.htm).
Cáceres O, Kirschner R, Piepenbring M, Schofer H, Gene J (2006) Hormographiella verticillata and an Ozonium stage as anamorphs of Coprinellus domesticus. Antonie Van Leeuwenhoek 89:79–90
Castresana J (2000) Selection of conserved blocks from multiple alignments for their use in phylogenetic analysis. Mol Biol Evol 17:540–552
Guindon S, Gascuel O (2003) PhyML—A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol 52:696–704
Hopple JS, Vilgalys R (1994) Phylogenetic relationships among coprinoid taxa and allies based on data from restriction site mapping of nuclear rDNA. Mycologia 86:96–107
Hopple JS, Vilgalys R (1999) Phylogenetic relationships in the mushroom genus Coprinus and dark-spored allies based on sequence data from the nuclear gene coding for the large ribosomal subunit RNA: divergent domains, outgroups, and monophyly. Mol Phylogenet Evol 13:1–19
Keirle MR, Hemmes DE, Desjardin DE (2004) Agaricales of the Hawaiian Islands 8: Agaricaceae. Coprinus and Podaxis, Psathyrellaceae: Coprinellus, Coprinopsis and Parasola. Fungal Divers 15:33–124
Kemp RFO (1975) Breeding biology of Coprinus speciesin the section Lanatuli. Trans Br Mycol Soc 65:375–388
Kits van Waveren E (1985) The Dutch, French and British species of Psathyrella. Persoonia Suppl. 2:1–300
Kües U (2000) Life history and developmental processes in the basidiomycete Coprinus cinereus. Microb Mol Biol Rev 64:316–353
Kühner R (1928) Le développement et la position taxonomique de l’Agaricus disseminatus Pers. Le Botaniste 20:147–195
Kühner R, Romagnesi H (1953) Flore Analytique des Champignons Supérieurs. Paris, pp 556
Larsson E, Örstadius L (2008) Fourteen coprophilous species of Psathyrella identified in the Nordic countries using morphology and nuclear rDNA sequence data. Mycol Res 112:1165–1185
Maddison WP, Maddison DR (2009) Mesquite: a modular system for evolutionary analysis. Version 2.6. http://mesquiteproject.org
Matheny PB, Curtis JM, Hofstetter V, Aime MC, Moncalvo JM, Ge ZW, Slot JC, Ammirati JF, Baroni TJ, Bougher NL, Hughes KW, Lodge DJ, Kerrigan RW, Seidl MT, Aanen DK, DeNitis M, Daniele GM, Desjardin DE, Kropp BR, Norvell LL, Parker A, Vellinga EC, Vilgalys R, Hibbett DS (2006) Major clades of Agaricales: a multilocus phylogenetic overview. Mycologia 98:982–995
Moncalvo JM, Vilgalys R, Redhead SA, Johnson JE, James TY, Aime CM, Hofstetter V, Verduin SJ, Larsson E, Baroni TJ, Greg Thorn R, Jacobsson S, Clémençon H, Miller OK Jr (2002) One hundred and seventeen clades of euagarics. Mol Phylogenet Evol 23:357–400
Moser M (1983) Die Röhrlinge und Blätterpilze. in GAMS H.: Kleine Kryptogamenflora IIb/2. Gustav-Fischer-Verlag, Jena, p 532
Nagy GL, Kocsubé S, Papp T, Vágvölgyi C (2009) Phylogeny and character evolution of the coprinoid mushroom genus Parasola as inferred from LSU and ITS nrDNA sequence data. Persoonia 22:28–37
Nagy GL, Urban A, Örstadius L, Papp T, Larsson E, Vágvölgyi C (2010) The evolution of autodigestion in the mushroom family Psathyrellaceae (Agaricales) inferred from Maximum Likelihood and Bayesian methods. Mol Phylogenet Evol 57:1037–1048
Nagy GL, Walther G, Házi J, Vágvölgyi C, Papp T (2011) Understanding the evolutionary processes of fungal fruiting bodies: correlated evolution and divergence times in the Psathyrellaceae. Syst Biol 60:303–317
Nieves–Rivera AM (2001) The edible Psathyrellas of Haiti. Inoculum 52:1–3
Padamsee M, Matheny PB, Dentinger BT, McLaughlin DJ (2008) The mushroom family Psathyrellaceae: evidence for large-scale polyphyly of the genus Psathyrella. Mol Phylogenet Evol 46:415–429
Pagel M, Meade A (2006) BayesPhylogenies 1.0. Software distributed by the authors
Rambaut A, Drummond A (2008) Tracer v 1.4.1. Soeftware distributedby the authors at http://beast.bio.ed.ac.uk/
Rampazzo A, Kuhnert P, Howard J, Bornand V (2009) Hormographiella aspergillata keratomycosis in a dog. Vet Ophthalmol 12:43–47
Redhead SA, Vilgalys R, Moncalvo JM, Johnson J, Hopple JS (2001) Coprinus Persoon and the disposition of Coprinus species sensu lato. Taxon 50:203–241
Reijnders AFM (1979) Developmental anatomy of Coprinus. Persoonia 10:383–424
Roshan U, Livesay DR (2006) Probalign: multiple sequence alignment using partition function posterior probabilities. Bioinformatics 22:2715–2721
Shimodaira H, Hasewaga M (2001) CONSEL: for assessing the confidence of phylogenetic tree selection. Bioinformatics 17:1246–1247
Simmons MP, Ochoterena H (2000) Gaps as characters in sequence-based phylogenetic analyses. Syst Biol 49:369–381
Singer R (1986) The Agaricales in modern taxonomy, 4th edn. Koeltz Scientific Books, Koenigstein, pp 912
Smith AH (1972) The North American species of Psathyrella. Mem NY Bot Gard 24:1–633
Staden R, Beal KF, Bonfield JK (2000) The staden package. Methods Mol Biol 132:115–130
Stajich JE, Wilke SK, Ahren D, Au CH, Birren BW, Borodovsky M, Burns C, Canback B, Casselton LA, Cheng CK, Deng J, Dietrich FS, Fargo DC, Farman ML, Gathman AC, Goldberg J, Guigo R, Hoegger PJ, Hooker JB, Huggins A, James TY, Kamada T, Kilaru S, Kodira C, Kues U, Kupfer D, Kwan HS, Lomsadze A, Li W, Lilly WW, Ma LJ, Mackey AJ, Manning G, Martin F, Muraguchi H, Natvig DO, Palmerini H, Ramesh MA, Rehmeyer CJ, Roe BA, Shenoy N, Stanke M, Ter-Hovhannisyan V, Tunlid A, Velagapudi R, Vision TJ, Zeng Q, Zolan ME, Pukkila PJ (2010) Insights into evolution of multicellular fungi from the assembled chromosomes of the mushroom Coprinopsis cinerea (Coprinus cinereus). Proc Natl Acad Sci USA 107:11889–11894
Stamatakis A (2006) RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690
Swofford DL (2002) PAUP: phylogenetic analysis using parsimony (and other methods) version 4.0b10. Sinauer Associates, Sunderland
Thompson JD, Gibson TJ, Higgins DG (2002) Multiple sequence alignment using ClustalW and ClustalX. Curr Protoc Bioinformatics Chapter 2, Unit 2 3
Vasutova M, Antonin V, Urban A (2008) Phylogenetic studies in Psathyrella focusing on sections Pennatae and Spadiceae–new evidence for the paraphyly of the genus. Mycol Res 112:1153–1164
Vellinga EC (2004) Genera in the family Agaricaceae—Evidence from nrITS and nrLSU sequences. Mycol Res 108:354–377
Verweij PE, van Kasteren M, van de Nes J, de Hoog GS, de Pauw BE, Meis JF (1997) Fatal pulmonary infection caused by the basidiomycete Hormographiella aspergillata. J Clin Microbiol 35:2675–2678
Walther G, Garnica S, Weiss M (2005) The systematic relevance of conidiogenesis modes in the gilled Agaricales. Mycol Res 109:525–544
White TJ, Bruns T, Lee S, Taylor J (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds) PCR Protocols: a guide to methods and applications pp 315–322
Acknowledgements
The authors would like to express their gratitude to Ellen Larsson and Leif Örstadius for valuable discussions and exchange of specimens and voucher information. Sándor Kocsubé is thanked for his generous help in designing the figures. This study has been supported via a grant to LGN from the Fungal Research Trust.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Nagy, L.G., Vágvölgyi, C. & Papp, T. Morphological characterization of clades of the Psathyrellaceae (Agaricales) inferred from a multigene phylogeny. Mycol Progress 12, 505–517 (2013). https://doi.org/10.1007/s11557-012-0857-3
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11557-012-0857-3