
Abstract
Purpose
This paper presents the results of a large study involving fusion prostate biopsies to demonstrate that temporal ultrasound can be used to accurately classify tissue labels identified in multi-parametric magnetic resonance imaging (mp-MRI) as suspicious for cancer.
Methods
We use deep learning to analyze temporal ultrasound data obtained from 255 cancer foci identified in mp-MRI. Each target is sampled in axial and sagittal planes. A deep belief network is trained to automatically learn the high-level latent features of temporal ultrasound data. A support vector machine classifier is then applied to differentiate cancerous versus benign tissue, verified by histopathology. Data from 32 targets are used for the training, while the remaining 223 targets are used for testing.
Results
Our results indicate that the distance between the biopsy target and the prostate boundary, and the agreement between axial and sagittal histopathology of each target impact the classification accuracy. In 84 test cores that are 5 mm or farther to the prostate boundary, and have consistent pathology outcomes in axial and sagittal biopsy planes, we achieve an area under the curve of 0.80. In contrast, all of these targets were labeled as moderately suspicious in mp-MR.
Conclusion
Using temporal ultrasound data in a fusion prostate biopsy study, we achieved a high classification accuracy specifically for moderately scored mp-MRI targets. These targets are clinically common and contribute to the high false-positive rates associated with mp-MRI for prostate cancer detection. Temporal ultrasound data combined with mp-MRI have the potential to reduce the number of unnecessary biopsies in fusion biopsy settings.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Prostate cancer (PCa) is the second leading cause of cancer-related death in North American men. According to the American and Canadian Cancer Societies, PCa accounts for 24 % of all new cancer cases and results in 33,600 deaths per year in North America.Footnote 1 The PCa-related death rate has declined significantly (almost 4 % per annum) between 2001 and 2009 due to improved testing and better treatment options. The majority of the cases diagnosed today are the early-stage disease where several treatment options are available, including surgery, brachytherapy, thermal ablation, external beam therapy and active surveillance. Early detection and accurate staging of PCa are essential to the selection of optimal treatment options, hence reducing the disease-associated morbidity and mortality [25].
The current standard of care for diagnosis of PCa is the histopathological analysis of biopsy samples acquired under transrectal ultrasound (TRUS) guidance. The sensitivity of conventional systematic biopsy under TRUS guidance, for detection of PCa, has been reported to be as low as 40 % [5, 9, 25]. Significant improvement of TRUS-guided PCa biopsy is required to decrease the rate of over-treatment for low-risk disease while preventing the under-treatment of high-risk cancer [16]. Several methods that enable patient-specific targeting have been proposed to improve the detection rate of PCa. Ultrasound (US)-based tissue typing techniques for characterization of PCa include analysis of single radio-frequency (RF) US frame data [8, 22, 27], elastography [18, 21, 24] and Doppler imaging [23, 30]. A shortcoming of these methods that have limited their clinical uptake is the challenge of identifying a globally effective, tissue-associated threshold that can reliably identify cancerous tissue in the analyzed images, and be ubiquitously generalized to prospective patient data. For instance, determining such preset threshold for separation of cancer from benign tissue has been difficult for shear wave elastography, one of the most advanced imaging methods to date with large clinical feasibility studies [3]. Magnetic resonance (MR) imaging, an emerging approach for performing targeted biopsies, uses co-alignment of multi-parametric MRI (mp-MRI) and TRUS and has reported negative predictive values as high as 94 % [26]. It has been shown that mp-MRI improves the detection of aggressive PCa, but it is not as sensitive for detection of low-grade cancers and smaller sizes of high-grade cancer. Moreover, mp-MRI leads to a high number of false positives and hence unnecessary biopsies [16, 26].
Recently, temporal US data captured from a stationary tissue location have been proposed for tissue characterization in our group. In this technology, a series of US frames is obtained from a stationary tissue position without any intentional mechanical excitation. This approach is a significant departure from traditional US tissue typing techniques. Features extracted from the temporal US data have been used in a machine learning framework to predict labels provided by histopathology as the ground truth. This approach has been used successfully for depiction of cancerous and non-cancerous prostate tissue in ex vivo [19, 22] and in vivo [12–15, 20] studies.
In previous implementations of temporal RF data technology, features were heuristically determined from spectral analysis of US image sequences, rather than through a systematic approach [1, 12, 14, 15]. Recently [1], we proposed an automatic feature selection framework for analyzing temporal US signals of prostate tissue. We addressed the so-called cherry picking of the features [15] by a deep learning-based feature selection framework. This framework exploits deep belief networks (DBN) [2] to automatically learn a high-level latent feature representation of the temporal US data. We demonstrated that this approach is an effective method in identifying both benign and cancerous biopsy cores in TRUS-guided biopsy [1] in a preliminary study containing 36 biopsy cores. In this paper, in our largest clinical study to date involving 255 TRUS-guided biopsy cores, we investigate the factors that affect the classification accuracy within a targeted biopsy interface. Furthermore, we demonstrate that temporal US data can be used to accurately classify tissue labels that were identified in mp-MRI as suspicious for cancer. Our results indicate that temporal US analysis can complement mp-MR imaging and together they can be an effective tool for cancer detection.
Materials
Data acquisition
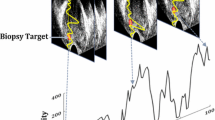
The study was approved by the ethics review board of the National Cancer Institute, National Institutes of Health (NIH) in Bethesda, Maryland, and all subjects provided informed consent to participate. One hundred and fifty-eight subjects were enrolled in the study where they underwent preoperative mp-MRI examination with three pulse sequences: T2-weighted, diffusion-weighted imaging (DWI) and dynamic contrast-enhanced (DCE) imaging. Prior to biopsy, suspicious lesions in mp-MRI were identified and scored by two independent radiologists, according to a previously published protocol [15, 29]. In this protocol, an overall score in the range from 1 (no cancer) to 5 (aggressive cancer) is assigned to a suspicious area. Scores are grouped into three descriptors of “low” (score of \(\le \) 2), “moderate” (score of 3) and “high” (score of \(\ge \) 4) and referred to as the MR suspicious level assigned to the area. Using the UroNav MR/US fusion system (Invivo Inc., a subsidiary of Philips Healthcare), targeted biopsy was performed with the identified mp-MRI lesions registered to the real-time 3D TRUS images [17, 31]. The clinician navigates the prostate volume to identify the labeled target for acquiring a core and holds the TRUS transducer steady for about 5 s to obtain 100 frames of temporal US data. Two biopsies are then taken from a target, one in the axial imaging plane and one in the sagittal imaging plane. For each subject, temporal US data are collected from either one or two MR-identified targets only in the axial plane to minimize the disruption to the clinical work flow. Temporal US data were collected from 255 biopsy cores of 158 subjects for this study. Tissue biopsy is followed by histopathology analysis, and results are used as the ground-truth labels for evaluation of cancer detection as described below (Fig. 1).

a Training data set: 32 biopsy cores from 27 patients used for model generation. b Test data set: 223 biopsy cores from 138 patients used for analysis of the temporal US approach
Histopathology and ground-truth labeling
The Gleason grade scale, ranging from 1 (resembling normal prostate tissue) to 5 (aggressive cancerous tissue), is the most common system to describe the level of abnormality for prostate tissue. The Gleason score (GS) is reported as the summation of the two most common Gleason grade patterns in a specimen. Following histopathology examination of the 255 collected cores, 83 were cancerous and their GS were as follows: 26 GS of 3 + 3, 26 GS of 3 + 4, five GS of 4 + 3, 21 GS of 4 + 4, and five GS of 4 + 5. The remaining 172 cores were non-cancerous with histologies including fibromuscular tissue, chronic inflammation, atrophy and prostatic intraepithelial neoplasia (PIN). As mentioned above, for each MRI-identified target in a patient, two biopsies are obtained from the axial and sagittal TRUS planes, respectively. For some cores, the histopathology reported from the two planes were not in agreement. Taking this into account, we only considered the histopathology of the axial plane for generating the ground-truth labels, the same plane as that of temporal US imaging. Among 255 cores from 158 patients, 55 cores from 45 patients have mismatches between axial and sagittal histopathology, where one biopsy is reported as benign and the other as cancerous tissue.
Data for model generation and testing
The reported registration accuracy for the Philips UroNav MR/US fusion system is \(2.4 \pm 1.2\) mm [31]. However, mis-registration is usually more prominent for targets close to the segmented boundary of the prostate. For biopsy cores taken far away from the boundary, we assume that the target is in the center of the core. However, clinicians normally adjust the needle penetration depth for targets that are close to the boundary, especially in the anterior region, so that the core sample is not taken beyond the prostate. To generate our model, we aim to use homogeneous prostate tissue regions with reliable ground-truth labels. Therefore, we select cores for training if they meet all of the following three selection criteria, similar to our previous work [1]: (i) located more than 3 mm distance to the prostate boundary in TRUS images; (ii) have matching histopathology labels between axial and sagittal biopsies; and (iii) have a tumor length larger than 7 mm if cancerous. We select 32 cores from 27 patients, which fulfill the above criteria, and use the temporal US data from them to generate our model. These 32 training cores are labeled as dataset D1, where 13 cores are cancerous and 19 cores are benign. The distribution of the histopathology labels of this data is presented in Fig. 1a.
The remaining 223 biopsy cores (distributed as presented in Fig. 1b) are divided into three subgroups based on the distance of the target to prostate boundary and agreement between axial and sagittal histopathology labels: (1) dataset \(D2-A\), consisting of 156 cores from 150 patients whose target distance to prostate boundary (d) is \(\ge \) 3 mm; (2) dataset \(D2-B\), consisting of 117 cores from dataset \(D2-A\) that also have agreement in histopathology labels of axial and sagittal biopsy cores; and (3) dataset D3, consisting of 67 cores whose target distance to prostate boundary is \(<\)3 mm. A flowchart summarizing the training and test data is shown in Fig. 2.

Flowchart of data division to training data and four subgroups of test data

An illustration of the proposed framework for prostate cancer detection using temporal US data
Region of interest
We define regions of interest (ROI) in the TRUS image to associate histopathology of each biopsy core to temporal US data. Each ROI is an area of \(2\times 10\) mm\(^{2}\) in the lateral and axial directions, respectively, along the projected needle path centered on the biopsy target. The width of this area is close to the width of the biopsy core, but the length is approximately half of the typical biopsy core length. Given the very large variability of biopsy core lengths in histopathology, selection of ROI length was based on a reasonable assumption that the center of a cancerous tissue reported in histopathology should be close to the target identified in MR. The selected area is divided into 20 ROIs of size 1 mm\(^{2}\), resulting a total of 5100 ROIs from all biopsy cores. To build the feature extraction and classification model, we used 640 ROIs from 32 training cores in the dataset D1. The number of RF samples within an ROI varies between 90 and 480 due to scan conversion and the depth of biopsy cores, while the length of temporal RF data is 100 time points. Therefore, each ROI can be thought of as 90–480 signals each with 100 time samples. We analyze the spectral component of the temporal US data in order to determine the characteristics of the non-cancerous and cancerous cores. To generate features for each ROI, we take the Fourier transform of the time course signals in the ROI, normalized to the frame rate. For each ROI, we generate 50 positive frequency components by averaging the absolute values of the discrete Fourier transform (DFT) of the zero-mean temporal US data [1, 15].
Method
Figure 3 shows a schematic diagram of our method. In this framework, we use deep belief networks (DBN) [2] to automatically learn a high-level latent feature representation of the temporal US data that can detect prostate cancer. We then use the hidden activations of the DBN as the input features to a support vector machine (SVM) classifier to generate a cancer likelihood map. Details of the training and testing have been described previously [1].

An illustration of the proposed feature visualization method
Automatic feature learning
For automatic feature learning, we first pre-train a DBN using our training dataset, D1, by performing a greedy layer-wise unsupervised pre-training approach [2]. Initially, we set our DBN structure including the number of hidden layers and nodes configuration, as well as the numerical meta-parameters according to the default values of the DBN library [28]. We then heuristically searched for the number of hidden layers and node configuration so that lowest reconstruction errors with the default library parameters were obtained in the training data. Using trial and error, and the guidelines provided by [10, 32], we can determine the meta-parameters [10] of the deep network such as the learning rate, the momentum, the weight cost, the size of mini-batch and the number of passes to achieve lower reconstruction error in the training data. The finalized pre-trained DBN is composed of a real-valued visible layer with 50 units, and three hidden layers consist of 100, 50 and 6 hidden units, respectively. The learning rate is fixed at 0.001, mini-batch size is 5 and the number of passes is 100 epochs. The momentum and the weight cost do not change from the default values (\(0.9, 2 \times 10^{-4}\)). This unsupervised pre-training guides learning to better generalize from training data [6]. Using standard training schemes based on random initialization, the AUC for the test dataset, \(D2-B\), is dropped to 0.53, which suggests that we are unable to effectively learn the model. This model also leads to poor generalization.
Following the pre-training step, we perform a fine-tuning step using the same training set in a supervised manner. The fine-tuning is done by stacking another output node as the last layer of the DBN. This node is used to represent the class label of the input data. For supervised fine-tuning, we use contrast divergence approximation [11] to learn the weighted mixture of activations. The numerical meta-parameters are optimized according to reconstruction error. In the supervised fine-tuning step, we ran 70 epochs with a fixed learning rate of 0.01 and a mini-batch size of 10. After completion of the learning procedure, the last hidden layer of the DBN produces the latent feature representation.
An essential point in using DBN is to build models that generalize well to unseen data. The greedy layer-wise learning algorithm is a powerful tool that enables us to identify the parameters of our model fast, even in this deep network configuration [11]. Bengio et al. [2] report that using this approach, weights are initialized near a good local optimum, leading to a better generalization of the model. The key concept is to train one layer at a time and to use the representation of the previous hidden layer as the input to the next hidden layer. Following fine-tuning through back propagation, the weights of the deep network are optimized to their final values.
In order to test the generalization of the trained DBN, we make certain that testing data are never used to pre-train or fine-tune the network parameters. In addition, we visualize the activation of hidden neurons in different layers [32] after training (see “Results and discussion” section). If only a small fraction of the connections is active, then we can indicate that our network requires few parameters for classification.

Performance of the proposed method across Gleason scores in dataset: a \(D2-A\), b \(D2-B\)
Classification
We use training dataset D1 to obtain the learned features from the last hidden layer of the trained DBN. Then, we use these features as inputs to a nonlinear SVM classifier. We have six learned features corresponding to the activations of the six hidden units in the last hidden layer. The SVM classifier uses a radial basis function (RBF) kernel; we determine the parameters of the classifier through a grid-search approach [15]. Following training, we use the SVM classifier on the test data to derive the tissue type labels for each ROI.
Feature visualization
To determine the characteristics of the non-cancerous and cancerous cores in the temporal US data and their correlation with learned features, we propose a feature visualization approach (Fig. 4). This approach is used to identify the most discriminative features of the time series (i.e., frequency components as introduced in “Region of interest” section), as learned by the classifier. We implement feature visualization for both training and testing data. First, data are propagated through the trained DBN and the activations of the last hidden layer, i.e., the learned latent features are computed. To examine the significance of an individual learned latent feature, the activations of all other hidden units in the third layer are set zero. The activation of the nonzero learned feature is back-propagated to the input layer. The resulting signal, displayed in the input layer as a series of frequency components, highlights those components that contribute to the activation of the nonzero learned feature. By comparing the components activated for benign and cancerous cores, we can identify those frequency ranges that are different between two tissue types. This process is performed for all latent features.
Results and discussion
To assess the performance of our method, sensitivity, specificity and accuracy were calculated. We also report the overall performance using the area under the receiver operating characteristic curve (AUC). To define the sensitivity and specificity, we consider cancerous cores as the positive class and the no-cancerous cores as the negative class. The sensitivity or recall is the percentage of cancerous cores that are correctly identified as cancerous, and the specificity is the percentage of non-cancerous cores that are correctly classified. The accuracy is the proportion of true results (both true positives and true negatives) among the total number of cores. Receiver operating characteristic curves (ROC) are two-dimensional graphs in which sensitivity is plotted on the y-axis and (1-specificity) is plotted on the x-axis. An ROC graph depicts relative trade-offs between sensitivity and specificity, where accuracy is reported based on a specific threshold. The maximum AUC is 1, where larger AUC values indicate better classification performance [7]. Table 1 shows the classification results for test dataset D2. Our results indicate that dataset \(D2-B\) has consistently higher classification results than dataset \(D2-A\) across all MR suspicious levels. A closer look at cores in dataset \(D2-A\) also shows that for those samples that are farther from the prostate boundary (at least 5 mm away) and have moderate MR suspicious level (53 cores), we achieve AUC of 0.89, irrespective of mismatch between axial and sagittal histopathology. In comparison, only 26 % of those cores are identified as cancerous after biopsy which means our approach can effectively complement mp-MRI to reduce the number of false positives for those targets with moderate MR suspicious level.
We also perform similar analysis for dataset D3, where we achieve AUC of 0.36. There are various factors that may have contributed to this drop in classification accuracy, including higher registration error among mp-MRI, TRUS and histopathology for targets close to the segmented prostate boundary, and the inclusion of US signal from tissue outside the prostate. Moreover, based on the clinical protocol, for targets that are close to the prostate boundary, the biopsy core is not centered on the target location to minimize the penetration of needle in tissue surrounding the prostate. A more accurate ground-truth data needs to be obtained to further validate our approach on targets that are close to the prostate boundary.

Cancer probability maps overlaid on B-mode US image, along the projected needle path in the temporal US data and centered on the target. The ROIs for which the cancer likelihood are more than 70 % are colored in red; otherwise, they are colored as blue. Green boundary shows the segmented prostate in MRI projected in TRUS coordinates, dashed line shows needle path and the arrow pointer shows the target: a correctly identified benign core; b correctly identified cancerous core
Moreover, we perform analysis on dataset \(D2-A\) without the elements of \(D2-B\). This analysis was done on 39 cores from 34 patient whose target distance to the boundary is more than 3 mm and has the disagreement in histopathology labels of axial and sagittal biopsy cores. Our results show that by using axial plane histopathology as the ground-truth label, we achieve an AUC of 0.73. On the other hand, by using sagittal plane histopathology as the ground-truth label, we obtained an AUC of 0.60. One of the factors that may have contributed to this performance is the fact that temporal US data, which carries tissue typing information, are obtained from the axial plane.
We also investigate the performance of our model on dataset \(D2-A\) and \(D2-B\) across various Gleason scores (Fig. 5). Results show that 59/83 of benign cores and 26/34 cancerous cores in dataset \(D2-B\) are correctly identified. Most importantly, all cases of clinically aggressive cancer grades with GS of 4 + 5 are correctly labeled. The accuracy of our method in detection of lower grade cancer with GS of 3 + 4 and 3 + 3 is 82 % in dataset \(D2-B\). It should be noted that in some GS categories there is not a sufficient number of samples for definitive conclusion.
Figure 6 shows examples of the cancer likelihood maps from dataset \(D2-B\), derived from the output of SVM, overlaid on B-mode US image. We use the approach described in our earlier publication [22] for this purpose. In the colormaps, red regions belong to ROIs for which the cancer likelihood are more than or equal 70 %. We found that with this threshold, the visualized maps demonstrated all the major tumors in the dataset without a large number of false positives.
To investigate the effect of the size of the tumor on our detection accuracy, we analyze the AUC against the greatest length of the tumor in MRI (ranging from 0.3 to 3.8 cm) for \(D2-B\). We obtained average AUC of 0.77 for cores with MR tumor size smaller than 1.5 cm and average AUC of 0.93 for cores with MR tumor size larger than 2 cm. The results show our method has a higher performance for larger tumors.
We also performed an additional sensitivity analysis by permuting the training and testing data. To create new training and testing sets, in each permutation, we exchanged a randomly selected cancerous or benign core in the training and testing data. The cores are selected from dataset \(D2-B\). This resulted in 32 different permutations given the distribution of cores in our training data. On average, we achieved AUC, accuracy, sensitivity and specificity of 0.70, 71, 68, and 70 %, respectively. In another experiment, to ensure that the classification model does not over-fit to the training data, we trained our SVM classification model using dataset D2 in a fold validation manner. We obtained AUC of 0.71 for \(D2-A\) and AUC of 0.73 for \(D2-B\) in a leave-one-out cross-validation analysis. We also obtained AUC of 0.71 and 0.70 for \(D2-A\) in threefold and 13-fold cross-validation analysis, respectively. The averaged AUC of leave-one-out cross-validation analysis follows our previous performance results, which supports the generalization of the classification model (Table 2).
We visualize the hidden activation of neurons in different layers in the testing dataset. As seen in Fig. 7, only a small fraction of the connections (less than 10 %) in the first and second hidden layers of the deep network are highly probable to be activated.

Hidden neuron activation probabilities (p) for 100 neurons and 2200 test data points after training. Black represents \(p = 0\) and white, \(p = 1\). Each row shows different neurons activations for a given input example, and each column shows a given neurons activations across many samples
For the feature visualization experiment, by subtracting the distributions of benign and cancerous tissue in the input layer as discussed in “Feature visualization” section, we found that feature six, corresponding to hidden activity of the sixth neuron of the third layer, along with features one and four, is those that maximally differentiate cancerous and benign tissue, especially in lower frequency range. Figure 8 shows the visualization of distribution differences for cancerous and benign tissue related to the first and sixth learned features of the third hidden layer, back- propagated to the input layer.

Differences of distributions between cancerous and benign tissue back projected in the input neurons: a corresponds to the first neuron in the third hidden layer; b corresponds to the sixth neuron in the third hidden layer. Results are shown in the frequency range of temporal ultrasound data analyzed in this paper. It is clear that frequencies between 0 and 2 Hz provide the most discriminative features for distinguishing cancerous and benign tissue
Conclusion and future work
In this paper, we presented an approach for accurate classification of tissue labels obtained in MR-TRUS-guided targeted prostate biopsy using temporal US data. We utilized a DBN [1] for systematic learning of discriminant latent features from high-dimensional temporal US features for characterizing prostate cancer. We then applied an SVM classifier along with the activation of the trained DBN to characterize the prostate tissue. In our largest clinical study to date, in 255 TRUS-guided biopsy cores, we identified two important factors that affect our classification performance: (i) distance of the target to the segmented prostate boundary which is correlated with the registration error between mp-MRI, TRUS, and histopathology, and (ii) disagreement between the axial and sagittal histopathology results.
We built our classification model using a fixed training data set consisting of temporal US data of 32 biopsy cores assessed the performance of the model on the remaining 223 biopsy cores. The latter is divided into three subgroups according to the distance of the target to the prostate boundary and agreement between axial and sagittal histopathology labels. We achieved an AUC of 0.73 for \(D2-A\) and 0.77 for \(D2-B\). For cores from targets with moderate MR suspicious level in \(D2-B\), we achieved AUC of 0.80, where mp-MRI has low positive predictive value. Our results show that analysis of temporal US data is a promising technology for accurate classification of tissue labels that were identified in mp-MRI as suspicious and can potentially complement mp-MRI for TRUS-guided biopsy.
While the physical phenomenon governing temporal ultrasound/tissue interaction is the subject of ongoing investigation in our group, several hypotheses have been explored so far. It has been proposed that the acoustic radiation force of the transmit US signal increases the temperature and changes the speed of sound in different tissue types [4]. It has also been suggested that a combination of micro-vibration of acoustic scatters in microstructures and the density of cells play a role [21]. Our results showed a consistently high classification accuracy in a large dataset in this paper. These results suggest that the phenomenon is consistent for the two independent training and test datasets in clinical settings. Interestingly, the range of frequencies that we have identified as most discriminative between cancerous and benign tissue (\(0-2\) Hz in Fig. 8) is also consistent with the ranges we have observed in our previous independent studies [14, 15]. Currently, we are performing controlled laboratory experiments to further investigate this phenomenon.
Since DBN is a computationally expensive method, we plan to use graphics processing unit (GPU) parallelization to optimize our proposed method for real-time display of cancer likelihood maps. Currently, the execution time for generating a cancer probability map overlaid on a B-mode US image using an Intel Core i7 CPU with 16 GB RAM is approximately 6 minutes. Integration with the UroNav targeted biopsy interface is also underway to run a prospective clinical study for cancer localization.
Notes
Canadian cancer society: http://www.cancer.ca/, and American cancer society: http://www.cancer.org/.
References
Azizi S, Imani F, Zhuang B, Tahmasebi A, Kwak JT, Xu S, Uniyal N, Turkbey B, Choyke P, Pinto P, Wood B (2015) Ultrasound-based detection of prostate cancer using automatic feature selection with deep belief networks. Med Image Comput Comput-Assist Interv—MICCAI 2015, pp 70–77. Springer (2015)
Bengio Y, Lamblin P, Popovici D, Larochelle H (2007) Greedy layer-wise training of deep networks. Adv Neural Inf Process Syst 19:153
Correas JM, Tissier AM, Khairoune A, Khoury G, Eiss D, Hélénon O (2013) Ultrasound elastography of the prostate: state of the art. Diagn Interv Imaging 94(5):551–560
Daoud MI, Mousavi P, Imani F, Rohling R, Abolmaesumi P (2013) Tissue classification using ultrasound-induced variations in acoustic backscattering features. IEEE Trans Biomed Eng 60(2):310–320
Epstein JI, Feng Z, Trock BJ, Pierorazio PM (2012) Upgrading and downgrading of prostate cancer from biopsy to radical prostatectomy: incidence and predictive factors using the modified Gleason grading system and factoring in tertiary grades. Eur Urol 61(5):1019–1024
Erhan D, Bengio Y, Courville A, Manzagol PA, Vincent P, Bengio S (2010) Why does unsupervised pre-training help deep learning? J Mach Learn Res 11:625–660
Fawcett T (2006) An introduction to roc analysis. Pattern Recognit Lett 27(8):861–874
Feleppa, E., Porter, C., Ketterling, J., Dasgupta, S., Ramachandran, S., Sparks, D.: Recent advances in ultrasonic tissue-type imaging of the prostate. In: Acoustical imaging, pp 331–339. Springer (2007)
Goossen T, Wijkstra H (2003) Transrectal ultrasound imaging and prostate cancer. Archivio Italiano di Urologia Andrologia 75(1):68–74
Hinton G (2010) A practical guide to training restricted Boltzmann machines. Momentum 9(1):926
Hinton GE, Osindero S, Teh YW (2006) A fast learning algorithm for deep belief nets. Neural Comput 18(7):1527–1554
Imani F, Abolmaesumi P, Gibson E, Khojaste A, Gaed M, Moussa M, Gomez JA, Romagnoli C, Siemens DR, Leviridge M, Chang S, Fenster A, Ward AD, Mousavi P (2013) Ultrasound-based characterization of prostate cancer: an in vivo clinical feasibility study. Med Image Comput Comput-Assist Interv–MICCAI 2013, pp 279–286, Springer
Imani F, Abolmaesumi P, Gibson E, Khojaste A, Gaed M, Moussa M, Siemens DR, Fenster A, Ward A, Mousavi P (2015) Computer-aided prostate cancer detection using ultrasound RF time series: in vivo feasibility study. IEEE Trans Med Imaging 34(11):2248–2257
Imani F, Ramezani M, Nouranian S, Gibson E, Khojaste A, Gaed M, Moussa M, Gomez J, Romagnoli C, Leveridge M, Chang S (2015) Ultrasound-based characterization of prostate cancer using joint independent component analysis. IEEE Trans Biomed Eng 62(7):1796–1804
Imani, F., Zhuang, B., Tahmasebi, A., Kwak, J.T., Xu S, Agarwal H, Bharat S, Uniyal N, Turkbey I, Choyke P, Pinto P (2015) Augmenting MRI-transrectal ultrasound-guided prostate biopsy with temporal ultrasound data: a clinical feasibility study. Int J Comput Assist Radiol Surg, pp 1–9 (2015)
Kuru TH, Roethke MC, Seidenader J, Simpfendörfer T, Boxler S, Alammar K, Rieker P, Popeneciu V, Roth W, Pahernik S, Schlemmer H (2013) Critical evaluation of magnetic resonance imaging targeted, transrectal ultrasound guided transperineal fusion biopsy for detection of prostate cancer. J Urol 190(4):1380–1386
Marks L, Young S, Natarajan S (2013) MRI-ultrasound fusion for guidance of targeted prostate biopsy. Curr Opin Urol 23(1):43
Miyagawa T, Tsutsumi M, Matsumura T, Kawazoe N, Ishikawa S, Shimokama T, Miyanaga N, Akaza H (2009) Real-time elastography for the diagnosis of prostate cancer: evaluation of elastographic moving images. Jpn J Clin Oncol 39(6):394–398
Moradi M, Abolmaesumi P, Mousavi P (2010) Tissue typing using ultrasound RF time series: experiments with animal tissue samples. Med Phys 37(8):4401–4413
Moradi, M., Mahdavi, S.S., Nir, G., Jones, E.C., Goldenberg, S.L., Salcudean, S.E.: Ultrasound RF time series for tissue typing: first in vivo clinical results. In: SPIE Medical Imaging, pp. 86,701I–86,701I. International Society for Optics and Photonics (2013)
Moradi M, Mahdavi SS, Nir G, Mohareri O, Koupparis A, Gagnon L, Fazli L, Casey R, Ischia J, Jones E, Goldenberg S (2014) Multiparametric 3D in vivo ultrasound vibroelastography imaging of prostate cancer: Preliminary results. Med Phys 41(7)
Moradi M, Mousavi P, Boag A, Sauerbrei EE, Siemens D, Abolmaesumi P (2009) Augmenting detection of prostate cancer in transrectal ultrasound images using SVM and RF time series. IEEE Trans Biomed Eng 56(9):2214–2224
Nelson ED, Slotoroff CB, Gomella LG, Halpern EJ (2007) Targeted biopsy of the prostate: The impact of color Doppler imaging and elastography on prostate cancer detection and Gleason score. Urology 70(6):1136–1140
Park S, Aglyamov SR, Emelianov SY (2007) Elasticity imaging using conventional and high-frame rate ultrasound imaging: Experimental study. IEEE Trans Ultrason Ferroelectr Freq Control 54(11):2246–2256
Rapiti E, Schaffar R, Iselin C, Miralbell R, Pelte MF, Weber D, Zanetti R, Neyroud-Caspar I, Bouchardy C (2013) Importance and determinants of Gleason score undergrading on biopsy sample of prostate cancer in a population-based study. BMC Urol 13(1):19
de Rooij M, Hamoen EH, Fütterer JJ, Barentsz JO, Rovers MM (2014) Accuracy of multiparametric MRI for prostate cancer detection: a meta-analysis. Am J Roentgenol 202(2):343–351
Schmitz G, Ermert H, Senge T (1999) Tissue-characterization of the prostate using RF ultrasonic signals. IEEE Trans Ultrason Ferroelectr Freq Control 46(1):126–138
Tanaka M, Okutomi M (2014) A novel inference of a restricted Boltzmann machine. In: International conference on pattern recognition (ICPR), 2014 22nd, pp 1526–1531. IEEE (2014)
Turkbey B, Mani H, Aras O, Ho J, Hoang A, Rastinehad A, Agarwal H, Shah V, Bernardo M, Pang Y, Daar D (2013) Prostate cancer: Can mp-MR imaging help identify patients who are candidates for active surveillance? Radiology 268(1):144–152
Xie SW, Li HL, Du J, Xia JG, Guo YF, Xin M, Li FH (2013) Influence of serum PSA level, prostate volume, and PSA density on prostate cancer detection with contrast-enhanced sonography using contrast-tuned imaging technology. J Ultrasound Med 32(5):741–748
Xu S, Kruecker J, Turkbey B, Glossop N, Singh AK, Choyke P, Pinto P, Wood B (2008) Real-time MRI-TRUS fusion for guidance of targeted prostate biopsies. Comput Aided Surg 13(5):255–264
Yosinski J, Lipson H (2012) Visually debugging restricted Boltzmann machine training with a 3D example. In: 29th international conference on machine learning, representation learning workshop 2012
Acknowledgments
This work was supported in part by the Natural Sciences and Engineering Research Council of Canada (NSERC) and the Canadian Institutes of Health Research (CIHR).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards.
Rights and permissions
About this article

Cite this article
Azizi, S., Imani, F., Ghavidel, S. et al. Detection of prostate cancer using temporal sequences of ultrasound data: a large clinical feasibility study. Int J CARS 11, 947–956 (2016). https://doi.org/10.1007/s11548-016-1395-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11548-016-1395-2