
Abstract
Increasing knowledge of the underlying signaling pathways and molecular defects involved in colorectal cancer growth or progression enabled the discovery of several prognostic and predictive biomarkers, leading to the development of novel molecularly targeted therapies. The mitogen-activated protein kinase (MAPK) signaling pathway plays a critical role in colorectal cancer progression. Mutations in BRAF, a principal effector of Ras in this signaling cascade, are found in 10 % of colorectal cancer and play a clear pathogenic role, particularly in patients with metastatic disease. Intense efforts have therefore focused on targeting BRAF as an oncogenic driver, with mixed early results. This article summarizes the molecular and clinical features of BRAF mutant colorectal cancer, the prognostic and predictive role of BRAFV600E mutation in colorectal cancer, initial clinical trial results in targeting BRAFV600E, and the more recent preclinical insights into potential mechanisms of resistance to BRAF inhibition that have now led to a number of rationale-driven combination therapeutic strategies.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Colorectal cancer is a major cause of cancer death in developed countries. Increasing knowledge of the underlying signaling pathways and molecular defects involved in carcinogenesis has led to the development of several novel target-based therapeutics, resulting in encouraging improvements in patient outcome. In parallel with these therapeutic advances, a wide range of prognostic and predictive biomarkers of anticancer treatment have been investigated, most of which have failed to translate into clinical utility. A search of PubMed for “colorectal cancer biomarker” yields over 16,400 entries as of December 2013. The majority of these publications report positive results. Yet despite so many potential candidates, RAS is the only predictive biomarker to achieve significant clinical utility, albeit as a negative predictive marker to agents targeting the epidermal growth factor receptor (EGFR).
A major challenge in colorectal cancer research is the identification of other prognostic and predictive biomarkers which will allow clinicians to better tailor treatment for individual patients. The Ras/Raf/MEK/ERK mitogen-activated protein kinase (MAPK) signaling pathway plays a critical role in colorectal cancer progression and has therefore been a high priority for the development of molecularly targeted agents. BRAF, a principal effector of Ras in this signaling cascade, was found to be mutated in a variety of cancers, including a small percentage (~10 %) of colorectal cancers [1–4]. Antiproliferative and antitumor activity of BRAF inhibitors has been observed in most of the BRAFV600E-bearing colorectal cancer cell lines tested and in the HT29 BRAFV600E-expressing colorectal cancer xenograft model, suggesting that BRAFV600E is a viable therapeutic target in colorectal cancer [5]. Efficacy results from randomized trials and initial results from an early-phase trial of the specific BRAFV600E inhibitor in melanoma and colorectal cancer, respectively, confirmed the validity of BRAFV600E as a therapeutic target as well as a predictive molecular biomarker for response to BRAF inhibition. This article will review the role of BRAFV600E in colorectal cancer, with emphasis on molecular and clinicopathological associations, prognostic and predictive implications of BRAFV600E, and finally some emerging therapeutic targeting strategies for BRAF mutant metastatic colorectal cancers.
BRAF oncogenic signaling pathway
The first mammalian effector of RAS to be characterized, and the most intensively studied, is the protein serine/threonine kinase v-raf murine sarcoma viral oncogene homolog (RAF). The Raf family of genes were first identified as potent retrovirus oncogenes in 1984 [6, 7]. RAF protein contains three conserved regions: CR1, CR2, and CR3 [8]. CR1 and CR2 in the N terminus are largely regulatory, whereas CR3 at the C terminus encompasses the catalytic kinase domain. RAF regulation is a complex process involving many steps. Upon activation, GTP-bound RAS recruits RAF protein to the cell membrane and binds it directly to activate RAF kinase, with one of the essential steps being the phosphorylation of two amino acids (T599 and S602 of BRAF) within the activation segment of the kinase domain. RAF then phosphorylates and activates its downstream effectors, including mitogen-activated protein kinase kinase (MAPKK or MEK)-1, MEK-2, ERK1, and ERK2, eventually leading to cell growth and proliferation.
The BRAF serine/threonine kinase is a member of the Raf kinase family consisting of A-RAF, B-RAF, and C-RAF or RAF-1 [9]. Although all three Raf isoforms share considerable sequence homology and exhibit the same substrate specificity (MEK1 and MEK2), they do differ in their biological functions and regulations, which have not been fully elucidated [10]. Raf remains the best characterized activator of MEK, with BRAF being the most potent MEK activator of the Raf isoforms. In BRAF, S446 is constitutively phosphorylated, priming the N-region for activation with a constitutive negative charge, and this occurs when BRAF is recruited to the plasma membrane for activation segment phosphorylation. In contrast, A-RAF and C-RAF activation require N-region phosphorylation in addition to activation segment phosphorylation.
Among the Raf kinase family members, high-throughput genomic sequencing has identified activating mutations in BRAF as the predominant genetic alterations in human cancers [8, 11]. Our work and that of others have found that KRAS and BRAF mutations almost never occur in the same tumor, suggesting not only that BRAF is the principal effector of KRAS in the MAPK pathway but also that they may be equally important in their tumorigenic effects [1, 2, 12, 13].
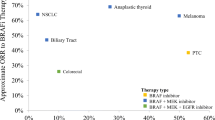
The discovery of BRAF mutations in various human cancers has stimulated intensive research of this gene. In solid tumors, the highest incidence of BRAF mutations is in malignant melanoma (27–70 %), papillary thyroid cancer (36–53 %), colorectal cancer (5–22 %), and serous ovarian cancer (~30 %), but they also occur at a lower frequency (1–3 %) in a wide variety of other cancers including NSCLC [14–16]. Over 40 missense mutations in BRAF have been reported, but most of these are extremely rare. The most common mutation in BRAF, accounting for up to 90 % of all BRAF mutations in human cancers, is a thymidine-to-adenine transversion at nucleotide 1799 in the kinase domain of the protein resulting in a V600E amino acid (valine to glutamate) exchange [17, 18]. This mutation leads to constitutive activation of the MAPK-signaling cascade with the mutated protein demonstrating greatly elevated kinase activity and potently transforms rodent fibroblasts and other cell types [11, 19]. Furthermore, while mutant BRAFV600E cells were shown to be dependent on continued BRAF activity for their tumorigenic growth [20–22], they do not require Ras function for proliferation [11].
Molecular and clinico-pathological characteristics
Molecular
BRAF mutations have been shown to be an early event in colorectal tumorigenesis and are associated with a specific pattern of tumor growth known as the serrated pathway [23, 24]. Consistent with the theory that BRAF mutation occurs early in the adenoma–carcinoma sequence, our work and a recent systematic review of published data demonstrated a very high concordance rate (97 %) of BRAF mutations in matched primary tumor and metastatic tissue [2, 25].
It has been consistently demonstrated that BRAF mutations are strongly associated with the microsatellite instability (MSI) and CpG island methylator phenotypes (CIMP). In sporadic colorectal cancers, BRAF mutation is found in about 40 to 60 % of MSI tumors, while only approximately 5 to 10 % of microsatellite stable (MSS) tumors harbor this mutation [2, 11–13, 26–28]. When tumors’ methylation status was measured at specific promoter loci, BRAF mutant tumors were more commonly found to be highly methylated (CIMP-high) compared to BRAF wild-type tumors [29–34]. Analysis of a recent series of 649 colon cancers demonstrated that 70 % of BRAF mutant tumors exhibited CIMP-high, and 18 % of BRAF wild-type tumors exhibited CIMP-low profile [35].
It is well established that sporadic MSI tumors are related to the loss of MLH1 expression through hypermethylation of the MLH1 gene promoter, which is strongly associated with BRAFV600E mutation [26, 36]. Conversely, germline MMR gene mutation (or Lynch syndrome) is almost never present when a BRAFV600E mutation is identified. As such, various groups have suggested adding BRAFV600E mutation analysis as further step to characterize colorectal cancer found to have loss of MLH1 expression [37, 38]. The finding of a BRAFV600E mutation, consistent with sporadic MSI colorectal cancer rather than a germline defect, then reduces the number of patients with an abnormal MMR IHC requiring follow-up with genetic counseling and MLH1 sequencing.
Clinico-pathological
The clinical development of BRAF inhibitors in colorectal cancer carrying the BRAFV600E mutation, the enriched population, will be challenging due to the low BRAFV600E mutation rate in this tumor type. Previous studies have reported an association between BRAF mutation and older age, female gender, and right-sided tumor location. Our group investigated a strategy to enrich the colorectal cancer population with BRAFV600E mutation by selecting patients with clinical features associated with this mutation [2]. In addition to potentially reducing the cost of screening, understanding this clinico-molecular correlation will also allow for more informed clinical development of BRAF inhibitors.
Our study demonstrated that subsets of colorectal patients with a higher prevalence of BRAFV600E can, in principle, be identified using a combination of basic clinical features including female gender, age > 70 years and right-sided tumor location, all of which were found to be independent predictors of BRAFV600E. BRAF mutation was rarely found in left-sided colon (4 %) and rectal cancers (2 %) when compared to right-sided cancers (22 %, P < 0.0001). Enrichment could further be improved by inclusion of KRAS mutation status; for example, the prevalence of BRAFV600E increased from 10 % in unselected patients to 37 % in females over the age of 70 years with right-sided cancer and to 50 % when KRAS wild-type status was included. Importantly, we have identified a subpopulation of patients (male patients under 70 years with left-sided or rectal cancers), where BRAFV600E was rarely found. While this knowledge is unlikely to replace mutation testing in the setting of clinical trials using BRAF inhibitors, it can help improve the efficiency of prescreening for BRAFV600E by defining a subpopulation of patient, where the yield from mutation testing will be very low.
A higher frequency of BRAF mutation has also been reported in poorly differentiated and mucinous tumor [1, 2, 31]. BRAF mutant tumors were reported to be poorly differentiated in 40 to 50 % of cases and well to moderately differentiated only in 6 to 16 % of cases. Mucinous cancers have a BRAF mutation rate of approximately 22 to 67 % compared to only 6 to 21 % in non-mucinous tumors [2, 31, 32, 39, 40].
As has been well described in some other cancer subtypes, specific oncogenic drivers may result in differences in phenotypic behavior of that subtype. For example, HER-2/neu overexpressing breast cancer is further defined by an increased incidence of central nervous system (CNS) metastases [41]. BRAF mutant metastatic colorectal cancer also appears to be a discrete disease subtype with a distinct patient population and significantly poorer survival. Tran et al. [42] investigated whether BRAF mutant colorectal cancer has a distinct pattern of metastatic spread, further defining this discrete disease subtype. This study of 524 metastatic colorectal cancer patients from both the Royal Melbourne Hospital and The University of Texas MD Anderson Cancer Center reported, for the first time, a significantly increased rate of peritoneal and distant lymph node metastases and a significantly decreased rate of lung metastases in BRAF mutant tumors compared with BRAF wild-type tumors. Although this study does not confirm peritoneal metastases as a poor prognostic factor, this may be because of small numbers and should not detract from existing reports that identify peritoneal metastases as an independent and significant poor prognostic factor in colorectal cancer [43, 44]. The strong association between BRAF mutant tumors and peritoneal metastases observed in this study may partially explain the poorer outcomes in BRAF mutant tumors.
Prognostic and predictive implications of BRAFV600E mutation
Prognostic Role
To date, the BRAFV600E mutation is the only oncogenic mutation that has been consistently shown to be associated with a poor prognosis in metastatic colorectal cancer, including data from several retrospective population-based studies and clinical trials [1, 2, 4, 35, 45–47]. Samowitz et al. [1] examined a large (n = 911) population-based sample of individuals with stage I to IV colon cancer and found BRAFV600E mutation to be associated with a poor 5-year overall survival (5-year overall survival: BRAF mutant vs. wild-type, 47.5 vs. 60.7 %, log-rank P < 0.01). Importantly, this study noted that BRAFV600E mutation was associated with a poor prognosis in microsatellite-stable tumors only (5-year overall survival: BRAF mutant versus wild-type, 16.7 vs. 60.0 %, log-rank P < 0.01). This was true in a multivariate analysis after adjustment for age, stage, and tumor site, in stage-stratified analyses for AJCC stages II to IV cancers, and in a Kaplan–Meier analysis for stages II to IV cancers. The V600E mutation did not have the same effect on survival in tumors with MSI, as microsatellite-unstable tumors with or without the BRAFV600E mutation were associated with an excellent percent 5-year survival (76.2 and 75.0 %, respectively).
As observed in the CAIRO-2 study, a phase III randomized trial in which capecitabine, oxaliplatin, and bevacizumab (CB group) was compared with the same combination plus cetuximab (CBC group) as first-line treatment, BRAF mutation seems to confer a bad prognosis independent of anti-EGFR therapy [4]. Patients with BRAF-mutated tumors had a significantly shorter median progression-free survival (PFS) and median overall survival (OS) than patients with BRAF wild-type tumors, both in the CB group and in the CBC group. Similarly, in the recent pooled analysis of the CRYSTAL (cetuximab combined with irinotecan in first-line therapy for metastatic colorectal cancer) and OPUS (oxaliplatin and cetuximab in first-line treatment of metastatic colorectal cancer) studies, 800 of 845 KRAS wild-type tumor samples were assessable for BRAF mutation [48]. BRAF mutation was detected in 9 % (70/800) of the KRAS wild-type patient population in this pooled analysis. BRAF mutation is associated with a poor prognosis in both the chemotherapy and the chemotherapy in combination with cetuximab arms, with median overall survivals of 9.9 and 14.1 months, respectively, as compared to median overall survivals of 21.1 and 24.8 months in the BRAF wild-type patients (Table 1).
Several studies have examined the prognostic significance of BRAF mutation in stage II and III colon cancers [3, 28, 49]. In the first of these studies, French et al. examined the prognostic significance of tumor MSI status and the presence of mutation in BRAFV600E mutation in a group of patients (n = 533) who participated in a randomized prospective clinical trial through the North Central Cancer Treatment Group. BRAFV600E mutation did not impart any significant impact on disease-free or overall survival in this group of patients with curatively resected colon cancer. In contrast to Samowitz et al.’s finding, within the subgroup of patients with MSI tumors, the 23 patients with BRAF wild-type tumors had significantly better OS than the 35 BRAF mutant patients (100 % versus 77 %, P = 0.001); no difference was observed by BRAF status in the 432 MSS patients for DFS or OS (P ≥ 0.57). The observed discrepancies between these two studies may be due to the differences in the two study populations. The Samowitz et al. study included patients with all stages of disease (stage I to IV) while the French et al. study included only high-risk stage II and stage III disease. Subjects included in the former study were derived from a routine care population rather than a controlled clinical trial setting as in the latter study.
Other subsequent studies including patients with stages II and III colon cancer have however failed to reproduce French et al.’s findings. The biomarker sub-study of the PETACC-3 trial reported on the prognostic value of KRAS and BRAF mutations in 1,404 patients with stages II and III colon cancers [3]. In both univariate and multivariate analysis, BRAF mutation was not prognostic for recurrence-free survival, but was for overall survival, particularly in patients with MSS tumors (hazard ratio 2.2, 95 % CI 1.4 to 3.4, P = 0.0003). Farina-Sarasqueta et al. [49] examined the impact on clinical outcome of KRAS, BRAF mutations, and MSI status in 106 stage II and 258 stage III curatively resected colon cancers. In both groups, there was a trend toward a longer OS for BRAF wild-type patients (P = 0.194 stage II and 0.069 stage III) compared to BRAF mutant. DFS was not significantly different between BRAF mutants and wild-type tumors. Consistent with Samowitz et al.’s finding, when stratifying for MSI status, BRAF mutation resulted in shorter survival in MSS patients in both stage II and stage III disease but not in the MSI group. In a multivariate analysis by the Cox proportional hazards model including differentiation grade, age as a continuous variable, sex, tumor location, T stage, N stage, KRAS status, BRAF status and MSI status, BRAF mutation was an independent factor for a shorter OS [hazards ratio (HR) = 0.45, 95 % confidence interval (CI) 0.25–0.8].
Predictive role
A predictive molecular biomarker defines a subpopulation of patients who are more or less likely to gain benefit from a specific therapeutic intervention, using underlying genetic or epigenetic changes in colorectal cancer to guide personalized treatment decisions. Other than KRAS, there are a couple of strong rationales to explore BRAFV600E mutation as a candidate additional biomarker of anti-EGFR mAb resistance: BRAF is the immediate downstream effector of KRAS in the Ras/Raf/MAPK-signaling pathway (Fig. 1); KRAS and BRAFV600E mutations are almost always mutually exclusive, suggesting that activation of only one of these genetic alterations is sufficient for tumorigenesis and cancer progression.

Oncogenic signaling of the MAPK and PI3K/mTOR pathways in colorectal cancer. EGFR over-expression and mutation in KRAS and BRAF result in constitutive activation of this signaling cascade in colorectal cancer, ultimately affecting nuclear targets involved in regulating cell proliferation, differentiation, survival, migration, and angiogenesis. KRAS and BRAFV600E mutation rarely co-exist in the same tumor suggesting that Raf is the principal effector of Ras in tumorigenesis
Indeed, several studies have suggested that the presence of the BRAFV600E mutation is associated with resistance to anti-EGFR mAb therapy in chemo-refractory metastatic colorectal cancer [50–52]. Di Nicolantonio et al. [50] in their retrospective analysis of 113 colorectal tumors from patients who received anti-EGFR mAbs in second or subsequent lines of treatment showed that 0/11 (0 %) tumors with BRAF mutation responded to anti-EGFR treatment compared to 22/68 (32 %) of BRAF wild-type/KRAS wild-type tumors. Patients with BRAFV600E mutations also had statistically significant shorter progression-free survival (P = 0.001) and overall survival (P < 0.001) than patients whose tumors harbored BRAF wild-type. Similar results were seen for patients treated with a combination of cetuximab and irinotecan. None of the patients with BRAF mutant tumors (0/13) responded compared to 24/74 (32 %) patients with BRAF wild-type/KRAS wild-type tumors [51].
De Roock et al. [52] examined the effect of downstream mutations of KRAS on the efficacy of cetuximab in the largest cohort to date of patients with chemotherapy–refractory metastatic colorectal cancer treated with cetuximab plus chemotherapy (n = 773). They found that in KRAS wild-type patients, carriers of BRAF mutation had a significantly lower response rate than did patients with BRAF wild-type tumors, with a response rate of 8.3 % (2/24) in BRAF mutant versus 38 % (124/326) in BRAF wild-types (OR 0.15, 95 % CI 0.02–0.51; P = 0.0012).
Nonetheless, not all studies have found a negative relationship between BRAF mutation status and anti-EGFR mAb response, particularly in the first-line setting. In an analysis of 231 tumors from patients treated with first-line cetuximab plus capecitabine, oxaliplatin, and bevacizumab in the CAIRO-2 study, there was no significant difference in response rate between the BRAF mutant and wild-type subgroups (39 vs. 48 %; P = 0.43) [4]. In the recent pooled analysis of the CRYSTAL and OPUS studies [48], there was a nonsignificant trend toward a survival benefit from adding cetuximab to first-line chemotherapy in BRAF mutant/KRAS wild-type patients (9.9 months versus 14.1 months, P = 0.0764). It would appear that BRAF mutations does not have a strong predictive role for cetuximab in combination with a standard cytotoxic treatment in the first-line setting, but these data need to be interpreted with caution due to the strong prognostic impact of BRAF mutations, the potential interactions with chemotherapy, and the small number of patients with BRAF mutations.
BRAF as a therapeutic target in colorectal cancer
The “oncogenic addiction” of the MAPK pathway in colorectal cancer is reflected by the frequent perturbation of this pathway in this malignancy, with EGFR amplification and expression reported in up to 18 % [53, 54] and 62 % of cases [54–56], respectively, and activating mutations in KRAS and BRAF reported in 30–50 and 10 % of cases, respectively [2, 12, 13, 57].
The challenges of directly targeting KRAS in cancer has shifted the focus of therapeutic development to its downstream effector, BRAF. Moreover, KRAS is central to a complex network of signal transduction pathways, including the phosphatidylinositol 3-kinase (PI3K) pathway, characterized by cross-talk and feedback loops [18]. BRAF, on the other hand, would appear to be a “purer” target with a relatively unidirectional MEK–ERK effector pathway. The finding that tumor cells and xenografts harboring the BRAFV600E mutation were extremely sensitive to MEK inhibition compared with those without this mutation or those bearing the Ras mutation further highlights the dependency of BRAFV600E mutant cancer cells on MEK–ERK signaling [58]. Consistent with this is the preclinical work with vemurafenib (RG7204; PLX4032; RO5185426), a first-in-class, specific small molecule inhibitor of BRAFV600E, which demonstrated the selective sensitivity of colorectal cancer cell lines and xenografts harboring BRAFV600E mutation to BRAFV600E inhibition compared with BRAF wild-type cells or xenografts [5, 59]. As a single agent, vemurafenib shows dose-dependent inhibition of ERK and MEK phosphorylation, thereby arresting cell proliferation in BRAFV600E-expressing cell lines and inhibiting tumor growth in BRAFV600E-bearing xenograft models.
The first “proof-of-concept” clinical data validating BRAFV600E mutation as a therapeutic target came from the preliminary result of the phase I study of PLX4032 in advanced melanoma that was first presented at the 45th American Society of Clinical Oncology (ASCO) annual meeting [60, 61]. Notably, patients in the initial cohorts were not stratified by BRAFV600E mutational status; of the 21 melanoma patients treated at the ≥240 mg twice daily dose level (minimum target dose for tumor regression), 16 carried the BRAFV600E mutation and five did not. The efficacy data were extremely promising with nine partial responses, all seen in tumors carrying the BRAFV600E mutation. The interim progression-free survival (PFS) for patients with BRAF-mutated melanoma was 6 months, with many patients still on treatment, while all five patients with BRAF wild-type melanomas had progressive disease. The safety data were encouraging, with the majority of adverse events being mild and transient. The most common side effects were rash, fatigue, and photosensitivity. Cutaneous squamous cell carcinoma following chronic dosing was also observed. Initial data suggesting a favorable therapeutic index and selectivity of this BRAFV600E inhibitor therefore provide an appealing therapeutic strategy for BRAFV600E-mutated cancers.
A subsequent randomized open-label trial that involved 675 patients with unresectable or metastatic melanoma carrying the BRAFV600E mutation showed a superior overall survival and progression-free survival benefit in patients treated with vemurafenib than patients treated with dacarbazine (control group) [61]. The confirmed, investigator-assessed best overall response rate was 48 % for patients receiving vemurafenib (including 2 patients with complete responses) compared to 5 % for patients receiving dacarbazine. On the basis of its treatment effects, vemurafenib received FDA approval in August 2011 for the treatment of patients with unresectable or metastatic melanoma with the BRAFV600E mutation, establishing a new treatment standard for patients with metastatic melanoma.
Disappointingly, the clinical activity with BRAFV600E inhibition seen in metastatic colorectal cancer is more modest than had been observed in melanoma. Results from a phase I extension study, where 21 patients with BRAFV600E mutant metastatic colorectal cancer were treated with PLX 4032, demonstrated one confirmed partial response and four minor responses among 19 evaluable patients. 5 patients showed a mixed response pattern (i.e., with both regressing and progressing lesions), and 2 of the 3 patients who underwent repeat FDG-PET imaging achieved a metabolic response [62]. Nonetheless, this modest activity does confirm mutant BRAF as a potential therapeutic target in colorectal cancer, assuming the mechanisms underlying the variations in patient activity could be identified. Additionally, it would be reasonable to assume that the functional biology of BRAF activation in these two tumor types is quite different, based on the marked differences in responses to BRAF inhibition. Efforts have since focused on better understanding what additional mechanisms of dependence on BRAFV600E as well as potential causes of early resistance in colorectal cancer. Much of the preclinical work to date has focused on genetic (e.g., other mutated kinases), epigenetic (e.g., methylation), and functional (e.g., activation of other pathways in response to BRAF inhibition) mechanisms that are both shared with those seen in melanoma as well as those that might be unique to colorectal cancer.
Mechanisms of resistance to BRAF inhibitors
Recently, a number of groups of investigators independently reported their preclinical findings on the potential mechanisms responsible for the de novo resistance of BRAFV600E colorectal cancer to BRAF inhibition [59, 63]. Prahallad et al. [59] confirmed, as seen clinically, that BRAFV600E mutant melanoma cells are more sensitive to BRAF inhibition than BRAFV600E mutant colorectal cancer cells. Notably, they found that treatment of several BRAF mutant colorectal cell lines with PLX4032 resulted in significant increase in EGFR phosphorylation, reflecting activation of the EGFR receptor. This strong feedback activation of EGFR incited by BRAF inhibition was shown to be ligand-dependent. Critically, they were able to demonstrate how this feedback loop was activated. They showed that ERK, which is normally activated (pERK) in BRAF mutant tumors, regulates production of a phosphatase (CD25c) that, in turn, negatively regulates EGFR signaling. Inhibition of pERK with vemurafenib stops that negative regulatory process, leading to rapid activation of EGFR, and therefore continued cell proliferation via multiple other downstream pathways. Interestingly, EGFR activation was not seen in melanoma cell lines treated with vemurafenib, using the same experimental approach, providing further evidence that BRAF mutant CRC is functionally different to melanoma. The use of an EGFR inhibitor with vemurafenib effectively abrogated this resistance mechanism. In these studies, both EGFR antibodies (cetuximab) and small molecules (erlotinib) proved effective as combinations indicating that the activation of the EGFR pathway was ligand-dependent.
Further supportive experimental work was published at a similar time. Corcoran et al. [63] similarly observed an EGFR-mediated rapid reactivation of the MAPK pathway in BRAF mutant colorectal cancer cells to BRAF inhibition with PLX4032. However, this resistance mechanism appears to involve the activation of RAS by EGFR and induction of phospho-CRAF rather than an increased EGFR phosphorylation per se as seen in Prahallad et al.’s study. Previous studies have shown that in the face of BRAF inhibition, RAS activation can lead to MAPK pathway activation via direct activation of CRAF or by the transactivation of BRAF-CRAF heterodimers [64–66]. Once again, it was observed that BRAF mutant colorectal cancer cells expressed higher levels of EGFR and phospho-EGFR compared to BRAF mutant melanoma cells, suggesting that colorectal cancer may be more susceptible to EGFR-mediated resistance than melanoma. This is not altogether surprising, given the importance of signaling through EGFR in colorectal cancer, not just BRAF mutant colorectal cancer.
Two other studies have recently suggested that PI3K/AKT pathway activation is an alternative resistance mechanism to BRAF inhibition in BRAF mutant colorectal cancer [67, 68]. PI3K signaling is activated in human cancers via several different mechanisms: direct mutational activation or amplification of PIK3CA and AKT1, or loss of PTEN. PI3K can also be activated by genetic mutation and/or amplification of upstream RTKs and possibly by mutationally activated Ras [69]. Nearly 40 % of colorectal cancer have been shown to have alterations in one of eight PI3K pathway genes and these mutations are almost always mutually exclusive of one another [70]. Additionally, BRAF mutation co-exists with PIK3CA and PTEN mutations in 13 % and 22 % of colorectal cancers [71, 72], suggesting perhaps activation of the PI3K/AKT axis may partly account for resistance to BRAF inhibition in BRAF mutant colorectal cancer.
Mao et al. [67] recently reported that BRAF mutant colorectal cancer cell lines showed a higher levels of PI3K/AKT pathway activation compared to BRAF mutant melanoma cell lines. Importantly, BRAF mutant CRC cell lines with mutation in PTEN and PIK3CA were less sensitive to growth inhibition by a potent BRAF inhibitor, PLX4720. Using multiple novel genetically engineered mouse models for sporadic colorectal cancer, Corcoran et al. demonstrated sustained PI3K/mTOR activity in BRAFV600E mutant colorectal cancer upon BRAF inhibition.
Combination therapeutic strategies
Elucidating the resistance mechanisms to BRAF inhibition has provided strong rationale for using combination therapeutic strategies in treating patients with BRAF mutant colorectal, for whom survival outcome is poor with no current effective targeted treatment. It has been shown that both in vivo and in vitro inhibition of EGFR with either anti-EGFR monoclonal antibody cetuximab or tyrosine kinase inhibitor gefitinib or erlotinib are strongly synergistic with BRAF inhibition, resulting in sustained MAPK pathway suppression and markedly increased therapeutic efficacy [59, 63]. Based on these compelling series of studies, clinical trials with BRAF and EGFR inhibitors (both small molecules and antibodies), as well as trials targeting the “triple combination” (BRAF/EGFR/PIK3CA inhibition) in BRAF mutant colorectal cancer are now in early stage of clinical development. Results from these trials are eagerly awaited.
Yang et al. [5] also explored a range of combination therapies with both standard agents and targeted inhibitors in preclinical xenograft models. Increased antitumor activity and improved survival in xenograft models has been demonstrated with the administration of vemurafenib in combination with an AKT inhibitor (MK-2206), capecitabine and/or bevacizumab, cetuximab and/or irinotecan, or erlotinib. Collectively, these findings suggest that the administration of vemurafenib in combination with standard cytotoxics or novel targeted therapies may result in more promising clinical efficacy than vemurafenib monotherapy in patients with BRAF mutant colorectal cancer.
Conclusions
Individualizing patient treatment based on their tumors’ molecular profile, i.e., biomarker-guided personalized cancer treatment, remains one of the most important aims in cancer management. One of the earliest, and arguably most successful examples of such personalized cancer therapy comes from the discovery of estrogen and/or progesterone receptor status as a biomarker for predicting benefit from the estrogen receptor α antagonist, tamoxifen, in breast cancer [73]. In 1998, a decade later, FDA approved the use of trastuzumab for the treatment of HER2-amplified metastatic breast cancer and, subsequently, in the adjuvant setting. This is based on numerous studies demonstrating that HER2 over-expression and amplification, which occurs in 15 to 30 % of breast cancers, is a strong predictor of benefit from treatment with trastuzumab, an antibody that targets the receptor tyrosine kinase HER2 [74]. Today, breast cancers are routinely screened for estrogen receptor status and HER2 over-expression/amplification, and treatment is tailored to tumors’ molecular changes accordingly. Other predictive biomarkers which have a significant impact on current clinical practice are the presence of an EGFR-activating mutation in metastatic non-small-cell lung cancer and KRAS mutations in metastatic colorectal cancer, which has been shown to correlate with responses to treatment with EGFR tyrosine kinase inhibitors [75] and resistance to anti-EGFR monoclonal antibodies [76–78], respectively.
Over the past decade, BRAF has emerged as an important molecular target in cancer treatment in view of its oncogenic potential in various tumor types and the impressive clinical efficacy seen in BRAF mutant melanoma treated with the BRAFV600E inhibitor, vemurafenib. Nevertheless, the potent antitumor activity observed in melanoma with single-agent BRAF inhibition has not been reproduced in metastatic colorectal cancer, despite the fact that this molecular alteration is a robust biomarker of poor prognosis in colorectal cancer. Using various preclinical models, researchers have recently reported convincing evidence on the underlying mechanisms of resistance to BRAF inhibition, thereby providing rationale for combination therapeutic strategies. Additionally, the consistent finding of BRAFV600E mutation as a negative prognostic biomarker for survival outcome in patients treated with conventional cytotoxic chemotherapy with or without anti-EGFR MoAb may justify the development of novel targeted agents in the first- or second-line setting in these patients.
Many challenges lie ahead in the development of agents targeting BRAFV600E mutant cancers. Robust preclinical analyses with well-validated models will be required to better inform clinical trial design. The tremendous pace of advancement in molecular technology and cancer genome biology is capable of transforming many aspects of oncology diagnosis, clinical trial design, and treatment. However, the major challenge in the future is how best to take advantage of these new insights and to apply it clinically. The most efficient and promising way to move the biomarker field forward is to continue to improve our understanding of the inter-related network of cell signaling pathways by using well-annotated human tissue, correlating molecular changes with patient treatment and outcome data, ideally in the context of clinical trials. This, in turn, could be aided substantially by the development of targeted therapeutics. To this end, exploration of in vivo biology with pharmacodynamic markers (such as pre- and post-treatment biopsies) in patients treated with these agents could be highly informative and should be strongly encouraged when possible.
References
Samowitz WS, Sweeney C, Herrick J, Albertsen H, Levin TR, Murtaugh MA et al (2005) Poor survival associated with the BRAF V600E mutation in microsatellite-stable colon cancers. Cancer Res 65(14):6063–6069
Tie J, Gibbs P, Lipton L, Christie M, Jorissen RN, Burgess AW et al (2011) Optimizing targeted therapeutic development: analysis of a colorectal cancer patient population with the BRAF(V600E) mutation. Int J Cancer 128(9):2075–2084
Roth AD, Tejpar S, Delorenzi M, Yan P, Fiocca R, Klingbiel D et al (2010) Prognostic role of KRAS and BRAF in stage II and III resected colon cancer: results of the translational study on the PETACC-3, EORTC 40993, SAKK 60–00 trial. J Clin Oncol 28(3):466–474. doi:10.1200/jco.2009.23.3452. Epub 2009 Dec 14
Tol J, Nagtegaal I, Punt C (2009) BRAF mutation in metastatic colorectal cancer. N Engl J Med 361(1):98–99
Yang H, Higgins B, Kolinsky K, Packman K, Bradley WD, Lee RJ et al (2012) Antitumor activity of BRAF inhibitor vemurafenib in preclinical models of BRAF-mutant colorectal cancer. Cancer Res 72(3):779–789
Jansen HW, Lurz R, Bister K, Bonner TI, Mark GE, Rapp UR (1984) Homologous cell-derived oncogenes in avian carcinoma virus MH2 and murine sarcoma virus 3611. Nature 307(5948):281–284
Sutrave P, Bonner TI, Rapp UR, Jansen HW, Patschinsky T, Bister K (1984) Nucleotide sequence of avian retroviral oncogene v-mil: homologue of murine retroviral oncogene v-raf. Nature 309(5963):85–88
Michaloglou C, Vredeveld LC, Mooi WJ, Peeper DS (2008) BRAF(E600) in benign and malignant human tumours. Oncogene 27(7):877–895
Chong H, Vikis HG, Guan KL (2003) Mechanisms of regulating the Raf kinase family. Cell Signal 15(5):463–469
Schreck R, Rapp UR (2006) Raf kinases: oncogenesis and drug discovery. Int J Cancer 119(10):2261–2271
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S et al (2002) Mutations of the BRAF gene in human cancer. Nature 417(6892):949–954
Fransen K, Klintenas M, Osterstrom A, Dimberg J, Monstein HJ, Soderkvist P (2004) Mutation analysis of the BRAF, ARAF and RAF-1 genes in human colorectal adenocarcinomas. Carcinogenesis 25(4):527–533
Rajagopalan H, Bardelli A, Lengauer C, Kinzler KW, Vogelstein B, Velculescu VE (2002) Tumorigenesis: RAF/RAS oncogenes and mismatch-repair status. Nature 418(6901):934
Garnett MJ, Marais R (2004) Guilty as charged: B-RAF is a human oncogene. Cancer Cell 6(4):313–319
Marchetti A, Felicioni L, Malatesta S, Grazia Sciarrotta M, Guetti L, Chella A et al (2011) Clinical features and outcome of patients with non-small-cell lung cancer harboring BRAF mutations. J Clin Oncol 29(26):3574–3579
Cardarella S, Ogino A, Nishino M, Butaney M, Shen J, Lydon C et al (2013) Clinical, pathologic, and biologic features associated with BRAF mutations in non-small cell lung cancer. Clin Cancer Res 19(16):4532–4540
Dhillon AS, Hagan S, Rath O, Kolch W (2007) MAP kinase signalling pathways in cancer. Oncogene 26(22):3279–3290
Beeram M, Patnaik A, Rowinsky EK (2005) Raf: a strategic target for therapeutic development against cancer. J Clin Oncol 23(27):6771–6790
Roberts PJ, Der CJ (2007) Targeting the Raf-MEK-ERK mitogen-activated protein kinase cascade for the treatment of cancer. Oncogene 26(22):3291–3310
Hingorani SR, Jacobetz MA, Robertson GP, Herlyn M, Tuveson DA (2003) Suppression of BRAF(V599E) in human melanoma abrogates transformation. Cancer Res 63(17):5198–5202
Sharma A, Trivedi NR, Zimmerman MA, Tuveson DA, Smith CD, Robertson GP (2005) Mutant V599EB-Raf regulates growth and vascular development of malignant melanoma tumors. Cancer Res 65(6):2412–2421
Sumimoto H, Hirata K, Yamagata S, Miyoshi H, Miyagishi M, Taira K et al (2006) Effective inhibition of cell growth and invasion of melanoma by combined suppression of BRAF (V599E) and Skp2 with lentiviral RNAi. Int J Cancer 118(2):472–476
Lee EJ, Choi C, Park CK, Maeng L, Lee J, Lee A et al (2005) Tracing origin of serrated adenomas with BRAF and KRAS mutations. Virchows Arch 447(3):597–602
Chan TL, Zhao W, Leung SY, Yuen ST (2003) BRAF and KRAS mutations in colorectal hyperplastic polyps and serrated adenomas. Cancer Res 63(16):4878–4881
Italiano A, Hostein I, Soubeyran I, Fabas T, Benchimol D, Evrard S et al (2010) KRAS and BRAF mutational status in primary colorectal tumors and related metastatic sites: biological and clinical implications. Ann Surg Oncol 17(5):1429–1434
Wang L, Cunningham JM, Winters JL, Guenther JC, French AJ, Boardman LA et al (2003) BRAF mutations in colon cancer are not likely attributable to defective DNA mismatch repair. Cancer Res 63(17):5209–5212
Oliveira C, Pinto M, Duval A, Brennetot C, Domingo E, Espin E et al (2003) BRAF mutations characterize colon but not gastric cancer with mismatch repair deficiency. Oncogene 22(57):9192–9196
French AJ, Sargent DJ, Burgart LJ, Foster NR, Kabat BF, Goldberg R et al (2008) Prognostic significance of defective mismatch repair and BRAF V600E in patients with colon cancer. Clin Cancer Res 14(11):3408–3415
Ogino S, Cantor M, Kawasaki T, Brahmandam M, Kirkner GJ, Weisenberger DJ et al (2006) CpG island methylator phenotype (CIMP) of colorectal cancer is best characterised by quantitative DNA methylation analysis and prospective cohort studies. Gut 55(7):1000–1006
Slattery ML, Curtin K, Sweeney C, Levin TR, Potter J, Wolff RK et al (2007) Diet and lifestyle factor associations with CpG island methylator phenotype and BRAF mutations in colon cancer. Int J Cancer 120(3):656–663
Li WQ, Kawakami K, Ruszkiewicz A, Bennett G, Moore J, Iacopetta B (2006) BRAF mutations are associated with distinctive clinical, pathological and molecular features of colorectal cancer independently of microsatellite instability status. Mol Cancer 5:2
Kambara T, Simms LA, Whitehall VL, Spring KJ, Wynter CV, Walsh MD et al (2004) BRAF mutation is associated with DNA methylation in serrated polyps and cancers of the colorectum. Gut 53(8):1137–1144
Barault L, Charon-Barra C, Jooste V, de la Vega MF, Martin L, Roignot P et al (2008) Hypermethylator phenotype in sporadic colon cancer: study on a population-based series of 582 cases. Cancer Res 68(20):8541–8546
Weisenberger DJ, Siegmund KD, Campan M, Young J, Long TI, Faasse MA et al (2006) CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nat Genet 38(7):787–793
Ogino S, Nosho K, Kirkner GJ, Kawasaki T, Meyerhardt JA, Loda M et al (2009) CpG island methylator phenotype, microsatellite instability, BRAF mutation and clinical outcome in colon cancer. Gut 58(1):90–96
Veigl ML, Kasturi L, Olechnowicz J, Ma AH, Lutterbaugh JD, Periyasamy S et al (1998) Biallelic inactivation of hMLH1 by epigenetic gene silencing, a novel mechanism causing human MSI cancers. Proc Natl Acad Sci U S A 95(15):8698–8702
Jensen LH, Lindebjerg J, Kolvraa S, Cruger DG (2009) Molecular screening for Lynch syndrome: from bench to bedside. J Clin Oncol 27(34):e224, author reply e5
Palomaki GE, McClain MR, Melillo S, Hampel HL, Thibodeau SN (2009) EGAPP supplementary evidence review: DNA testing strategies aimed at reducing morbidity and mortality from Lynch syndrome. Genet Med 11(1):42–65
Ogino S, Brahmandam M, Cantor M, Namgyal C, Kawasaki T, Kirkner G et al (2006) Distinct molecular features of colorectal carcinoma with signet ring cell component and colorectal carcinoma with mucinous component. Mod Pathol 19(1):59–68
Naguib A, Mitrou PN, Gay LJ, Cooke JC, Luben RN, Ball RY et al (2010) Dietary, lifestyle and clinicopathological factors associated with BRAF and K-ras mutations arising in distinct subsets of colorectal cancers in the EPIC Norfolk study. BMC Cancer 10:99
Leyland-Jones B (2009) Human epidermal growth factor receptor 2-positive breast cancer and central nervous system metastases. J Clin Oncol 27(31):5278–5286
Tran B, Kopetz S, Tie J, Gibbs P, Jiang ZQ, Lieu CH et al (2011) Impact of BRAF mutation and microsatellite instability on the pattern of metastatic spread and prognosis in metastatic colorectal cancer. Cancer 117(20):4623–4632
Assersohn L, Norman A, Cunningham D, Benepal T, Ross PJ, Oates J (1999) Influence of metastatic site as an additional predictor for response and outcome in advanced colorectal carcinoma. Br J Cancer 79(11–12):1800–1805
Catalano V, Loupakis F, Graziano F, Torresi U, Bisonni R, Mari D et al (2009) Mucinous histology predicts for poor response rate and overall survival of patients with colorectal cancer and treated with first-line oxaliplatin- and/or irinotecan-based chemotherapy. Br J Cancer 100(6):881–887
Richman SD, Seymour MT, Chambers P, Elliott F, Daly CL, Meade AM et al (2009) KRAS and BRAF mutations in advanced colorectal cancer are associated with poor prognosis but do not preclude benefit from oxaliplatin or irinotecan: results from the MRC FOCUS trial. J Clin Oncol 27(35):5931–5937
Bokemeyer C, Kohne C, Rougier P, Stroh C, Schlichting M, Cutsem EV (2010) Cetuximab with chemotherapy (CT) as first-line treatment for metastatic colorectal cancer (mCRC): analysis of the CRYSTAL and OPUS studies according to KRAS and BRAF mutation status. J Clin Oncol 28(15 s):abstr 3506
Souglakos J, Philips J, Wang R, Marwah S, Silver M, Tzardi M et al (2009) Prognostic and predictive value of common mutations for treatment response and survival in patients with metastatic colorectal cancer. Br J Cancer 101(3):465–472
Bokemeyer C, Van Cutsem E, Rougier P, Ciardiello F, Heeger S, Schlichting M et al (2012) Addition of cetuximab to chemotherapy as first-line treatment for KRAS wild-type metastatic colorectal cancer: pooled analysis of the CRYSTAL and OPUS randomised clinical trials. Eur J Cancer 48(10):1466–1475
Farina-Sarasqueta A, van Lijnschoten G, Moerland E, Creemers GJ, Lemmens VE, Rutten HJ et al (2010) The BRAF V600E mutation is an independent prognostic factor for survival in stage II and stage III colon cancer patients. Ann Oncol 21(12):2396–2402
Di Nicolantonio F, Martini M, Molinari F, Sartore-Bianchi A, Arena S, Saletti P et al (2008) Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol 26(35):5705–5712
Loupakis F, Ruzzo A, Cremolini C, Vincenzi B, Salvatore L, Santini D et al (2009) KRAS codon 61, 146 and BRAF mutations predict resistance to cetuximab plus irinotecan in KRAS codon 12 and 13 wild-type metastatic colorectal cancer. Br J Cancer 101(4):715–721
De Roock W, Claes B, Bernasconi D, De Schutter J, Biesmans B, Fountzilas G et al (2010) Effects of KRAS, BRAF, NRAS, and PIK3CA mutations on the efficacy of cetuximab plus chemotherapy in chemotherapy–refractory metastatic colorectal cancer: a retrospective consortium analysis. Lancet Oncol 11(8):753–762
Laurent-Puig P, Cayre A, Manceau G, Buc E, Bachet JB, Lecomte T et al (2009) Analysis of PTEN, BRAF, and EGFR status in determining benefit from cetuximab therapy in wild-type KRAS metastatic colon cancer. J Clin Oncol 27(35):5924–5930
Tol J, Dijkstra JR, Klomp M, Teerenstra S, Dommerholt M, Vink-Borger ME et al (2010) Markers for EGFR pathway activation as predictor of outcome in metastatic colorectal cancer patients treated with or without cetuximab. Eur J Cancer 46(11):1997–2009
Theodoropoulos GE, Karafoka E, Papailiou JG, Stamopoulos P, Zambirinis CP, Bramis K et al (2009) P53 and EGFR expression in colorectal cancer: a reappraisal of ‘old’ tissue markers in patients with long follow-up. Anticancer Res 29(2):785–791
McKay JA, Murray LJ, Curran S, Ross VG, Clark C, Murray GI et al (2002) Evaluation of the epidermal growth factor receptor (EGFR) in colorectal tumours and lymph node metastases. Eur J Cancer 38(17):2258–2264
Nagasaka T, Sasamoto H, Notohara K, Cullings HM, Takeda M, Kimura K et al (2004) Colorectal cancer with mutation in BRAF, KRAS, and wild-type with respect to both oncogenes showing different patterns of DNA methylation. J Clin Oncol 22(22):4584–4594
Solit DB, Garraway LA, Pratilas CA, Sawai A, Getz G, Basso A et al (2006) BRAF mutation predicts sensitivity to MEK inhibition. Nature 439(7074):358–362
Prahallad A, Sun C, Huang S, Di Nicolantonio F, Salazar R, Zecchin D et al (2012) Unresponsiveness of colon cancer to BRAF(V600E) inhibition through feedback activation of EGFR. Nature 483(7387):100–103
Flaherty K, Puzanov I, Sosman J, Kim K, Ribas A, McArthur G, et al. (2009) Phase I study of PLX4032: proof of concept for V600E BRAF mutation as a therapeutic target in human cancer. J Clin Oncol (suppl; abstr 9000) 27:15 s
Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J et al (2011) Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med 364(26):2507–2516
Kopetz S, Desai J, Chan E, Hecht JR, O’Dwyer PJ, Lee RJ, et al. (2010) PLX4032 in metastatic colorectal cancer patients with mutant BRAF tumors. J Clin Oncol 28(15 s):suppl; abstr 3534
Corcoran RB, Ebi H, Turke AB, Coffee EM, Nishino M, Cogdill AP et al (2012) EGFR-mediated re-activation of MAPK signaling contributes to insensitivity of BRAF mutant colorectal cancers to RAF inhibition with vemurafenib. Cancer Discov 2(3):227–235
Poulikakos PI, Zhang C, Bollag G, Shokat KM, Rosen N (2010) RAF inhibitors transactivate RAF dimers and ERK signalling in cells with wild-type BRAF. Nature 464(7287):427–430
Hatzivassiliou G, Song K, Yen I, Brandhuber BJ, Anderson DJ, Alvarado R et al (2010) RAF inhibitors prime wild-type RAF to activate the MAPK pathway and enhance growth. Nature 464(7287):431–435
Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N et al (2010) Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell 140(2):209–221
Mao M, Tian F, Mariadason JM, Tsao CC, Lemos R Jr, Dayyani F et al (2013) Resistance to BRAF inhibition in BRAF-mutant colon cancer can be overcome with PI3K inhibition or demethylating agents. Clin Cancer Res 19(3):657–667
Coffee EM, Faber AC, Roper J, Sinnamon MJ, Goel G, Keung L et al (2013) Concomitant BRAF and PI3K/mTOR blockade is required for effective treatment of BRAF(V600E) colorectal cancer. Clin Cancer Res 19(10):2688–2698
Yuan TL, Cantley LC (2008) PI3K pathway alterations in cancer: variations on a theme. Oncogene 27(41):5497–5510
Parsons DW, Wang TL, Samuels Y, Bardelli A, Cummins JM, DeLong L et al (2005) Colorectal cancer: mutations in a signalling pathway. Nature 436(7052):792
Bamford S, Dawson E, Forbes S, Clements J, Pettett R, Dogan A et al (2004) The COSMIC (catalogue of somatic mutations in cancer) database and website. Br J Cancer 91(2):355–358
Ogino S, Nosho K, Kirkner GJ, Shima K, Irahara N, Kure S et al (2009) PIK3CA mutation is associated with poor prognosis among patients with curatively resected colon cancer. J Clin Oncol 27(9):1477–1484
Early Breast Cancer Trialists’ Collaborative Group (1988) Effects of adjuvant tamoxifen and of cytotoxic therapy on mortality in early breast cancer. An overview of 61 randomized trials among 28,896 women. N Engl J Med 319(26):1681–1692
Slamon DJ, Leyland-Jones B, Shak S, Fuchs H, Paton V, Bajamonde A et al (2001) Use of chemotherapy plus a monoclonal antibody against HER2 for metastatic breast cancer that overexpresses HER2. N Engl J Med 344(11):783–792
Sequist LV, Martins RG, Spigel D, Grunberg SM, Spira A, Janne PA et al (2008) First-line gefitinib in patients with advanced non-small-cell lung cancer harboring somatic EGFR mutations. J Clin Oncol 26(15):2442–2449
Amado RG, Wolf M, Peeters M, Van Cutsem E, Siena S, Freeman DJ et al (2008) Wild-type KRAS is required for panitumumab efficacy in patients with metastatic colorectal cancer. J Clin Oncol 26(10):1626–1634
Lievre A, Bachet JB, Le Corre D, Boige V, Landi B, Emile JF et al (2006) KRAS mutation status is predictive of response to cetuximab therapy in colorectal cancer. Cancer Res 66(8):3992–3995
Karapetis CS, Khambata-Ford S, Jonker DJ, O’Callaghan CJ, Tu D, Tebbutt NC et al (2008) K-ras mutations and benefit from cetuximab in advanced colorectal cancer. N Engl J Med 359(17):1757–1765
Acknowledgments
JT and JD are supported by the Victorian Government through a Victorian Cancer Agency Clinical Researcher Fellowship.
Conflict of interest
JT: Nil. JD’s research funding: Roche, GSK, and Novartis; Advisory Board: GSK, Novartis, and Merck Serono.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Tie, J., Desai, J. Targeting BRAF mutant metastatic colorectal cancer: clinical implications and emerging therapeutic strategies. Targ Oncol 10, 179–188 (2015). https://doi.org/10.1007/s11523-014-0330-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-014-0330-0