
Abstract
Mammalian target of rapamycin (mTOR) has emerged as an important target for cancer therapy. Rapamycin has a distinct, well-documented toxicity profile and most of the toxicity data has been reported in patients with organ transplantation. Newer mTOR inhibitors have slightly different pharmacokinetic properties, yet they present toxicity profiles similar to rapamycin. Most of these toxicities are mild to moderate in severity and can be managed clinically by dose modification and supportive measures. Mucositis and pneumonitis are the most commonly reported toxicities, but they rarely lead to treatment discontinuation. Pathogenesis of pneumonitis is uncertain, but various hypotheses have been suggested, including cell-mediated immune response to the drug.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
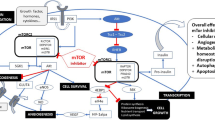
The mammalian target of rapamycin (mTOR) has emerged as an important target for anticancer therapy in various tumor types. mTOR is an intracellular protein with a central role in the synthesis of key cellular proteins that influence many aspects of cell growth and proliferation including differentiation, cell cycle progression, angiogenesis, protein degradation, and apoptosis [1]. mTOR is a downstream effector of the phosphatidylinositol 3-kinase (PI3K)-Akt pathway, a major cell-survival pathway known to be deregulated in many cancers. Mechanisms underlying aberrant PI3K/AKT pathway activation include mutation and silencing of the PTEN tumor suppressor gene, activating mutations in the PI3K catalytic subunit, and Akt amplification [2, 3]. The PI3K-Akt pathway has also been shown to be activated by upstream signals, such as growth factors that drive tumor proliferation through membrane-bound receptor tyrosine kinases, which are frequently overexpressed and/or mutated in cancers. These receptors include the epidermal growth factor receptor (EGFR), the insulin-like growth factor 1 receptor (IGF-1R), estrogen and progesterone receptors, vascular endothelial growth factor receptors (VEGFRs), and c-Kit [4].
Rapamycin was identified from soil samples containing the bacterial strain Streptomyces hygroscopicus and initially found to have potent antifungal properties. Studies subsequently showed that rapamycin has antitumor and immunosuppressive properties. Rapamycin interacts with the cytosolic protein FK-binding protein 12 (FKBP12), and subsequently the rapamycin-FKBP12 complex interacts directly with mTOR complex-1 (mTORC1). This causes inhibition of PI3K-Akt pathway leading to inhibition of cell growth and proliferation [5]. Although rapamycin has been shown to possess intrinsic antitumor activity, its poor aqueous solubility and chemical stability has precluded its utilization at doses necessary for anticancer treatment. This led to development of new analogues of rapamycin (rapalogues). Those currently in clinical development as anticancer agents include temsirolimus (cell cycle inhibitor-779 or CCI-779), everolimus (RAD-001), and deforolimus (AP23573). These agents have demonstrated anti-proliferative activity against a diverse range of malignancies in preclinical studies, and clinical evaluations have been very encouraging thus far. In 2007, temsirolimus was approved by the US Food and Drug Administration (FDA) for treatment of advanced renal cell cancer based on a phase III clinical trial [6]. Rapamycin has a distinct, well-described toxicity profile, mainly from the extensive experience in patients with organ transplant. Despite slightly different pharmacokinetic and pharmacodynamic properties, rapamycin analogues have shown similar toxicity profiles to that of the parent compound. Most of the toxicities of this class of compounds are mild to moderate in severity and can be managed clinically by dose modification and supportive measures.
In this review, we will first outline the main toxicities associated with rapamycin and its analogs in general and will thereafter discuss the specific toxicity profile of each mTOR inhibitor as reported in various recent clinical studies.
Common toxicities of mTOR inhibitors
There is an abundance of clinical data available about the toxicity of rapamycin in transplant patients. By contrast, fully reported toxicity profiles with the novel rapamycin analogues are still relatively limited to date. Therefore, based on their striking similarities as well as on emerging data, it is likely that the safety profile of rapamycin derivatives will mimic to some extent that of the parent compound.
Mucositis, stomatitis and mouth sores
Mucositis, stomatitis and mouth sores have been reported in almost all the clinical studies with mTOR inhibitors and have been one of the most common adverse events associated with these agents (75% with temsirolimus [7], 78% with deforolimus [8], and 41% with everolimus [9]). In some studies, they were the main dose-limiting toxicity [9]. Oral mucositis with mTOR inhibitors is distinct from typical radiation or chemotherapy induced stomatitis. Typically, it has a rapid onset (usually within 5 days) and mild to moderate severity (grade 1–2). Mouth sores are usually found on the mucosa of the lips, lateral tongue, buccal mucosa and soft palate. Unlike viral-induced ulcers, they are not commonly seen on the hard palate or outer aspects of the lip. They present as 1–3 round ulcers and have similar appearance to aphthous ulcers (canker sores): distinct oval lesions with grayish-white necrotic centers surrounded by a ring of erythema. Unlike radiation/chemotherapy-associated mucositis, there is no pseudomembrane formation. Occasionally they are severe (grade 3), but generally reversible by withholding treatment. In many cases mucositis improves or resolves spontaneously despite treatment continuation. The exact mechanism of mucositis is unknown. Antiseptic mouthwashes, chewing ice cubes and other methods were inconsistently effective in preventing stomatitis, but play a definite role in symptom palliation. Increased incidences of stomatitis with dose escalation and decrease in frequency and severity with subsequent cycles of treatment have been reported [8]. Mucositis associated with mTOR inhibitor deforolimus and due to radiation is shown in Fig. 1.

Mucositis associated with the mTOR inhibitor deforolimus (left) and with radiation (right). mTOR inhibitor-related mucositis appears as a distinct oval lesion with grayish-white necrotic centers surrounded by a ring of erythema and no pseudomembrane formation as with radiation or chemotherapy induced mucositis (photographs curtesy of Stephen Sonis DMD).
Pulmonary toxicity
Pulmonary toxicity is an often-misdiagnosed toxicity of mTOR inhibitors. Pneumonitis has been reported since the early of use of rapamycin [10], and has been further explored in subsequent studies in transplant patients and other studies with rapamycin analogues [7, 11–13]. The incidences of pneumonitis with mTOR inhibitors have been reported with the frequency ranging from 5% [13] to as high as 36% [14]. The incidences reported in various studies largely depend on the frequency and type of imaging studies performed (chest X-rays or CT scans). Although variable sample size of these trials (22 to 111 patients) has also been a confounding factor, it seems that in studies with higher incidences, routine screening identified radiological changes of pneumonitis in many asymptomatic patients.
Duran et al described pulmonary toxicity in 22 patients treated with temsirolimus [14]. The most common clinical presentation was dyspnea on exertion and dry cough followed by fatigue and fever, which were reported in around 50% of the patients diagnosed with pneumonitis. Pneumonitis generally occurs after prolonged exposure to mTOR inhibitors (6 months to 1 year). Two different radiological patterns have been described: ground glass opacities and lung parenchymal consolidation. In a few patients, pulmonary function tests revealed a restrictive pulmonary disease pattern or isolated reduction in the diffusing capacity for carbon monoxide. The risk of developing this toxicity with mTOR inhibitors may be increased among subjects with abnormal pre-treatment pulmonary functions or history of lung disease. Discontinuation or dose reduction of sirolimus resulted in clinical and radiological improvement in most cases within 3 weeks.
Histopathological features of pneumonitis induced by mTOR inhibitors revealed several distinct features, including lymphocytic alveolitis, lymphocytic interstitial pneumonitis, bronchoalveolar obliterans organizing pneumonia, focal fibrosis, pulmonary alveolar hemorrhage, or a combination thereof [15]. The diagnosis of rapalogue-associated pulmonary toxicity should be made after a work-up to exclude infectious causes and other pulmonary disease.
The physiopathologic mechanism of pulmonary toxicity is still not clearly defined, with several hypotheses being proposed. Morelon et al [10] suggested a cell-mediated autoimmune response based on analysis of bronchoalveolar lavage (BAL). BAL fluid analysis in these patients displayed lymphocytic alveolitis with a majority of CD4-positive cells and a significantly increased number of eosinophils and mast cells. As CD8-positive but not CD4-positive cells are usually found in drug-induced alveolitis, these findings support the hypothesis that a cell-mediated immune response is one of the factors generating sirolimus-induced pneumonitis. To account for both types of causative mechanism (immune mediated and direct toxic), it was speculated that sirolimus pulmonary toxicity might expose cryptic antigens, which could in turn induce an autoimmune response leading to lymphocytic alveolitis and interstitial pneumonitis.
Pham et al [15] hypothesized that T-cell-mediated, delayed-type hypersensitivity may be an alternative pathogenic mechanism. Sirolimus alone may not be capable of inducing an immune response, but its high affinity for plasma proteins may render it immunogenic as a hapten. It is conceivable that antigen-presenting cells in the lungs, such as alveolar type 2 lining cells, cause an initial immune response when processing the sirolimus-protein complex, with subsequent T-cell recognition of the processed antigen complex, release of cytokines, and preferential differentiation of Th0 to Th1, and to a lesser extent, Th2 cells. Repeated exposure to sirolimus may result in antigen presentation predominantly to Th1 cells, leading to Th1 activation, release of Th1 cytokines, and recruitment and activation of macrophages and other inflammatory cells. This hypothesis is supported by the findings of striking alveolitis (with moderate to marked alveolar lymphocytosis) and a predominance of CD4-positive T-cells on flow cytometry analysis of BAL fluid in affected patients.
Variable incidences of pulmonary toxicity have been reported with newer mTOR inhibitors. Although no significant pulmonary toxicity was reported in two phase I studies of temsirolimus, another study reported an incidence as high as 36% [14]. Again, frequency and type of imaging techniques used, as well as different doses and duration of drug exposure, may be factors explaining this variation. Similarly, although only one patient was reported to have drug-related pneumonitis in a phase I study of everolimus, a phase III study reported incidences up to 8% [16]. In a phase I study with deforolimus, pneumonitis occurred with an incidence of 16%, typically after extended exposure (>4 months) and with no apparent dose relationship [8].
There are no specific guidelines for management of pneumonitis associated with mTOR inhibitors. As mentioned in case series and based on the experience with various mTOR inhibitors in cancer patients at our institution, a thorough work up should be performed to rule out any other cause of lung injury (especially infectious or cancer related) before making a diagnosis of mTOR inhibitor-associated pneumonitis. Baseline lung imaging is recommended before initiating treatment with mTOR inhibitors for comparison at onset of respiratory symptoms. It is also preferred to monitor pulmonary function tests in patients who have baseline pulmonary conditions. It has been proposed that treatment for this toxicity should be directed towards managing the patient symptoms, changes in chest imaging and pulmonary function tests. Asymptomatic patients with minor radiological changes may not require interruption of drug treatment or specific therapy. Patients with respiratory symptoms and lower results on pulmonary function tests should have the drug withheld, and brief steroid therapy should be initiated promptly. In majority of these patients, the drug can be resumed once they are asymptomatic and pulmonary function tests and imaging studies return to baseline. Chest CT scans of a patient on treatment with deforolimus is shown in Fig. 2.

Pneumonitis associated with deforolimus (left) resolved radiologically ten days after withholding treatment (right) [M. Mita]
Skin toxicity
Skin toxicity is commonly reported with mTOR inhibitors and manifests typically as maculopapular or acneform rash, but also as dryness, eczema, skin discoloration, as well as nail dystrophy. The maculopapular rash mainly occurs on the face and neck, generally during the first few weeks of treatment and is occasionally associated with acne-type lesions with erythematous base. Histopathologic examination as reported in one study revealed a nonspecific accumulation of neutrophils in the dermis and epidermis [7]. The skin rash and acne are generally mild (grade 1–2) and resolve spontaneously or with a brief course of topical steroid cream or systemic antibiotics. The skin rash has been reported with an incidence up to 66% with deforolimus [8], 50–60% with temsirolimus [7], and 48% with everolimus [9].
Hyperlipidemia
Rapamycin has been associated with elevation of serum lipid levels [17]. Rapamycin decreases lipoprotein lipase activity in cell cultures [18]. However, similar effects were not documented in humans [19]. In humans, rapamycin significantly raises the levels of serum high-density lipoproteins (HDL), but also induces significant increases in low-density lipoproteins (LDL), cholesterol, and particularly, triglycerides. Metabolic studies suggest that rapamycin treatment increases circulating intermediate, low, and very-low-density fractions primarily caused by delayed clearance of lipoprotein remnants [20]. In two different phase III studies, patients treated with rapamycin required more frequent treatment with lipid lowering drugs [4, 21]. Although hyperlipidemia associated with rapamycin treatment is common, it rarely requires drug discontinuation and has not been generally associated with any of the serious adverse clinical events associated with hyperlipidemia, such as pancreatitis, cerebrovascular accident, or myocardial infarction, or with patient death.
Significant hyperlipidemia (hypercholesteremia and/or hypertryglyceredemia) was reported in various studies with mTOR inhibitors, with incidences of 21–37% for temsirolimus [7, 22], 8–44% for everolimus [9, 23], and 28–41% for deforolimus [8]. Treatment with statins and/or gemfibrozil resulted in good recovery and subsequent control while continuing treatment.
Hyperglycemia
In a preclinical study [24], rapamycin was shown to decrease muscle insulin sensitivity paralleled by increased glycogen synthase kinase-3β activity. In diabetic animals, rapamycin reduced β-cell mass by 50% through increased apoptosis. Rapamycin increased the stress-responsive c-Jun NH2-terminal kinase pathway in muscle and islets, which could account for its effect on insulin resistance and β-cell apoptosis. Moreover, glucose-stimulated insulin secretion and biosynthesis were impaired in islets treated with rapamycin. The animal study concluded that rapamycin induces fulminant diabetes by increasing insulin resistance and reducing β-cell function and mass. These findings emphasize the essential role and interaction of mTOR/S6K1 in glucose metabolism, which may cause hyperglycemia and exacerbation of pre-existing diabetes in patients on mTOR inhibitors. Other studies highlight the importance of the mTOR/p70 S6 kinase signaling pathway as a modulator of insulin-stimulated glucose transport in skeletal muscle cells [25]. The incidences of hyperglycemia reported in various studies with mTOR inhibitors are between 8–22% [8, 9, 13, 23, 26, 27]. Although in most patients the severity is not clinically significant, occasional cases require more stringent control of hyperglycemia, especially in patients with diabetes. Therefore, close monitoring of glucose levels is recommended in all patients receiving the mTOR inhibitors.
Bone marrow suppression
The effect of rapamycin on bone marrow cell lines is mediated by cytokines or vascular growth factors in a similar fashion as its effect on lymphocytes in the immune suppression process in transplantation, though less potently. In a single center study, it was shown that the suppression is reversible and tends to be predominant for megakaryocytes, and was dose and concentration dependent [28]. Thrombocytopenia and leucopenia have been reported with novel mTOR inhibitors with varying incidences. Thrombocytopenia was reported about 29–33% with temsirolimus [7, 22], 10% for everolimus [9], and 20–25% for deforolimus [8, 29]. Leucopenia has also been reported up to 27% for temsirolimus [22] and 38% for deforolimus [8], but significant neutropenia is less common. In a phase I study, grade 3 neutropenia was reported in only 8% of patients receiving temsirolimus [22]. Thrombocytopenia and neutropenia are rarely complicated by bleeding or infection and usually do not require hospitalization, platelet transfusion or growth factor support. Treatment can usually be resumed with dose adjustment in majority of patients.
Infection and malignancy
A phase III study compared two different doses (2 and 5 mg/day) of rapamycin with placebo to investigate its ability to prevent acute rejection in recipients of primary mismatched renal allograft when added to a regimen of cyclosporine and corticosteroids. There was no significant difference in infections among all three groups at 12 months [4, 30].
In the same study, the incidence of post-transplantation lympho-proliferative disorders (PTLD) in the 5 mg rapamycin group was 1.4%, a value numerically higher than in other groups, but neither statistically significant nor outside of the range observed in numerous studies of other immunosuppressive agents. At the time of diagnosis of PTLD, most patients had discontinued rapamycin and switched to treatment with other immunosuppressants. On the other hand, other studies reported an in vitro anti-proliferative effect on variety of neoplastic cell lines. Rapamycin also reported to be beneficial for patients undergoing transplantation as treatment for liver cancer. Similarly it was also reported to inhibit the progression of dermal Kaposi's sarcoma in kidney-transplant recipients, while providing effective immunosuppression [31, 32]. So far, there is no evidence of significant immune suppression or increase in second malignancies in clinical studies with the newer mTOR inhibitors reported to date.
Renal function abnormalities
In studies with animal models, rapamycin was not reported to have any effect on glomerular filtration rate (GFR). To avoid confounding factors associated with transplantation, effects of rapamycin on renal function was assessed in 117 patients with refractory psoriasis treated with 0, 1, 3, or 5 mg/m of rapamycin for 12 weeks. No difference in mean serum creatinine values were seen in these groups [33]. Other studies involving renal transplant patients revealed significantly better mean creatinine values at 12 and 24 months among patients treated with rapamycin compared to those treated with cyclosporine [34, 35]. However, renal tubular abnormalities have been observed among patients treated with rapamycin, leading to hypokalemia and hypophosphatemia [36].
Toxicity profiles of newer mTOR inhibitors in clinical development
Temsirolimus (Torisel, CCI-779; Wyeth Pharmaceuticals, Collegeville, PA)
Temsirolimus, a water-soluble ester of sirolimus, was recently approved as first-line treatment for renal cell carcinoma with poor prognostic features. Two schedules of administration of temsirolimus were explored in phase 1 clinical trials. In one phase I study, 24 patients with advanced cancers were treated at the doses ranging from 7.5 to 220 mg/m² intravenously (i.v.) on once a weekly schedule [7]. At 220 mg/m², dose-limiting toxicities consisted of manic-depressive syndrome, stomatitis, and asthenia in two of nine patients. The most frequently reported drug-related toxicities (including grade 1 to 4) were acne-like, maculopapular rash and mucositis or stomatitis occurring in 75% of patients enrolled. Other frequent side effects were asthenia (46%), nausea (42%) and vomiting (21%). Hematological toxicity, mainly thrombocytopenia, occurred in only 29% of patients and was mild to moderate in intensity. Other side effects reported in fewer than 25% of patients included anorexia (21%), diarrhea (21%), hypercholesteremia (21%), hypertriglycerridemia (21%), weight loss (21%), and peripheral edema (21%). All toxicities were reversible on treatment discontinuation. Although the formal definition of maximum tolerated dose (MTD) was not met, further dose escalation was stopped and weekly doses of 25, 75, and 250 mg CCI-779 were tested in phase II trials in patients with breast and renal cancer.
In another phase I study, temsirolimus was administered i.v. once daily for 5 days every 2 weeks [22]. Sixty-three patients were treated at doses ranging from 0.75 to 24 mg/m²/d. In the 24 mg/m²/d cohort, one patient developed a dose-limiting toxicity of grade 3 stomatitis and two patients required dose reductions. At the 19 mg/m²/d cohort, two patients had dose-limiting toxicities, one with grade 3 vomiting, diarrhea, and asthenia, and one with elevated transaminases; three additional patients required dose reductions therefore establishing the MTD at 15 mg/m²/d. Frequently occurring toxicities were asthenia (56%), mucositis (54%), nausea (41%), cutaneous toxicity (41%), hypertriglyceridemia (37%), thrombocytopenia (33%), hypercholesterolemia (22%), elevated transaminases (19%), and hyperglycemia (17%). Interestingly, no pulmonary toxicity was reported in these phase I studies.
The experience from phase II trials confirmed the overall favorable safety profile of temsirolimus. A phase II study was conducted in 109 patients with advanced breast cancer with two weekly dose levels, 75 or 250 mg/m² [37]. The study reported that efficacy was similar for both dose levels, but toxicity was more common with the higher dose level, especially grade 3 or 4 depression (10% of patients at the 250-mg dose level, 0% at the 75-mg dose level). The most common temsirolimus-related adverse events were mucositis (70%), maculopapular rash (51%), and nausea (43%). The most common, clinically significant (grade 3 or 4) adverse events were mucositis (9%), leucopenia (7%), hyperglycemia (7%), somnolence (6%), thrombocytopenia (5%), and depression (5%). Other phase II studies in patients with glioblastoma multiforme (65 patients treated at 250 mg/m²/week dose) [26], relapsed mantle cell lymphoma (35 patients treated with 250 mg/m²/week doses) [38], and renal cell carcinoma (111 patients randomly assigned to 25, 75, or 250 mg/m² weekly doses) [13] showed similar incidences of the above-mentioned toxicities.
Everolimus (RAD001; Novartis; Basel, Switzerland)
Everolimus is an orally bioavailable mTOR inhibitor. A phase-1 dose-escalation study in advanced cancer patients was performed administering oral everolimus on two schedules, 5 to 70 mg weekly and 5 and 10 mg daily [9]. Dose-limiting toxicities were stomatitis and fatigue at 50 mg/wk and hyperglycemia at 10 mg/d dose levels on the two schedules, respecrively. MTD was not defined. The significant toxicities were skin rash and erythema (48%), stomatitis and mucositis (41%), fatigue (34%), thrombocytopenia (10%), anemia (7%), hyperlipidemia (8%) and hyperglycemia (8%). Gastrointestinal toxicities reported in 66% of patients included stomatitis, nausea, vomiting, anorexia, constipation, and abdominal pain or distension. Pulmonary toxicity as bronchiolitis obliterans organizing pneumonia was reported in one patient (70 mg/wk) after 4 to 6 weeks of therapy, and resolved completely after drug discontinuation and glucocorticoid therapy.
Various phase II studies of everolimus reported a similar toxicity profile [23, 39]. A phase-III, randomized, double-blind, placebo-controlled trial of everolimus in patients with metastatic renal cell carcinoma who progressed on sunitinib, sorafanib or both showed stomatitis in 40% patients in the everolimus group vs 8% in the placebo group, rash 25% vs 4%, and fatigue 20% vs 16% as most commonly reported side effects. These toxicities were mostly mild or moderate in severity. Pneumonitis (any grade) was detected in 22 (8%) patients in the everolimus group, and was severe (grade 3) in eight of them [16].
Deforolimus (AP23573; MK-8669; Ariad Pharmaceuticals; Cambridge MA)
Deforolimus is a non pro-drug analog of rapamycin. The drug is available in both i.v. and oral formulations. A phase I study in patients with advanced solid malignancies was performed and enrolled 32 patients [8]. Deforolimus was administered as a 30-minute i.v. infusion once daily for five consecutive days every 2 weeks in a 28-day cycle. The MTD was 18.75 mg/d. Dose-limiting toxicity (DLT) was oral mucositis. Commonly reported toxicities were mild to moderate and reversible. Drug-related side effects included mouth sores (79%), skin rash (66%), anemia (53%), fatigue (45%), hypertriglyceridemia (41%), hypercholesterolemia (28%), hyperglycemia (28%), leucopenia (38%), and thrombocytopenia (25%). gastrointestinal-related toxicities were common but generally mild and included nausea (41%), vomiting (28%), diarrhea (25%) and constipation (22%).
A second phase I study with an oral formulation of deforolimus in patients with refractory malignancies was recently presented. Seven regimens, all over a 28-day cycle were investigated in 147 patients. The DLT for all regimens was aphthous, ulcer-like mouth sores that were reversible with dose reduction or symptomatic therapy. Common toxicities reported were similar to the i.v. formulation and included mouth sores (79%), fatigue (49%), rash (45%), anemia (24%), diarrhea (23%), nausea (21%) and thrombocytopenia (20%) [29].
Phase II clinical studies of deforolimus reported similar toxicities both in solid tumors [40] and in relapsed or refractory hematologic malignancies [41].
Comparative toxicities of these three agents in published phase II clinical trials is shown in Table 1.
Conclusion
mTOR has emerged as an important target in cancer therapy. Sirolimus in use since 2001 as an immunosuppressant in transplant patients and its toxicity profile has been well documented since. With the development of newer mTOR inhibitors, the toxicity profile of these agents is gradually being characterized. Overall, the side effects of these newer agents are similar to sirolimus. Although it is likely that the mTOR inhibitors do exhibit certain particularities in their safety profile, the differences in reported incidences of toxicities can be attributed, at least in part, to the variable number of patients and different dosing schedules in these studies. In general, no significant immune suppression has been noticed with any of these anticancer agents. Oral agents seem to have increased gastrointestinal toxicities. The majority of adverse events are dose dependent; however pneumonitis and mucositis were reported even at lower doses. The toxicities are typically reversible and are rarely serious, and therefore generally manageable in an outpatient setting. In most cases, mTOR inhibitors can be resumed on resolution of toxicity. Since the antitumor effects with mTOR inhibitors are mainly tumor growth inhibition, continuing treatment after toxicity resolution is important for maximum clinical benefit. Further understanding of the physiopathologic mechanism underlying the toxicities of mTOR inhibitors may make it possible to predict which patients are at risk and may also help to develop optimal prevention and treatment strategies.
References
Hennessy BT et al (2005) Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat Rev Drug Discov 4(12):988–1004
Scaltriti M, Baselga J (2006) The epidermal growth factor receptor pathway: a model for targeted therapy. Clin Cancer Res 12(18):5268–5272
Yu H, Rohan T (2000) Role of the insulin-like growth factor family in cancer development and progression. J Natl Cancer Inst 92(18):1472–1489
MacDonald AS (2001) A worldwide, phase III, randomized, controlled, safety and efficacy study of a sirolimus/cyclosporine regimen for prevention of acute rejection in recipients of primary mismatched renal allografts. Transplantation 71(2):271–280
Huang S, Houghton PJ (2002) Inhibitors of mammalian target of rapamycin as novel antitumor agents: from bench to clinic. Curr Opin Investig Drugs 3(2):295–304
Hudes G et al (2007) Temsirolimus, interferon alfa, or both for advanced renal-cell carcinoma. N Engl J Med 356(22):2271–2281
Raymond E et al (2004) Safety and pharmacokinetics of escalated doses of weekly intravenous infusion of CCI-779, a novel mTOR inhibitor, in patients with cancer. J Clin Oncol 22(12):2336–2347
Mita MM et al (2008) Phase I trial of the novel mammalian target of rapamycin inhibitor deforolimus (AP23573; MK-8669) administered intravenously daily for 5 days every 2 weeks to patients with advanced malignancies. J Clin Oncol 26(3):361–367
O'Donnell A et al (2008) Phase I pharmacokinetic and pharmacodynamic study of the oral mammalian target of rapamycin inhibitor everolimus in patients with advanced solid tumors. J Clin Oncol 26(10):1588–1595
Morelon E et al (2001) Characteristics of sirolimus-associated interstitial pneumonitis in renal transplant patients. Transplantation 72(5):787–790
Lennon A et al (2001) Interstitial pneumonitis associated with sirolimus (rapamycin) therapy after liver transplantation. Transplantation 72(6):1166–1167
Atkins MB, Stadler W et al (2002) A randomized double blind phase 2 study of intravenous CCI-779 administered weekly to patients with advanced renal cell carcinoma. Proc Am Soc Clin Oncol 21:36 Abstract
Atkins MB et al (2006) Randomized phase II study of multiple dose levels of CCI-779, a novel mammalian target of rapamycin kinase inhibitor, in patients with advanced refractory renal cell carcinoma. J Clin Oncol 22(5):909–918
Duran I et al (2004) Characterisation of the lung toxicity of the cell cycle inhibitor temsirolimus. Eur J Cancer 42(12):1875–1880
Pham PT et al (2004) Sirolimus-associated pulmonary toxicity. Transplantation 77(8):1215–1220
Motzer RJ et al (2008) Efficacy of everolimus in advanced renal cell carcinoma: a double-blind, randomised, placebo-controlled phase III trial. The Lancet 372(9637):449–456
Brattstrom C et al (1998) Hyperlipidemia in renal transplant recipients treated with sirolimus (rapamycin). Transplantation 65(9):1272–1274
Kraemer FB et al (1998) Insulin regulates lipoprotein lipase activity in rat adipose cells via wortmannin- and rapamycin-sensitive pathways. Metabolism 47(5):555–559
Massy ZA et al (2000) Hyperlipidaemia and post-heparin lipase activities in renal transplant recipients treated with sirolimus or cyclosporin A. Nephrol Dial Transplant 15(6):928
Hoogeveen RC et al (2001) Effect of sirolimus on the metabolism of apoB100- containing lipoproteins in renal transplant patients. Transplantation 72(7):1244–250
Kahan BD (2000) Efficacy of sirolimus compared with azathioprine for reduction of acute renal allograft rejection: a randomised multicentre study. The Rapamune US Study Group. Lancet 356(9225):194–202
Hidalgo M et al (2006) A phase I and pharmacokinetic study of temsirolimus (CCI-779) administered intravenously daily for 5 days every 2 weeks to patients with advanced cancer. Clin Cancer Res 12(19):5755–5763
23. Yee KW et al Phase I/II study of the mammalian target of rapamycin inhibitor everolimus (RAD001) in patients with relapsed or refractory hematologic malignancies. Clin Cancer Res 12(17):5165–5173
Fraenkel M et al (2008) mTOR inhibition by rapamycin prevents beta-cell adaptation to hyperglycemia and exacerbates the metabolic state in type 2 diabetes. Diabetes 57(4):945–957
Tremblay F, Marette A (2001) Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. J Biol Chem 276(41):38052–38060
Galanis E et al (2005) Phase II trial of temsirolimus (CCI-779) in recurrent glioblastoma multiforme: a north central cancer treatment group study. J Clin Oncol 23(23):5294–5304
Bellmunt J et al (2008) Temsirolimus safety profile and management of toxic effects in patients with advanced renal cell carcinoma and poor prognostic features. Ann Oncol 19(8):1387–1392
Hong JC, Kahan BD (2000) Sirolimus-induced thrombocytopenia and leukopenia in renal transplant recipients: risk factors, incidence, progression, and management. Transplantation 69(10):2085–2090
Mita MM, Poplin E, Tap WD, Carmona A, Yonemoto L, Wages DS, Bedrosian CL, Rubin EH, Tolcher AW (2008) Deforolimus trial 106- A Phase I trial evaluating 7 regimens of oral Deforolimus (AP23573, MK-8669). J Clin Oncol 26:3509 abstract
Kneteman N, Babini R et al (2000) Sirolimus immunosuppression for liver transplantation in the presence of malignancy. XVIII International Congress of the Transplantation Society: Abstract
Stallone G et al (2005) Sirolimus for Kaposi's sarcoma in renal-transplant recipients. N Engl J Med 352(13):1317–1323
Stallone G et al (2008) Kaposi's sarcoma and mTOR: a crossroad between viral infection neoangiogenesis and immunosuppression. Transpl Int 21(9):825–832
Kahan BD, Camardo JS (2001) Rapamycin: clinical results and future opportunities. Transplantation 72(7):1181–1193
Groth CG et al (1999) Sirolimus (rapamycin)-based therapy in human renal transplantation: similar efficacy and different toxicity compared with cyclosporine. Sirolimus European renal transplant study group. Transplantation 67(7):1036–1042
Kreis H et al (2000) Sirolimus in association with mycophenolate mofetil induction for the prevention of acute graft rejection in renal allograft recipients. Transplantation 69(7):1252–1260
Morales JM, Wramner H, Kreis D et al (2000) Sirolimus vs cyclosporin: a comparision of renal function over two years. XVIII International Congress of the Transplantation Society 140:0428 Abstract
Chan S et al (2005) Phase II study of temsirolimus (CCI-779), a novel inhibitor of mTOR, in heavily pretreated patients with locally advanced or metastatic breast cancer. J Clin Oncol 23(23):5314–5322
Witzig TE et al (2005) Phase II trial of single-agent temsirolimus (CCI-779) for relapsed mantle cell lymphoma. J Clin Oncol 23(23):5347–5356
Amato RJ et al (2006) A phase II trial of RAD001 in patients (Pts) with metastatic renal cell carcinoma (MRCC). J Clin Oncol (Meeting Abstracts) 24((18_suppl)):4530
Chawla SP, Staddon AP, Schuetze SM, D'Amato GZ, Blay JY, Sankhala KK, Daly ST, Rivera VM, Demetri GD (2006) Updated results of a phase II trial of AP23573, a novel mTOR inhibitor, in patients (pts) with advanced soft tissue or bone sarcomas. J Clin Oncol 24(18S):9505
Rizzieri DA et al (2008) A phase 2 clinical trial of deforolimus (AP23573, MK-8669), a novel mammalian target of rapamycin inhibitor, in patients with relapsed or refractory hematologic malignancies. Clin Cancer Res 14(9):2756–2762
Conflict of interest statement
No funds were received in support of this study.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Sankhala, K., Mita, A., Kelly, K. et al. The emerging safety profile of mTOR inhibitors, a novel class of anticancer agents. Targ Oncol 4, 135–142 (2009). https://doi.org/10.1007/s11523-009-0107-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11523-009-0107-z