
Abstract
The mammalian target of rapamycin (mTOR) is a highly conserved serine/threonine kinase that belongs to the family of PI3K-related protein kinases (PIKKs). Dysregulation of mTOR signaling is associated with the development of cancers, including myeloid and lymphoid malignancies. Here, we will provide a brief overview of mTOR inhibitors and discuss the results obtained using these compounds in hematologic malignancies and especially in lymphomas. Moreover, mechanisms of drug resistance will be highlighted.
Access provided by CONRICYT-eBooks. Download chapter PDF
Similar content being viewed by others

Keywords
- Everolimus (RAD001)
- Lymphoid Malignancies
- mTOR inhibitors
- Rapamycin
- Ridaforolimus (MK-8669)
- Temsirolimus (CCI-779)
Introduction: Rapamycin and Rapalogs History
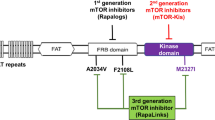
mTOR inhibitors comprise different compounds which have been developed starting from rapamycin, a macrolide antibiotic produced by the bacterium Streptomyces hygroscopicus. Rapamycin was isolated in a soil sample on Easter Island, also known as Rapa Nui, from where its name is derived [1] and firstly used as an antifungal agent [2]. However, shortly after, it was also shown to have strong immunosuppressive and antiproliferative properties due to its ability to inhibit mTOR [3,4,5]. Thus, FDA-approved Rapamycin use in transplantation to prevent allograft rejection and in coronary-artery stents to prevent restenosis in 1999 and 2003, respectively [6]. On the other hand, its application in cancer therapy started in the late 1990s, when several analogs of the drug, called rapalogs and including temsirolimus (CCI-779) , everolimus (RAD001) , and ridaforolimus (MK-8669) , were developed with the aim to improve its pharmacokinetics and stability (Fig. 1) [7]. Temsirolimus was the first mTOR inhibitor to gain FDA authorization for any malignancy, having been approved for the treatment of advanced renal cell carcinoma [8]. Moreover, temsirolimus is the only mTOR inhibitor approved for the treatment of lymphomas and in particular it is registered for the treatment of relapsed and/or refractory mantle cell lymphoma (MCL) in the European Union and several other countries. To date, all these agents, and the so called second generation mTOR inhibitors, are being investigated alone or in combination in solid as well as in hematologic malignancies.

Schematic representation of mTOR signaling pathways and mTOR inhibitors mechanisms of action. mTOR works through two distinct multiprotein complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). mTORC1 has 4E–BP1 and S6 K1 as its two major substrates by which promote the translation of key cell cycle regulators and transcription factors. mTORC2 is a key regulator of Akt full activation via phosphorylation of Ser473. Rapamycin interaction with the intracellular receptor FKBP12 as well as new generation mTOR inhibitor has been shown. The third-generation mTOR inhibitor is a molecule in which rapamycin is cross-linked with a kinase inhibitor of mTOR. Abbreviations: mTOR mammalian target of rapamycin, RTKs Receptor tyrosine kinases, PI3K phosphoinositide 3-kinase, TSC tuberous sclerosis, Rapa Rapamycin
The mTOR Pathway and mTOR Inhibitors
mTOR is a downstream effector of the PI3K/AKT pathway (Fig. 1). mTOR works through two distinct multiprotein complexes, mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2) [9] which are evolutionarily conserved from yeast to mammals [10, 11]. These two complexes consist of unique mTOR-interacting proteins that determine their substrate specificity and localize them to different subcellular compartments, thus affecting their activation and function [12].
mTORC1 recruits substrates through the regulatory-associated protein of mTOR (RAPTOR) that are then further aligned to the catalytic cleft of mTOR. Rapamycin inhibits mTOR complex 1 (mTORC1) through the interaction with the intracellular receptor FKBP12 forming an inhibitory complex, which binds a region in the C terminus of TOR proteins [13, 14]. However, the exact mechanism of how this interaction with the FRB domain leads to inhibition of mTOR signaling remains to be defined. It has been proposed that rapamycin does not inhibit initial substrate recruitment but blocks correct alignment of some substrates to the catalytic cleft [15]. This could explain why rapamycin is more effective in blocking the phosphorylation and activation of ribosomal protein S6 kinase 1 (S6 K1) than that of eIF4E–binding protein 1 (4E–BP1). On the other hand, mTORC2 was identified as a rapamycin-insensitive entity, as acute exposure to rapamycin did not affect mTORC2 activity or Akt phosphorylation. However, subsequent studies have shown that, at least in some cell lines, prolonged exposure with rapamycin seems to prevent also mTORC2 assembly by progressive sequestration of the intracellular pool of mTOR and subsequently led to inhibition of AKT-signaling [16].
Temsirolimus (CCI-779) , everolimus (RAD001), and ridaforolimus (MK-8669) are rapamycin analogs, called rapalogs, developed to overcome its limited pharmacological properties, such as poor water solubility and chemical stability [7] and to obtain drugs with improved pharmacokinetic (PK) properties and reduced immunosuppressive effects. However, they preserve the interactions with FKBP12 and mTOR maintaining a similar mechanism of action based on inhibition of mTORC1 and induction of cell cycle arrest in the G1 phase [17]. Unluckily, in clinical trials conducted in cancer patients they showed only limited benefits. Possible explanations could be not only due to the incomplete block of mTORC1 kinase towards its substrate 4E–BP1 and the rapalog’s inability to effectively inhibit mTORC2, but also related to the existence of feedback loops as well as the activation of mechanisms outside the mTOR pathway [18].
Besides rapalogs, second generation mTOR inhibitors have been developed with the aim to have a more potent anticancer activity (Table 1). One class is represented by the so-called selective mTORC1/2 inhibitors which are small molecules working like ATP-competitive inhibitors of mTOR. In particular, they block the phosphorylation of all known downstream targets of both mTORC complexes without inhibiting other kinases. It seems that the greater anti-proliferative and pro-apoptotic effects of these molecules compared to rapamycin and observed in preclinical studies are linked to the complete block of 4E–BP1 phosphorylation and to the decreased protein expression of cyclin D1 and D3 as well as to a significant induction of p27 [19, 20]. Another class of small molecules able to inhibit mTOR is the mTOR and PI3K dual-inhibitors. With respect to the other compounds they do have the advantage to target all the three key enzymes, PI3K, Akt, and mTOR. Thus, they potentially overcome the known feedback loops occurring with rapalogs and being active in tumors with alterations downstream of PI3K but upstream of mTOR [21]. Unluckily, the results in clinical trials are not consistent with the ones obtained in preclinical studies carried out in several types of cancers using these molecules [22].
Recently, mTOR resistance mutations to both rapalogs and kinase inhibitors of mTOR have been identified. To overcome this resistance, a third generation mTOR inhibitors have been developed. This compound was called Rapalink in order to create a bivalent interaction that exploits the unique juxtaposition of two drug-binding pockets that contain rapamycin cross-linked with a kinase inhibitor of mTOR in the same molecule [23].
Pharmacokinetics of Rapalogs
Rapamycin and rapalogs have complex pharmacokinetics [24]. The use of rapamycin in cancer treatment has been largely limited by its intrinsic chemical stability. Thus, rapamycin chemical structure has been modified to increase its water solubility and bioavailability by adding a moiety at position C43. In particular an ester, an ether, or a phosphonate group creates temsirolimus, everolimus, and ridaforolimus, respectively (Fig. 2).

Chemical structure of rapamycin and rapalogs
Rapamycin and its derivatives are substrates for the CYP3A4 pathway [25]. Temsirolimus is quickly metabolized through de-esterification in the liver to form its primary metabolite sirolimus. However, temsirolimus is not considered a prodrug for sirolimus, as both agents are pharmacologically active. Everolimus is also metabolized, mainly in the gut and liver, but even if six main metabolites have been identified following its administration, everolimus is the main circulating component in human blood. As a result of their metabolism by isoenzymes of the CYP3A pathway, drugs that are substrates, activators, and inhibitors of these enzymes like rifampicin, anticonvulsants, and immunosuppressive compounds such as cyclosporine could potentially interact with rapalogs [26]. Moreover, due to the liver metabolism, both temsirolimus and everolimus require dose adjustments in patients with hepatic impairment while no correction is required in the presence of renal function alteration. Liver metabolism interferes also with the route of administration. Indeed, while intravenous (i.v.) rapalogs like temsirolimus and ridaforolimus display predictable pharmacokinetics with a high distribution volume and low interpatient variability, the pharmacokinetics of everolimus may be subjected to first-pass metabolism in the liver as well as influenced for absorption and bioavailability by the gastrointestinal tract (i.e. expression of ATP-binding cassette membrane transporters in the gut) [27].
Toxicity
The use of mTOR inhibitors as all the anti-cancer agents has been linked to the possibility of developing adverse events (AEs) that require specific management [28]. They can be directly mediated by the mTOR inhibitors antiproliferative effect [29] or driven by their ability to block a specific pathway [30].
Pneumonitis
Pneumonitis , or interstitial lung disease (ILD), is a potential complication of mTOR inhibitors [31]. The reported incidence varies widely as a result of a non uniform diagnostic criterium and active surveillance. Two main mechanisms for the pathophysiology of mTORi-induced ILD have been proposed. First, a directly toxic effect has been suggested since pulmonary toxicity appears to be a dose-related effect. Alternatively, an immunological origin is suggested by the high numbers of CD4+ T cells and eosinophils found in the BAL fluid of patients with ILD. In particular, three mechanisms are proposed: exposition of cryptic antigens, delayed-type hypersensitivity reaction and cytokine production. The diagnosis of mTORi-induced ILD is often difficult as clinical, radiological and pathological features are nonspecific and often are not distinguishable from respiratory infections. Thus, ILD should be a diagnosis of exclusion and diagnostic work up cannot be limited to x-ray or CT-scan but needs to include bronchoalveolar lavage (BAL) and pulmonary function tests (PFT). The onset typically occurs within 2–6 months after treatment initiation. The most common symptoms of ILD are nonspecific and include dyspnea, (dry) cough, fever, fatigue, hypoxia and occasionally hemoptysis. PFTs should be performed prior to starting mTOR inhibitor therapy to confirm a normal baseline organ function. mTOR inhibitors should be avoided in patients with significant pulmonary fibrosis or severe chronic obstructive pulmonary disease. The optimal management of ILD is essential to balance the risk of iatrogenic morbidity with the maximum efficacy using mTORi in treating cancer patients.
Metabolic Adverse Events
Hyperglycemia and hyperlipidemia are the metabolic AEs registered in patients treated with mTOR inhibitors [32].
Mammalian target of rapamycin (mTOR) inhibitors are associated with a high incidence of hyperglycemia, ranging from 13% to 50%. In particular, Grade 3 to 4 hyperglycemic events occurred in 12% of patients treated with everolimus, and in 11% of patients treated with temsirolimus. The pathophysiology of mTOR inhibitor-induced hyperglycemia and new-onset diabetes mellitus (NODM) is complex and linked to the interaction between mTOR downstream target S6 K1 with growth factors, hormones, and nutrients.
mTOR inhibitors directly act on pancreatic β-cells with a reduction in glucose-stimulated insulin secretion. On the other hand, they also seem to improve peripheral insulin resistance. Preclinical data in muscle cells showed that long-term rapamycin treatment is able to promote β-oxidation of fatty acids while diminishing basal glucose transport and glycogen synthesis [33].
A similar mechanism has been proposed for hyperlipidemia. In primary cultures of rat hepatocytes, rapamycin has been shown to affect glucose uptake and glycogen synthesis switching the metabolic preference to fatty acids as a metabolic fuel, thus, stimulating lipolysis and producing high serum levels of fatty acids [34]. Another pathophysiologic mechanism through mTOR inhibitors that may cause hyperlipidemia is an impaired lipid clearance via inhibition of insulin-stimulated lipoprotein lipase (LPL) and a significant reduction in the fractional catabolic rate of very LDL apoB100 (a triglyceride-rich lipoprotein).
Levels of lipids and glucose (preferably fasting) should be performed before starting and regularly during treatment with mTOR inhibitors. In the case of onset of metabolic AEs management strategies are similar for all causes of diabetes and hyperlipidemia. Interventions such as diet, exercise, and specific drugs (lipid-lowering agents, oral antihyperglycemic agents or insulin) should be initiated based on lipids and glucose levels.
Hematological Toxicities
An alteration in the IL-10-dependent inflammatory auto-regulation seems to be responsible of mTOR inhibitor-related anemia. In particular, it may promote disruptions in iron homeostasis and gastrointestinal iron absorption as well as effects on erythroid progenitor cell differentiation and/or erythropoietin receptor-mediated proliferation [35]. Anemia is generally mild, dose-dependent, and reversible upon discontinuation of treatment. The onset is generally within a month of initiation and is sustained throughout treatment. If detected, other causes of anemia have to be screened (i.e. occult blood in stools and vitamin B12 and folate levels). Oral or intravenous iron supplementation and erythropoiesis-stimulating agents should be effective for managing mTOR inhibitor-associated anemia, if not treatment needs to be discontinued.
Thrombocytopenia and leucopenia/neutropenia have been reported with mTOR inhibitor therapy. These AEs frequently occur simultaneously and usually resolve spontaneously. Complete blood counts should be performed routinely. Management is similar to that used for chemotherapy related-hematological toxicities. Grade 3 or higher neutropenia or thrombocytopenia may require temporary interruption of mTOR.
mTOR Inhibitors Associated Stomatitis (mIAS)
The incidence of mIAS varies widely (2–78%). As reported across multiple mTOR inhibitor clinical trials, grade 3/4 toxicities occur in up to 9% of patients. mIAS typically presents as distinct, painful, ovoid, superficial ulcers surrounded by a characteristic erythematous margin and due to a direct toxic effects of mTOR inhibitors on oral and nasal mucous membranes [36]. It resembles recurrent aphthous ulceration not only in clinical presentation but also in response to therapy.
Prophylactic strategies, including oral hygiene and avoiding injury to the epithelium of the oral cavity, are recommended. Topical high-potency corticosteroids, nonsteroidal anti-inflammatory drugs (NSAIDs) and anesthetics can be used to promote healing and reduce pain, but severe resistant mIAS could require systemic corticosteroids. Moreover, if grade 2 or higher mIAS restricts oral intake of nutrients, in such cases, mTOR inhibitor dose reduction/discontinuation may be considered.
Mechanisms of Resistance
Mutations of mTOR
Similarly to what happen in patients treated with kinase inhibitors acquired resistance mutations have been reported in cells exposed to mTORC1 inhibitors [23]. The MCF-7 breast cancer cell line was exposed to high concentrations of either rapamycin or a second-generation mTOR inhibitor (AZD8055) for 3 months. Subsequent deep sequencing of the emerged resistant colonies revealed clones harbored mutations located in the FKBP12–rapamycin-binding domain (FRB domain) or in the kinase domain. The clinical relevance of these mutations is supported by a case report of a patient who acquired an identical mTOR mutation after relapse while under treatment with everolimus [37] as well as by their observation in untreated patients.
Mutations that conferred resistance to ATP-competitive inhibitors of mTOR did not alter binding of the drug to mTOR but generated a hyperactive state of the kinase that can affect both mTORC1 and mTORC2. On the other hand, some of the identified hyper-activating mutations of mTOR are associated with increased sensitivity to rapamycin, suggesting that cancer cells harboring such mutations have an mTOR-dependent proliferation pattern. Interestingly, in a case report of a primary refractory HL, damaging mutations of the TSC2 gene was considered responsible to the increased mTOR pathway activation and, thus, to the impressive clinical response observed using everolimus [38].
Genetic and Functional Heterogeneity
Genetic tumor heterogeneity is a well know concept in cancer biology. Both immunohistochemical staining and genome sequencing have been demonstrated that cancer cells displaying a high mTOR activity coexist with cancer cells having low mTORC1 activity in the same tumor. This observation has also been extended to primary tumor and distant metastases [39]. Moreover, genetic tumor heterogeneity has been reported for proteins that belong to signaling pathways upstream to mTOR such as PI3K/AKT and Ras/Raf/MEK/MAPK pathways.
Along with genetic alterations, a functional heterogeneity has been described for downstream effectors of mTOR like S6 K1 and 4E–BP1. An in vitro study on human colorectal cancer demonstrated that phosphorylation of S6 K1 and 4E–BP1 rarely occurs in the same cancer cell but rather shows mutual exclusivity [40]. Thus, since rapalogs do not block mTORC1-mediated 4EBP1 phosphorylation of cancer cells with a phospho-S6low/phospho-4E-BP1high pattern might be intrinsically resistant to rapalogs despite the presence of mTORC1 activity.
Finally, mTORC activity could be affected by micro-environmental conditions like oxygen levels [41] and pH values [42]. In both cases a downregulation of mTORC1 activity is registered, thus, cancer cells xhibit an mTORC1-independent growth and are therefore resistant to mTORC1 inhibition. Of note, hypoxia not necessarily leads to mTORC1 inhibition. For example, tumor cells harboring low levels of Ataxia Telangiectasia Mutated (ATM) protein , display a paradoxically elevated mTORC1 activity in hypoxic tumor regions. In particular, ATM is the driver of a cascade comprising HIF1𝛼 and REDD1 which inactivates mTORC1 activity in a TSC1/TSC2 dependent mechanism [43].
Alternative Proliferation Pathways
There is a complex network of regulatory feedback loops responsible for limiting the proliferative signals transmitted by upstream effectors once mTORC1 is activated. Thus, once mTORC1 is inhibited, these negative feedback loops are stopped and alternative proliferation pathways like PI3K/AKT and RAS/RAF/MEK/MAPK are free to contrast the anticancer efficacy of rapalogs. This concept has been demonstrated in the preclinical setting, in which some data showed that blocking AKT or MAPK potentiated the anticancer efficacy of rapalogs [44].
Molecular Mechanisms of mTOR Activation in Lymphomas
Aberrant activation of the mTOR pathway is a marker of more aggressive disease and poorer prognosis in both Hodgkin (HL) and non-Hodgkin lymphomas (NHLs). As already discussed, this condition can be related to mTOR specific biology but it is often linked to alterations in key upstream pathway(s) [45,46,47].
For example, in a subset of MCL, mTOR directly mediates Cyclin D1 downregulation trough glycogen synthase kinase (GSK)-3β [46], while other authors described PTEN inactivating phosphorylation as the key mechanism responsible for the PI3K/Akt/mTOR pathway activation. Moreover, a similar mechanism has been described in HL too [48].
Activated B-cell DLBCL (ABC-DLBCL) cell lines activate S6 K1, a downstream target of mTOR, independently from Akt either through up-regulation of PIM2 or through activation by B cell receptor (BCR) signaling components [47]. Conversely, loss of PTEN has been described to correlate with the PI3K/Akt/mTOR pathway activation in germinal center B-cell-like DLBCL (GCB-DLBCL). Of note, mTOR mutations have been described in DLBCL samples [49]. Instead, phosphorylation of Akt is common in T cell lymphoma [50].
Summary of Clinical Trials
Based on the encouraging preclinical in vitro and in vivo data [51,52,53] clinical trials using rapalogs have been carried out in hematological malignancies and, in particular, in lymphoproliferative disorders.
Temsirolimus
Temsirolimus has been widely investigated in hematological malignancies alone or in combinations. In lymphoma setting, it has been firstly used as single agent in a phase II trial at 250 mg/m2 weekly in 34 patients with relapsed MCL. The overall response rates (ORR) was 38% with 1 (3%) complete response (CR) and 12 (35%) partial response (PR). The median time-to-progression in all patients was 6.5 months and the duration of response for the 13 responders was 6.9 months. Hematological toxicities were the most common adverse events (AEs) with thrombocytopenia occurring in all patients and being the most frequent cause of dose reductions even if usually resolving in 1 week. Hyperglycemia, increased triglycerides, mucositis, and fatigue were also registered [54]. A lower dose of 25 mg/m2 weekly has been evaluated in a subsequent clinical trial with the aim to reduce the previous registered events. The ORR was similar (41%) and severe thrombocytopenia was less common (100% vs. 39%) [55]. The encouraging results of these phase II trials (RR of around 40%) pave the way for a large randomized phase III trial [56] in relapsed/refractory MCL patients. The higher doses in the temsirolimus arm (175 mg weekly for 3 weeks followed by 75 mg weekly) were significantly better than the investigator’s choice both in ORR (22.2% vs 2%) and progression free survival (PFS), but the results were poorer than those reported in the phase II trial. However, data were considered consistent enough to obtain the European license for this indication. Recently, it has been published another phase III trial in relapsed/refractory MCL patients in which the standard of care temsirolimus has been used as a control arm compared to ibrutinib [57]. The primary efficacy analysis showed a significant improvement in PFS and a safer profile for patients treated with ibrutinib (median PFS 14.6 months vs 6.2 months). Moreover, an independent review committee-assessed overall response rate (ORR) was significantly higher for ibrutinib (71.9% vs 40.4%; p < 0.0001) with a CR rate of 18.7% vs 1.4%, respectively. Median treatment duration was 14.4 months for ibrutinib and 3.0 months for temsirolimus. Safety profile was favorable for the ibrutinib arm too. Reported grade 3 or higher treatment-related adverse events were lower with 94 (68%) versus 121 (87%) patients involved. Moreover, less patients discontinued treatment due AEs in the ibrutinib arm (25.5% vs 6.5%). Single agent temsirolimus has also been investigated in relapsed/refractory (Rel/Ref) primary CNS Lymphoma (PCNSL). A relatively high RR (54%) was observed but PFS (median PFS 2.1 months) was comparable with other studies [58]. Of note, treatment-associated mortality was considerable (13.5%). The authors interpretation is that frequent administration of steroids before response assessment as well as compromised condition of enrolled patients could be potential confounding factors for response evaluation and outcome. The most common AEs ≥3 grade were hyperglycemia (29.7%), thrombocytopenia (21.6%), infection (19%), anemia (10.8%), and rash (8.1%). Interestingly, neither drug nor its main metabolites were found in the CSF except in one patient in the 75-mg cohort who had 2 ng/ml of temsirolimus.
Temsirolimus has been combined with different drugs in different settings.
Combination of temsirolimus and bortezomib has been assessed in heavily pretreated Multiple Myeloma [59] and B-Cell Non-Hodgkin Lymphoma [60] patients. In both studies, the enrolled subjects received i.v. bortezomib (1.6 mg/m2) weekly on days 1, 8, 15, and 22 along with i.v. temsirolimus (25 mg) weekly on days 1, 8, 15, 22, and 29 every 35 days. Fourteen of 43 (33%) MM patients had a PR or better. Moreover, the authors noted a difference in bortezomib-responsive versus refractory patients to previous treatment with bortezomib suggesting that the combination might not completely overcome resistance or re-sensitize MM cells that are resistant to the proteasome inhibitor. On the other hand, the ORR in the Lymphoma setting was 31% (12 of 39 patients; 3 CR and 9 PR) while the median PFS was 4.7 months. Although the patients with Diffuse Large B-Cell Lymphoma (DLBCL) had a low ORR, 2 heavily pretreated patients achieved a CR after 2 cycles of therapy and both maintained remission for 7 months after the completion of protocol therapy. The underlying genetic heterogeneity of DLBCL has been suggested by the authors as presumably responsible for the wide variation observed in responses. There were no unexpected toxicities from the combination. AEs were generally manageable and similar with those reported with temsirolimus and bortezomib alone, in both studies.
The incorporation of temsirolimus in the doublet rituximab/bendamustine has been recently reported in a phase I study of Rel/Ref FL and MCL [61] showing promising preliminary activity especially in MCL along with a safety profile. An objective response was observed in 14/15 patients (93%), including 5 CR (33%; all MCL). Ongoing studies are assessing the temsirolimus combination with Rituximab and DHAP in patients with Rel/Ref DLBCL (NCT01653067) [62] and temsirolimus plus lenalidomide in relapsed NHLs (NCT01076543).
Everolimus
Like temsirolimus, the oral drug everolimus has been used as single agent in Rel/Ref aggressive and indolent NHLs [53, 63,64,65] as well as HL [66]. Recently, a phase II study has been carried out using oral single-agent everolimus in relapsed/refractory indolent lymphomas, mostly chronic lymphocytic leukemia (CLL) and follicular lymphoma (FL) [67]. Eligible patients received oral everolimus 10 mg daily on a 28 day-cycle schedule. The ORR in all 55 patients was 35% (19/55) with 4% (2/55) CRu, and 31% (17/55) PR; 36% (20/55) had stable disease. The median time to response was 2.3 months (range, 1.4–14.1) and the median DR was 11.5 months (95% CI, 5.7–30.4). The ORR was higher in FL (61%) than in CLL/SLL (19%). The median PFS and OS were 7.2 months and 29.4 months, respectively. Everolimus was well-tolerated with modest hematologic toxicity. Of note, two patients died of sepsis related to the drug. Thus, the authors concluded by suggesting further studies with mTORC1 inhibitors such as everolimus as single agent, and in combination with other agents. The addition of alemtuzumab to everolimus in rel/ref CLL has been published recently too, but based on their results (33% partial responses, no complete responses) no further development of this regimen was recommended by the authors [68]. Another phase II trial evaluated the activity and safety of everolimus in Rel/Ref marginal zone lymphomas (MZLs) [69]. Thirty patients received everolimus for six cycles or until dose-limiting toxicity or progression. Twenty-four out of 30 patients were evaluable and a relevant proportion experienced side effects, resulting in dose reduction (9 patients) and/or early treatment discontinuation (10 patients). ORR was 25% (1 CR and 5 PRs). Moreover, one toxic death due to treatment-related pneumonia was recorded. Thus, due to the moderate antitumor activity and the observed toxicity, it seems that single agent everolimus has limited therapeutical space in this indolent setting. Of note, it has also been carried out a phase III trial of everolimus in monotherapy as maintenance (PILLAR-2; NCT00790036) providing 1 year of adjuvant everolimus to poor-risk (IPI] ≥3) in DLBCL patients who had achieved a CR with R-chemo. No differences have been observed in the 2-yr DFS rate (78% vs 77%) even if it seemed that everolimus had a trends toward OS and DFS in selected patient subgroups (males and IPI 4/5). However, also in this setting the responses were modest, transient and in some cases toxicity was relevant [70].
Conversely from what happened in CLL/SLL, the combination of everolimus with other drugs seems to be promising. Based on the encouraging results of the preclinical data [71] showing that combining panobinostat with the mTOR inhibitor everolimus inhibited panobinostat-induced mTOR activation and enhanced panobinostat antiproliferative effects in HL cell lines, a combination of these two drugs has been carried out in a phase I trial [72]. ORR 43% with CR 15% while the dose-limiting toxicity was thrombocytopenia (grade3/4 64%). Similarly, after a phase I trial, a phase II study of everolimus in combination with CHOP (cyclophosphamide, doxorubicin, vincristine, and prednisone) as a first-line treatment for patients with peripheral T-cell lymphoma (PTCL) has been published [73]. Five (5) mg everolimus per day from day 1 to 14 every 21 days for a total of six cycles has been administered. A difference in the CR rate among subtypes was observed and was associated with PTEN loss evaluated by immunohistochemistry. Objective response rate was very high (90%; CR (n = 17) and PR (n = 10)).
Another combination has been tested in a phase I/II trial in which everolimus has been added to rituximab with or without bortezomib in Rel/Ref Waldenström’s Macroglobulinemia (WM) [74]. Forty-six patients have received six cycles of both the combinations followed by maintenance with everolimus until progression. Thirty-six (78%) of the 46 patients enrolled received full dose therapy (FDT) of the three drugs. Promising results that deserve to be assessed in future trials on a larger randomized trial has been showed with an ORR of 89% (32/36 patients) with two CR (6%) and 19 PR or better response (53%). No dose-limiting toxicities have been observed in the phase I of the trial. No unexpected toxicities was recorded. Moreover, of note, 98% of registered patients had previously received rituximab, and 57% had received bortezomib.
Ridaforolimus
Only two clinical trials need to be cited on Ridaforolimus. In the first one drug has been investigated in a phase II clinical trial as monotherapy in 55 patients with Rel/Ref hematological malignancies. Drug was used as 30-min infusion on days 1–5 of a 2 weeks cycle. Of note best response was PR and it was observed only in two subsets of hematological malignancy, 29% in agnogenic myeloid metaplasia (AMM) and 33% in MCL. The most frequent grade 3/4 AEs were similar to those observed with other mTOR inhibitors, in particular mouth sores (15%), thrombocytopenia (15%), hyponatremia (7%) and hypokalemia (6%) [75]. On the other hand, the second one is a phase I study in which ridaforolimus is evaluated in combination with vorinostat in patients with advanced solid tumors or lymphoma (NCT01169532).
Second Generation mTOR Inhibitors
AZD8055 is a first-in-class dual mTORC1/mTORC2 inhibitor. In preclinical models it was shown to prevent the mTORC2-mediated AKT activation observed with rapalogs [76]. In a phase I study of 49 patients with advanced solid tumors or lymphomas (NCT00731263) [77]. MTD was 90 mg BID. The most frequent AEs were elevated transaminases (22%) and fatigue (16%). Interestingly, metabolic AEs like hypercholesterolemia nor hypertriglyceridemia were not registered as observed with other mTORC1/mTORC2 inhibitors [78, 79]. The best response was SD in 7 patients for ⩾4 months.
The results of Part A of a phase I/II study on the dual mTORC1/mTORC2 kinase inhibitor CC-223 in 28 pretreated patients with advanced solid tumors or MM has been recently published [78]. The MTD was 45 mg/d, although 11.1% of patients at the MTD required dose reductions and 55.6% required interruptions. Hyperglycemia was the most common grade 3 AE (18%). Substantial pS6 K1 (>70%), p4E–BP1 (>40%), and pAKT (>50%) inhibition was observed at ≥30 mg CC-223, although pS6 K1 and pAKT inhibition was more complete than p4E–BP1 inhibition. Additionally, preliminary evidence of inhibition of pS6 K1, p4E–BP1, pAKT, and proliferation marker Ki-67 was observed in paired tumor biopsies in 2 patients. The authors reported one PR (3.6%) lasting 220 days in 1 patient with breast cancer and 8 patients (29%) with SD (>100 days in 5 patients), including 2 patients with tumors exhibiting molecular abnormalities associated with mTORC pathway activation. Part B focused on dose expansion into parallel cohorts of selected tumor types (MM, DLBCL, and selected solid tumors) is ongoing (NCT01177397).
TAK-228, another dual mTORC1/mTORC2 kinase inhibitor, has been tested in a phase I study including 39 patients with MM (31), NHL (4), and WM (4) [79]. Drug has been administered once daily (QD) at 2, 4, 6, or 7 mg, or QD for 3 days on and 4 days off each week (QDx3d QW) at 9 or 12 mg, in 28-day cycles. Cycle 1 DLTs occurred in 5 QD patients (stomatitis, urticaria, blood creatinine elevation, fatigue, and nausea and vomiting) and 4 QDx3d QW patients (erythematous rash, fatigue, asthenia, mucosal inflammation, and thrombocytopenia). The MTDs were determined to be 4 mg QD and 9 mg QDx3d QW. Thirty-six patients (92%) reported at least one drug-related toxicity; the most common grade ≥3 drug-related toxicities were thrombocytopenia (15%), fatigue (10%), and neutropenia (5%). Of the 33 response-evaluable patients, one MM patient had a minimal response, one WM patient achieved PR, one WM patient had a minor response, and 18 patients (14 MM, 2 NHL, and 2 WM) had SD. Authors concluded saying that further studies including combination strategies need to be carried out.
Preliminary data on BEZ235, a dual PI3-Kinase/mTOR inhibitor in adult patients with RR acute leukemia showing a single-agent anti-leukemic efficacy most pronounced in ALL, with an overall response rate of 30% and a sustained molecular remission in one patient. Since results of PK analysis and assessment of PD markers associated with PI3K signaling did not correlate with response the authors concluded that a more comprehensive genomic analysis may help to identify a subset of patients likely to benefit from treatment with dual PI3K-mTOR inhibitors (NCT01756118) [80].
CC-115, a novel inhibitor of mTOR kinase and DNA-PK, was evaluated in primary CLL cells in vitro and in seven Rel/Ref CLL patients and one SLL patient harboring ATM deletions/mutations enrolled in a larger phase I clinical trial, including 110 additional patients with solid tumors (NCT01353625) [81]. All but one patient had a decrease in lymphadenopathy, resulting in one iwCLL partial response (PR) and three PRs with lymphocytosis. Moreover, the encouraging preclinical data on the ability of CC-115 to revert CD40-mediated resistance to chemotherapy or venetoclax as well as to overcome Idelalisib resistance makes this compound attractive for further combination studies in the clinical setting.
Summary
The PI3K/AKT/mTOR signaling pathway plays a central role in cell growth proliferation and survival controlling different processes in protein synthesis and angiogenesis. Deregulation of this pathway is commonly found in several types of tumors.
Currently, two mTOR inhibitors, everolimus and temsirolimus, are approved by the European Medicines Agency (EMA) and the US Food and Drug Administration to treat cancer patients in clinical practice.
Unluckily the promising results obtained in the preclinical settings using rapalogs did not translate into the expected benefits in clinical trials because response to mTOR inhibitors is not durable and patients ultimately progress because of various mechanisms of resistance. The so-called “second generation mTOR inhibitors” are small molecules developed with the aim to overcome rapalogs weaknesses. However clinical trials results do not seems to differ a lot from those obtained with rapalogs.
Common and serious mTOR inhibitors related side effects include non-infectious pneumonitis, metabolic disorders, hematological and mucosal toxicities. They require specific management in order to balance risk and benefit related to the specific treatment.
Looking forward correlative or translational sub-studies are needed to clearly and quickly identify biomarkers of response and emerging drug resistance in order to maximize the benefit linked to mTOR inhibitors treatment. Moreover, future approaches may consider combinational strategies as a way to overcome such resistance and therefore improve efficacy of mTOR targeting agents in the clinical context.
References
Vézina C, Kudelski A, Sehgal SN. Rapamycin (AY-22,989), a new antifungal antibiotic. I. Taxonomy of the producing streptomycete and isolation of the active principle. J Antibiot (Tokyo). 1975;28(10):721–6.
Sehgal SN, Baker H, Vézina C. Rapamycin (AY-22,989), a new antifungal antibiotic. II. Fermentation, isolation and characterization. J Antibiot (Tokyo). 1975;28:727–32.
Eng CP, Sehgal SN, Vézina C. Activity of rapamycin (AY-22,989) against transplanted tumors. J Antibiot (Tokyo). 1984;37(10):1231–7.
Yatscoff RW, LeGatt DF, Kneteman NM. Therapeutic monitoring of rapamycin: a new immunosuppressive drug. Ther Drug Monit. 1993;15(6):478–82.
Guertin DA, Sabatini DM. Defining the role of mTOR in cancer. Cancer Cell. 2007;12(1):9–22.
Abizaid A. Sirolimus-eluting coronary stents: a review. Vasc Health Risk Manag. 2007;3(2):191–201.
Ballou LM, Lin RZ. Rapamycin and mTOR kinase inhibitors. J Chem Biol. 2008;1(1–4):27–36.
Baldo P, Cecco S, Giacomin E, Lazzarini R, Ros B, Marastoni S. mTOR pathway and mTOR inhibitors as agents for cancer therapy. Curr Cancer Drug Targets. 2008;8(8):647–65.
Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149(2):274–93.
Helliwell SB, Wagner P, Kunz J, Deuter-Reinhard M, Henriquez R, Hall MN. TOR1 and TOR2 are structurally and functionally similar but not identical phosphatidylinositol kinase homologues in yeast. Mol Biol Cell. 1994;5(1):105–18.
Loewith R, Jacinto E, Wullschleger S, Lorberg A, Crespo JL, Bonenfant D, Oppliger W, Jenoe P, Hall MN. Two TOR complexes, only one of which is rapamycin sensitive, have distinct roles in cell growth control. Mol Cell. 2002;10(3):457–68.
Betz C, Hall MN. Where is mTOR and what is it doing there? J Cell Biol. 2013;203(4):563–74.
Chen J, Zheng XF, Brown EJ, Schreiber SL. Identification of an 11-kDa FKBP12-rapamycin-binding domain within the 289-kDa FKBP12-rapamycin-associated protein and characterization of a critical serine residue. Proc Natl Acad Sci U S A. 1995;92(11):4947–51.
Choi J, Chen J, Schreiber SL, Clardy J. Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science. 1996;273(5272):239–42.
Shimobayashi M, Hall MN. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol. 2014;15(3):155–62.
Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell. 2006;22(2):159–68.
Fingar DC, Blenis J. Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene. 2004;23(18):3151–71.
Calimeri T, Ferreri AJM. M-TOR inhibitors and their potential role in haematological malignancies. Br J Haematol. 2017;177(5):684–702.
Mi W, Ye Q, Liu S, She QBAKT. Inhibition overcomes rapamycin resistance by enhancing the repressive function of PRAS40 on mTORC1/4E-BP1 axis. Oncotarget. 2015;6(16):13962–77.
Thoreen CC, Kang SA, Chang JW, Liu Q, Zhang J, Gao Y, Reichling LJ, Sim T, Sabatini DM, Gray NS. An ATP-competitive mammalian target of rapamycin inhibitor reveals rapamycin-resistant functions of mTORC1. J Biol Chem. 2009;284(12):8023–32.
Dienstmann R, Rodon J, Serra V, Tabernero J. Picking the point of inhibition: a comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol Cancer Ther. 2014;13(5):1021–31.
Rodon J, Dienstmann R, Serra V, Tabernero J. Development of PI3K inhibitors: lessons learned from early clinical trials. Nat Rev Clin Oncol. 2013;10(3):143–53.
Rodrik-Outmezguine VS, Okaniwa M, Yao Z, Novotny CJ, McWhirter C, Banaji A, Won H, Wong W, Berger M, de Stanchina E, Barratt DG, Cosulich S, Klinowska T, Rosen N, Shokat KM. Overcoming mTOR resistance mutations with a new-generation mTOR inhibitor. Nature. 2016;534(7606):272–6.
Danesi R, Boni JP, Ravaud A. Oral and intravenously administered mTOR inhibitors for metastatic renal cell carcinoma: pharmacokinetic considerations and clinical implications. Cancer Treat Rev. 2013;39(7):784–92.
Klümpen HJ, Beijnen JH, Gurney H, Schellens JH. Inhibitors of mTOR. Oncologist. 2010;15(12):1262–9.
Boni J, Leister C, Burns J, Cincotta M, Hug B, Moore L. Pharmacokinetic profile of temsirolimus with concomitant administration of cytochrome p450-inducing medications. J Clin Pharmacol. 2007;47(11):1430–9.
Kirchner GI, Meier-Wiedenbach I, Manns MP. Clinical pharmacokinetics of everolimus. Clin Pharmacokinet. 2004;43(2):83–95.
Kaplan B, Qazi Y, Wellen JR. Strategies for the management of adverse events associated with mTOR inhibitors. Transplant Rev (Orlando). 2014;28(3):126–33.
Mahé E, Morelon E, Lechaton S, Sang KH, Mansouri R, Ducasse MF, Mamzer-Bruneel MF, de Prost Y, Kreis H, Bodemer C. Cutaneous adverse events in renal transplant recipients receiving sirolimus-based therapy. Transplantation. 2005;79(4):476–82.
Houde VP, Brûlé S, Festuccia WT, Blanchard PG, Bellmann K, Deshaies Y, Marette A. Chronic rapamycin treatment causes glucose intolerance and hyperlipidemia by upregulating hepatic gluconeogenesis and impairing lipid deposition in adipose tissue. Diabetes. 2010;59(6):1338–48.
Willemsen AE, Grutters JC, Gerritsen WR, van Erp NP, van Herpen CM, Tol J. mTOR inhibitor-induced interstitial lung disease in cancer patients: comprehensive review and a practical management algorithm. Int J Cancer. 2016;138(10):2312–21.
Busaidy NL, Farooki A, Dowlati A, Perentesis JP, Dancey JE, Doyle LA, Brell JM, Siu LL. Management of metabolic effects associated with anticancer agents targeting the PI3K-Akt-mTOR pathway. J Clin Oncol. 2012;30(23):2919–28.
Di Paolo S, Teutonico A, Leogrande D, Capobianco C, Schena PF. Chronic inhibition of mammalian target of rapamycin signaling downregulates insulin receptor substrates 1 and 2 and AKT activation: a crossroad between cancer and diabetes? J Am Soc Nephrol. 2006;17(8):2236–44.
Kraemer FB, Takeda D, Natu V, Sztalryd C. Insulin regulates lipoprotein lipase activity in rat adipose cells via wortmannin- and rapamycin-sensitive pathways. Metabolism. 1998;47(5):555–9.
Sofroniadou S, Kassimatis T, Goldsmith D. Anaemia, microcytosis and sirolimus--is iron the missing link? Nephrol Dial Transplant. 2010;25(5):1667–75.
Peterson DE, O'Shaughnessy JA, Rugo HS, Elad S, Schubert MM, Viet CT, Campbell-Baird C, Hronek J, Seery V, Divers J, Glaspy J, Schmidt BL, Meiller TF. Oral mucosal injury caused by mammalian target of rapamycin inhibitors: emerging perspectives on pathobiology and impact on clinical practice. Cancer Med. 2016;5(8):1897–907.
Wagle N, Grabiner BC, Van Allen EM, Amin-Mansour A, Taylor-Weiner A, Rosenberg M, Gray N, Barletta JA, Guo Y, Swanson SJ, Ruan DT, Hanna GJ, Haddad RI, Getz G, Kwiatkowski DJ, Carter SL, Sabatini DM, Jänne PA, Garraway LA, Lorch JH. Response and acquired resistance to everolimus in anaplastic thyroid cancer. N Engl J Med. 2014;371(15):1426–33.
Perini GF, Campregher PV, Ross JS, Ali S, Hamerschlak N, Santos FP. Clinical response to everolimus in a patient with Hodgkin’s lymphoma harboring a TSC2 mutation. Blood Cancer J. 2016;6:e420.
Gerlinger M, Rowan AJ, Horswell S, Math M, Larkin J, Endesfelder D, GronroosE MP, Matthews N, Stewart A, Tarpey P, Varela I, Phillimore B, Begum S, McDonald NQ, Butler A, Jones D, Raine K, Latimer C, Santos CR, Nohadani M, Eklund AC, Spencer-Dene B, Clark G, Pickering L, Stamp G, Gore M, Szallasi Z, Downward J, Futreal PA, Swanton C. Intratumor heterogeneity and branched evolution revealed by multiregion sequencing. N Engl J Med. 2012;366(10):883–92.
Gerdes MJ, Sevinsky CJ, Sood A, Adak S, Bello MO, Bordwell A, Can A, Corwin A, Dinn S, Filkins RJ, Hollman D, Kamath V, Kaanumalle S, Kenny K, Larsen M, Lazare M, Li Q, Lowes C, McCulloch CC, McDonough E, Montalto MC, Pang Z, Rittscher J, Santamaria-Pang A, Sarachan BD, Seel ML, Seppo A, Shaikh K, Sui Y, Zhang J, Ginty F. Highly multiplexed single-cell analysis of formalin-fixed, paraffin-embedded cancer tissue. Proc Natl Acad Sci U S A. 2013;110(29):11982–7.
Wouters BG, Koritzinsky M. Hypoxia signalling through mTOR and the unfolded protein response in cancer. Nat Rev Cancer. 2008;8(11):851–64.
Faes S, Duval AP, Planche A, Uldry E, Santoro T, Pythoud C, Dormond O. Acidic tumor microenvironment abrogates the efficacy of mTORC1 inhibitors. Mol Cancer. 2016;15(1):78.
Cam H, Easton JB, High A, Houghton PJ. mTORC1 signaling under hypoxic conditions is controlled by ATM-dependent phosphorylation of HIF-1α. Mol Cell. 2010;40(4):509–20.
Carracedo A, Ma L, Teruya-Feldstein J, Rojo F, Salmena L, Alimonti A, Pandolfi PP. Inhibition of mTORC1 leads to MAPK pathway activation through a PI3K-dependent feedback loop in human cancer. J Clin Invest. 2008;118(9):3065–74.
Abubaker J, Bavi PP, Al-Harbi S, Siraj AK, Al-Dayel F, Uddin S, Al-Kuraya K. PIK3CA mutations are mutually exclusive with PTEN loss in diffuse large B-cell lymphoma. Leukemia. 2007;21(11):2368–70.
Dal Col J, Zancai P, Terrin L, Guidoboni M, Ponzoni M, Pavan A, Spina M, Bergamin S, Rizzo S, Tirelli U, De Rossi A, Doglioni C, Dolcetti R. Distinct functional significance of Akt and mTOR constitutive activation in mantle cell lymphoma. Blood. 2008;111(10):5142–51.
Ezell SA, Wang S, Bihani T, Lai Z, Grosskurth SE, Tepsuporn S, Davies BR, Huszar D, Byth KF. Differential regulation of mTOR signaling determines sensitivity to AKT inhibition in diffuse large B cell lymphoma. Oncotarget. 2016;7(8):9163–74.
Dutton A, Reynolds GM, Dawson CW, Young LS, Murray PG. Constitutive activation of phosphatidyl-inositide 3 kinase contributes to the survival of Hodgkin's lymphoma cells through a mechanism involving Akt kinase and mTOR. J Pathol. 2005;205(4):498–506.
Zhang J, Grubor V, Love CL, Banerjee A, Richards KL, Mieczkowski PA, Dunphy C, Choi W, Au WY, Srivastava G, Lugar PL, Rizzieri DA, Lagoo AS, Bernal-Mizrachi L, Mann KP, Flowers C, Naresh K, Evens A, Gordon LI, Czader M, Gill JI, Hsi ED, Liu Q, Fan A, Walsh K, Jima D, Smith LL, Johnson AJ, Byrd JC, Luftig MA, Ni T, Zhu J, Chadburn A, Levy S, Dunson D, Dave SS. Genetic heterogeneity of diffuse large B-cell lymphoma. Proc Natl Acad Sci U S A. 2013;110(4):1398–403.
Cai Q, Deng H, Xie D, Lin T, Lin T. Phosphorylated AKT protein is overexpressed in human peripheral T-cell lymphomas and predicts decreased patient survival. Clin Lymphoma Myeloma Leuk. 2012;12(2):106–12.
Wanner K, Hipp S, Oelsner M, Ringshausen I, Bogner C, Peschel C, Decker T. Mammalian target of rapamycin inhibition induces cell cycle arrest in diffuse large B cell lymphoma (DLBCL) cells and sensitises DLBCL cells to rituximab. Br J Haematol. 2006;134(5):475–84.
Márk Á, Hajdu M, Váradi Z, Sticz TB, Nagy N, Csomor J, Berczi L, Varga V, Csóka M, Kopper L, Sebestyén A. Characteristic mTOR activity in Hodgkin-lymphomas offers a potential therapeutic target in high risk disease–a combined tissue microarray, in vitro and in vivo study. BMC Cancer. 2013;13:250.
Witzig TE, Reeder C, Han JJ, LaPlant B, Stenson M, Tun HW, Macon W, Ansell SM, Habermann TM, Inwards DJ, Micallef IN, Johnston PB, Porrata LF, Colgan JP, Markovic S, Nowakowski GS, M G. The mTORC1 inhibitor everolimus has antitumor activity in vitro and produces tumor responses in patients with relapsed T-cell lymphoma. Blood. 2015;126(3):328–35.
Witzig TE, Geyer SM, Ghobrial I, Inwards DJ, Fonseca R, Kurtin P, Ansell SM, Luyun R, Flynn PJ, Morton RF, Dakhil SR, Gross H, Kaufmann SH. Phase II trial of single-agent temsirolimus (CCI-779) for relapsed mantle cell lymphoma. J Clin Oncol. 2005;23(23):5347–56.
Ansell SM, Inwards DJ, Rowland KM Jr, Flynn PJ, Morton RF, Moore DF Jr, Kaufmann SH, Ghobrial I, Kurtin PJ, Maurer M, Allmer C, Witzig TE. Low-dose, single-agent temsirolimus for relapsed mantle cell lymphoma: a phase 2 trial in the North Central Cancer Treatment Group. Cancer. 2008;113(3):508–14.
Hess G, Herbrecht R, Romaguera J, Verhoef G, Crump M, Gisselbrecht C, Laurell A, Offner F, Strahs A, Berkenblit A, Hanushevsky O, Clancy J, Hewes B, Moore L, Coiffier B. Phase III study to evaluate temsirolimus compared with investigator's choice therapy for the treatment of relapsed or refractory mantle cell lymphoma. J Clin Oncol. 2009;27(23):3822–9.
Dreyling M, Jurczak W, Jerkeman M, Silva RS, Rusconi C, Trneny M, Offner F, Caballero D, Joao C, Witzens-Harig M, Hess G, Bence-Bruckler I, Cho SG, Bothos J, Goldberg JD, Enny C, Traina S, Balasubramanian S, Bandyopadhyay N, Sun S, Vermeulen J, Rizo A, Rule S. Ibrutinib versus temsirolimus in patients with relapsed or refractory mantle-cell lymphoma: an international, randomized, open-label, phase 3 study. Lancet. 2016;387(10020):770–8.
Korfel A, Schlegel U, Herrlinger U, Dreyling M, Schmidt C, von Baumgarten L, Pezzutto A, Grobosch T, Kebir S, Thiel E, Martus P, Kiewe P. Phase II trial of Temsirolimus for relapsed/refractory primary CNS lymphoma. J Clin Oncol. 2016;34(15):1757–63.
Ghobrial IM, Weller E, Vij R, Munshi NC, Banwait R, Bagshaw M, Schlossman R, Leduc R, Chuma S, Kunsman J, Laubach J, Jakubowiak AJ, Maiso P, Roccaro A, Armand P, Dollard A, Warren D, Harris B, Poon T, Sam A, Rodig S, Anderson KC, Richardson PG. Weekly bortezomib in combination with temsirolimus in relapsed or relapsed and refractory multiple myeloma: a multicentre, phase 1/2, open-label, dose-escalation study. Lancet Oncol. 2011;12(3):263–72.
Fenske TS, Shah NM, Kim KM, Saha S, Zhang C, Baim AE, Farnen JP, Onitilo AA, Blank JH, Ahuja H, Wassenaar T, Qamar R, Mansky P, Traynor AM, Mattison RJ, Kahl BS. A phase 2 study of weekly temsirolimus and bortezomib for relapsed or refractory B-cell non-Hodgkin lymphoma: a Wisconsin oncology network study. Cancer. 2015;121(19):3465–71.
Hess G, Keller U, Scholz CW, Witzens-Harig M, Atta J, Buske C, Kirschey S, Ruckes C, Medler C, van Oordt C, Klapper W, Theobald M, Dreyling M. Safety and efficacy of Temsirolimus in combination with Bendamustine and Rituximab in relapsed mantle cell and follicular lymphoma. Leukemia. 2015;29(8):1695–701.
Witzens-Harig M, Keller U, Viardot A, Buske C, Cromb A, Hoenig E, Meissner J, Ho AD, Marks R, Dreyling MH, Safety HG. Clinical activity of Temsirolimus in combination with rituximab and DHAP in patients with relapsed or refractory diffuse large B-cell lymphoma – results of the part I cohort of the STORM trial. Blood. 2015;120:2727.
Smith SM, van Besien K, Karrison T, Dancey J, McLaughlin P, Younes A, Smith S, Stiff P, Lester E, Modi S, Doyle LA, Vokes EE, Pro B. Temsirolimus has activity in non-mantle cell non-Hodgkin’s lymphoma subtypes: the University of Chicago phase II consortium. J Clin Oncol. 2010;28(31):4740–6.
Zent CS, LaPlant BR, Johnston PB, Call TG, Habermann TM, Micallef IN, Witzig TE. The treatment of recurrent/refractory chronic lymphocytic leukemia/small lymphocytic lymphoma (CLL) with everolimus results in clinical responses and mobilization of CLL cells into the circulation. Cancer. 2010;116(9):2201–7.
Witzig TE, Reeder CB, LaPlant BR, Gupta M, Johnston PB, Micallef IN, Porrata LF, Ansell SM, Colgan JP, Jacobsen ED, Ghobrial IM, Habermann TM. A phase II trial of the oral mTOR inhibitor everolimus in relapsed aggressive lymphoma. Leukemia. 2011;25(2):341–7.
Johnston PB, Inwards DJ, Colgan JP, Laplant BR, Kabat BF, Habermann TM, Micallef IN, Porrata LF, Ansell SM, Reeder CB, Roy V, Witzig TE. A phase II trial of the oral mTOR inhibitor everolimus in relapsed Hodgkin lymphoma. Am J Hematol. 2010;85(5):320–4.
Bennani NN, LaPlant BR, Ansell SM, Habermann TM, Inwards DJ, Micallef IN, Johnston PB, Porrata LF, Colgan JP, Markovic SN, Nowakowski GS, Macon WR, Reeder CB, Mikhael JR, Northfelt DW, Ghobrial IM, Witzig TE. Efficacy of the oral mTORC1 inhibitor everolimus in relapsed or refractory indolent lymphoma. Am J Hematol. 2017;92(5):448–53.
Zent CS, Bowen DA, Conte MJ, LaPlant BR, Call TG. Treatment of relapsed/refractory chronic lymphocytic leukemia/small lymphocytic lymphoma with everolimus (RAD001) and alemtuzumab: a phase I/II study. Leuk Lymphoma. 2016;57(7):1585–91.
Conconi A, Raderer M, Franceschetti S, Devizzi L, Ferreri AJ, Magagnoli M, Arcaini L, Zinzani PL, Martinelli G, Vitolo U, Kiesewetter B, Porro E, Stathis A, Gaidano G, Cavalli F, Zucca E. Clinical activity of everolimus in relapsed/refractory marginal zone B-cell lymphomas: results of a phase II study of the International Extranodal Lymphoma Study Group. Br J Haematol. 2014;166(1):69–76.
Witzig TE, Tobinai K, Rigacci L, Lin T, Ikeda T, Vanazzi A, Hino M, Shi Y, Mayer J, Costa LJ, Bermudez CD, Zhu J, Belada D, Bouabdallah K, Kattan JG, Wu C, Fan J, Louveau A-L, Voi M, Cavall F. PILLAR-2: a randomized, double-blind, placebo-controlled, phase III study of adjuvant everolimus (EVE) in patients (pts) with poor-risk diffuse large B-cell lymphoma (DLBCL). J Clin Oncol. 2016;34:7506.
Lemoine M, Derenzini E, Buglio D, Medeiros LJ, Davis RE, Zhang J, Ji Y, Younes A. The pan-deacetylase inhibitor panobinostat induces cell death and synergizes with everolimus in Hodgkin lymphoma cell lines. Blood. 2012;119(17):4017–25.
Oki Y, Buglio D, Fanale M, Fayad L, Copeland A, Romaguera J, Kwak LW, Pro B, de Castro Faria S, Neelapu S, Fowler N, Hagemeister F, Zhang J, Zhou S, Feng L, Younes A. Phase I study of panobinostat plus everolimus in patients with relapsed or refractory lymphoma. Clin Cancer Res. 2013;19(24):6882–90.
Kim SJ, Shin DY, Kim JS, Yoon DH, Lee WS, Lee H, Do YR, Kang HJ, Eom HS, Ko YH, Lee SH, Yoo HY, Hong M, Suh C, Kim WS. A phase II study of everolimus (RAD001), an mTOR inhibitor plus CHOP for newly diagnosed peripheral T-cell lymphomas. Ann Oncol. 2016;27(4):712–8.
Ghobrial IM, Redd R, Armand P, Banwait R, Boswell E, Chuma S, Huynh D, Sacco A, Roccaro AM, Perilla-Glen A, Noonan K, MacNabb M, Leblebjian H, Warren D, Henrick P, Castillo JJ, Richardson PG, Matous J, Weller E, Treon SP. Phase I/II trial of everolimus in combination with bortezomib and rituximab (RVR) in relapsed/refractory Waldenstrom macroglobulinemia. Leukemia. 2015;29(12):2338–46.
Rizzieri DA, Feldman E, Dipersio JF, Gabrail N, Stock W, Strair R, Rivera VM, Albitar M, Bedrosian CL, Giles FJ. A phase 2 clinical trial of deforolimus (AP23573, MK-8669), a novel mammalian target of rapamycin inhibitor, in patients with relapsed or refractory hematologic malignancies. Clin Cancer Res. 2008;14(9):2756–62.
Chresta CM, Davies BR, Hickson I, Harding T, Cosulich S, Critchlow SE, Vincent JP, Ellston R, Jones D, Sini P, James D, Howard Z, Dudley P, Hughes G, Smith L, Maguire S, Hummersone M, Malagu K, Menear K, Jenkins R, Jacobsen M, Smith GC, Guichard S, Pass M. AZD8055 is a potent, selective, and orally bioavailable ATP-competitive mammalian target of rapamycin kinase inhibitor with in vitro and in vivo antitumor activity. Cancer Res. 2010;70(1):288–98.
Naing A, Aghajanian C, Raymond E, Olmos D, Schwartz G, Oelmann E, Grinsted L, Burke W, Taylor R, Kaye S, Kurzrock R, Banerji U. Safety, tolerability, pharmacokinetics and pharmacodynamics of AZD8055 in advanced solid tumours and lymphoma. Br J Cancer. 2012;107(7):1093–9.
Bendell JC, Kelley RK, Shih KC, Grabowsky JA, Bergsland E, Jones S, Martin T, Infante JR, Mischel PS, Matsutani T, Xu S, Wong L, Liu Y, Wu X, Mortensen DS, Chopra R, Hege K, Munster PN. A phase I dose-escalation study to assess safety, tolerability, pharmacokinetics, and preliminary efficacy of the dual mTORC1/mTORC2 kinase inhibitor CC-223 in patients with advanced solid tumors or multiple myeloma. Cancer. 2015;121(19):3481–90.
Ghobrial IM, Siegel DS, Vij R, Berdeja JG, Richardson PG, Neuwirth R, Patel CG, Zohren F, Wolf JL. TAK-228 (formerly MLN0128), an investigational oral dual TORC1/2 inhibitor: a phase I dose escalation study in patients with relapsed or refractory multiple myeloma, non-Hodgkin lymphoma, or Waldenström’s macroglobulinemia. Am J Hematol. 2016;91(4):400–5.
Wunderle L, Badura S, Lang F, Wolf A, Schleyer E, Serve H, Goekbuget N, Pfeifer H, Safety BG. Efficacy of BEZ235, a dual PI3-kinase/mTOR inhibitor, in adult patients with relapsed or refractory acute leukemia: results of a phase I study. Blood. 2013;122:2675.
Thijssen R, Ter Burg J, Garrick B, van Bochove GG, Brown JR, Fernandes SM, Rodríguez MS, Michot JM, Hallek M, Eichhorst B, Reinhardt HC, Bendell J, Derks IA, van Kampen RJ, Hege K, Kersten MJ, Trowe T, Filvaroff EH, Eldering E, Kater AP. Dual TORK/DNA-PK inhibition blocks critical signaling pathways in chronic lymphocytic leukemia. Blood. 2016;128(4):574–83.
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2018 Springer International Publishing AG, part of Springer Nature
About this chapter

Cite this chapter
Calimeri, T., Ferreri, A.J.M. (2018). mTOR Inhibitors, with Special Focus on Temsirolimus and Similar Agents. In: Ferreri, A. (eds) Resistance of Targeted Therapies Excluding Antibodies for Lymphomas. Resistance to Targeted Anti-Cancer Therapeutics, vol 17. Springer, Cham. https://doi.org/10.1007/978-3-319-75184-9_5
Download citation
DOI: https://doi.org/10.1007/978-3-319-75184-9_5
Published:
Publisher Name: Springer, Cham
Print ISBN: 978-3-319-75183-2
Online ISBN: 978-3-319-75184-9
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)