
Abstract
Prediction of the stage of cancer plays an important role in planning the course of treatment and has been largely reliant on imaging tools which do not capture molecular events that cause cancer progression. Gene-expression data–based analyses are able to identify these events, allowing RNA-sequence and microarray cancer data to be used for cancer analyses. Breast cancer is the most common cancer worldwide, and is classified into four stages — stages 1, 2, 3, and 4 [2]. While machine learning models have previously been explored to perform stage classification with limited success, multi-class stage classification has not had significant progress. There is a need for improved multi-class classification models, such as by investigating deep learning models. Gene-expression-based cancer data is characterised by the small size of available datasets, class imbalance, and high dimensionality. Class balancing methods must be applied to the dataset. Since all the genes are not necessary for stage prediction, retaining only the necessary genes can improve classification accuracy. The breast cancer samples are to be classified into 4 classes of stages 1 to 4. Invasive ductal carcinoma breast cancer samples are obtained from The Cancer Genome Atlas (TCGA) and Molecular Taxonomy of Breast Cancer International Consortium (METABRIC) datasets and combined. Two class balancing techniques are explored, synthetic minority oversampling technique (SMOTE) and SMOTE followed by random undersampling. A hybrid feature selection pipeline is proposed, with three pipelines explored involving combinations of filter and embedded feature selection methods: Pipeline 1 — minimum-redundancy maximum-relevancy (mRMR) and correlation feature selection (CFS), Pipeline 2 — mRMR, mutual information (MI) and CFS, and Pipeline 3 — mRMR and support vector machine–recursive feature elimination (SVM-RFE). The classification is done using deep learning models, namely deep neural network, convolutional neural network, recurrent neural network, a modified deep neural network, and an AutoKeras generated model. Classification performance post class-balancing and various feature selection techniques show marked improvement over classification prior to feature selection. The best multiclass classification was found to be by a deep neural network post SMOTE and random undersampling, and feature selection using mRMR and recursive feature elimination, with a Cohen-Kappa score of 0.303 and a classification accuracy of 53.1%. For binary classification into early and late-stage cancer, the best performance is obtained by a modified deep neural network (DNN) post SMOTE and random undersampling, and feature selection using mRMR and recursive feature elimination, with an accuracy of 81.0% and a Cohen-Kappa score (CKS) of 0.280. This pipeline also showed improved multiclass classification performance on neuroblastoma cancer data, with a best area under the receiver operating characteristic (auROC) curve score of 0.872, as compared to 0.71 obtained in previous work, an improvement of 22.81%. The results and analysis reveal that feature selection techniques play a vital role in gene-expression data-based classification, and the proposed hybrid feature selection pipeline improves classification performance. Multi-class classification is possible using deep learning models, though further improvement particularly in late-stage classification is necessary and should be explored further.
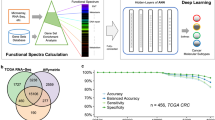
Graphical Abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
1 Introduction
Cancer is a genetic disease and globally one of the leading causes of death. In 2020, 2.3 million women were diagnosed with breast cancer and 685,000 died [35]. Charting out plans for treatment and prognoses have to factor in the stage of cancer. Automating this process using artificial intelligence methods is of great research interest as it would eliminate the need of assessments by professionals and would also ensure greater data collection from a single procedure. Cancer prediction models to date have depended on neural networks to uncover complex connections in the data. Depicting the metamorphosis of genotype into phenotype by inspecting the transcribed mRNA count in a genomic system is called gene expression. The most popular standardised ways to recognise gene expression variation are RNA sequencing and microarray data. RNA-Seq or RNA sequencing is a next-generation sequencing method that measures the presence and change in the RNA quantity in a sample at any given time [28]. Microarray-based gene expression profiles are widespread in cancer research for biomarker identification in the prediction of clinical endpoints like diagnosis, prognosis, and treatment response prediction. They use microarrays to calculate the relative activity of previously marked target genes [10, 17]. RNA-Seq gives greater coverage and resolution of the changing nature of the transcriptome compared to microarray-based techniques [25].
PET and MR imaging techniques, although widely available for the scope of early breast cancer detection, rely on physical features which do not provide insights into cancer progression causing molecular events [3]. On the other hand, gene expression analysis can capture early stage indicators as well as ascertain molecular events that show early to late stage disease advancement. So, gene-expression data can be used to identify and classify the stages of cancer. By nature, gene-expression cancer data brings with it some challenges, including high dimensionality and class imbalance. Hence, appropriate feature selection methods and class balancing techniques must be applied. While machine learning models for predicting the stage and type of cancer exist [36, 37], no attempts have been made to pre-process the gene expression data, apply deep learning models [8, 18], and determine the stage of cancer with high accuracy. Therefore, there is a need to explore multiclass classification of cancer stages, using a hybrid feature selection technique and deep learning models.
2 Related work
A survey of neural-network-based cancer prediction models from microarray data [7] surveyed papers published between 2003 and 2018 on neural networks, gene-expression data, and cancer prediction, and covered cancer classification, discovery, survivability prediction, and statistical analysis models. Pre-processing methods covered included affymetrix normalisation, fragments per kilobase per million (FPKM) normalisation, and zero mean one unit variance normalisation. Synthetic minority oversampling technique (SMOTE) and other oversampling techniques were used for class balancing. Deep MLP models, generative models, extreme learning machines, convoluted neural network (CNN), and genetic algorithms were used for classification of cancer and for cancer survivability prediction.
The initial findings of gene expression profile of peripheral blood mononuclear cells may contribute to the identification and immunological classification of breast cancer patients [30] which imply that evaluating gene expression trends of PMBCs can be a less invasive diagnostic method and helpful in giving insights into breast cancer biomarkers.
In [22], the authors propose a novel machine learning method using transfer learning for reconstructing gene regulatory networks (GRNs) from gene expression data. The method leverages knowledge from a source organism’s GRN to reconstruct the GRN of a target organism, and performs well in positive-unlabelled settings and demonstrates superior performance compared to state-of-the-art approaches, identifying previously unknown functional relationships among genes in the human GRN.
The combined pN stage and breast cancer subtypes in breast cancer: a better discriminator of outcome can be used to refine the 8th AJCC staging manual [38], suggests that the combined pN stage and breast cancer subtypescan predict and discriminate between breast cancer results.
KRAS expression is a prognostic indicator and associated with immune infiltration in breast cancer [20], concluded that KRAS expression can indicate the breast cancer prognoses and is closely linked to tumour immune infiltration.
Microarray cancer feature selection: Review, challenges, and research directions [17] present an extensive survey of studies on microarray cancer classification with a focus on feature selection methods. The use of filter, wrapper, and embedded and hybrid approaches to feature selection were covered. The list of techniques discussed are filter techniques: correlation-based feature selection, the fast correlation-based filter (FCBF) technique, the INTERACT algorithm, information gain, ReliefF, mRMR algorithm, consistency-based filter; wrapper techniques: ant colony optimization, distance sensitive rival penalised competitive learning–support vector machine (ADSRPCL-SVM) genetic algorithm with SVM; embedded techniques: SVM-RFE; hybrid approach: a combination of statistical and machine learning approaches, such as ANOVA and LDA coupled with SVM and filtering using mRMR followed by NB and SVM.
A hybrid gene selection method based on ReliefF and ant colony optimization algorithm for tumour classification [29] described an effective hybrid gene selection method based on ReliefF [33] and ant colony optimisation (ACO) algorithm called RFACO-GS for tumour classification. It was tested on four datasets — colon cancer, leukaemia, lung cancer, and prostate cancer. The classification accuracy of RFACO-GS, 94.3%, was found to be highest out of the algorithms implemented.
The authors in the work [39] propose a model called laminar augmented cascading flexible neural forest (LACFNForest) for the classification of cancer subtypes. The model utilises a cascading flexible neural forest with a hierarchical broadening ensemble method and an output judgment mechanism to improve classification accuracy and reduce computational complexity. Experimental results on RNA-seq gene expression data demonstrate that LACFNForest outperforms conventional methods in cancer subtype classification, offering a promising approach for ensemble learning of classifiers with improved accuracy and robustness.
Identification of gene-expression signatures and protein markers for breast cancer grading and staging [36] described a computational method for prediction of gene signatures for breast cancer stages based on RNA-seq data using the TCGA [31] breast cancer dataset. The Wilcoxon signed-rank test was applied to identify genes that are differentially expressed in cancer versus control samples. Spearman correlation coefficient was used to assess the level of correlation between the average gene expression and the sample stage for identifying genes whose expression change go up or down with respect to stages. The Mann Whitney test is then applied to identify the differentially expressed genes among the different stages. Pathway enrichment analysis was performed. SVM-RFE approach was applied to predict gene signatures for each breast cancer grade as well as stage. A 30-gene panel and a 21-gene panel are predicted as gene signatures for distinguishing advanced stage (stages 3—4) from early stage (stages I–II) cancer samples and for distinguishing stage 2 from stage 1 samples, respectively.
Classification models for invasive ductal carcinoma (IDC) progression, based on gene expression data-trained supervised machine learning [27], covered staging of IDC samples using machine learning algorithms. Samples bearing clinical stages of stages 1 and 2 were pooled together as ‘early stage’, while stages 3 and 4 were pooled together as ‘late stage’. Near zero variance features and features having correlation coefficients more than 80% were removed. The training datasets were standardised using z-score normalisation. Feature selection algorithms such as RFE, RLASSO, random forest, linear modelling, and linear regression were implemented. In order to get consensus ranking, the overall mean of each feature rank obtained from an individual method was calculated. Subsequently, the top 20, 30, 40, 50, 60, and 80 features were used to train and evaluate accuracy of models for binary classification of early vs late IDC, based on 5 machine-learning methods namely RF, Naive Bayes, SVM, logistic regression, and decision tree. The feature list which gave the highest accuracy for all the machine-learning methods was selected for model generation and evaluation. The classification accuracy of the generated prediction models ranges from 74 for SVM to 95% for random forest, and auROC value ranges from 0.76 for LR to 0.93 for the random forest trained model for complete gene expression-based model.
In deep learning for stage prediction in neuroblastoma using gene expression data [23], the dataset to build a model was obtained through the Gene Expression Omnibus (GEO) [4] and TCGA. DNN Classifier on TensorFlow was used to classify the neuroblastoma dataset into 5 stages — 1, 2, 3, 4, and 4S. Stages 1 and 4 could be distinguished in neuroblastoma patients. Considering the poor prediction of the other stages in the test set, it is likely that overfitting occurred for stages 2, 3, and 4S, small size of dataset (280 cases). The accuracy calculated from each training set and test set was found to be 100% and 55.56%, respectively. The stage wise (1, 2, 3, 4, and 4S) one-vs-rest (OVR) AUCs were 0.8, 0.66, 0.59, 0.85, and 0.58, respectively.
A novel machine learning method was proposed by exploiting the knowledge about the gene regulatory networks (GRNs) from gene expression data of a source organism for the reconstruction of the GRN of the target organism, by means of a novel transfer learning technique. The results of proposed methods outperform state-of-the-art approaches and identify previously unknown functional relationships among the analysed genes [22]. A laminar augmented cascading flexible neural forest (LACFNForest) model was proposed to complete the classification of cancer subtypes. This model is a cascading flexible neural forest using deep flexible neural forest (DFNForest) as the base classifier. A hierarchical broadening ensemble method was proposed, which ensures the robustness of classification results and avoids the waste of model structure and function as much as possible. The LACFNForest model effectively improves the accuracy of cancer subtype classification with good robustness. It provides a new approach for the ensemble learning of classifiers in terms of structural design [39].
The inference of gene regulatory networks (GRNs) is of great importance for understanding the complex regulatory mechanisms within cells. The emergence of single-cell RNA-sequencing (scRNA-seq) technologies enables the measure of gene expression levels for individual cells, which promotes the reconstruction of GRNs at single-cell resolution. The authors proposed a multi-view contrastive learning (DeepMCL) model to infer GRNs from scRNA-seq data collected from multiple data sources or time points. An attention mechanism is introduced to integrate the embeddings extracted from different data sources and different neighbour gene pairs [21].
In gene expression classification based on deep learning [2], gene expression data of 4 types of cancer were used: diffuse large B cell lymphoma, prostate cancer, leukaemia, and colon cancer. Min–max normalisation technique was applied. Four deep learning models were applied on the cancer classification task, and the results were compared. The models used were deep neural network, recurrent neural network (RNN), convolutional neural network, and modified DNN: DNN in combination with dropout. The performance of the models was evaluated using the accuracy measure. It was found that the modified DNN model performed best across the datasets.
In integration of RNA-Seq data with heterogeneous microarray data for breast cancer profiling [5], heterogeneous datasets of microarray data and RNA-Seq data were integrated to identify gene expressions and classify genes as possible biomarkers for breast cancer. Overall, classification models tend to perform poorly with respect to minority classes and usually overfit during training leading to incorrectly high accuracy. Gene expression data is characterised by high dimensionality and selecting the most important features from this data reduces computational cost. Hence, the construction of a hybrid model to use deep learning on gene-expression data, in order to gain insight into and improve the results of cancer staging prediction, has been proposed in the coming sections.
3 Materials and methods
3.1 Proposed methodology
The proposed methodology for multiclass classification of cancer stages has been detailed in Fig. 1.

Proposed methodology for cancer staging multiclass classification
3.2 Data extraction and pre-processing
Gene expression data is extremely high dimensional by nature. Often, the number of samples is in the order of tens and hundreds, while the number of features is close to 20,000. This poses serious computational challenges. Efficient methods that can capture the required information from a select group of features while not compromising on classification performance, computational, and time requirements are crucial. Gene expression data is mostly of 2 types, RNA-Seq and microarray. Having explored that study reproducibility and data-model sensitivity is an issue in medical datasets, the two chosen datasets from TCGA (RNA-Seq) and METABRIC (microarray) were combined. This is extremely important since most of the earlier studies would have used microarray, but more recent studies would be using RNA-Seq as it continues to rise in popularity. This would mean that the model, if trained properly, could accept any sample as input and classify it into the correct stage [32]. The steps involved in combining the datasets are detailed in Fig. 2.

Data extraction and pre-processing
3.3 Hybrid feature selection
High-dimensional data and class imbalance were two other issues that were identified in current work. As such, a good feature selection method would be crucial to the success of the task of staging cancer. Since each method has its own strengths and weaknesses, a combination of different types of feature selection methods might prove fruitful by utilising the advantages of each. It also adds a level of confidence since the selected features would be due to the consensus of the selected methods. Therefore, experiments were conducted to identify the optimal feature selection pipeline for a deep learning model. Based on available literature, possible choices and combinations of filter and embedded feature selection methods were selected. Three pipelines were built for multiclass classification, and two from those three were implemented [14, 16, 24] for binary classification. The filter methods used are mRMR [9], CFS and MI. SVM-RFE, an embedded technique, is also used. The three pipelines are described in Fig. 3.

Feature selection pipelines
The three methods were chosen for their performance on gene expression datasets in other works. mRMR has been shown to be successful in selecting features and hence was chosen for all three pipelines. In order to identify which combination performs best, the other methods chosen varied.
3.4 Deep learning models for classifying cancer stages
Finally, the choice of deep learning model was also made experimentally. Most previous works used machine learning methods, and only a handful used deep learning algorithms.
Therefore, combinations of feature selection and deep learning methods were executed to find the optimal combination. The deep architectures selected were deep neural network, convolutional neural network, deep neural network with dropout, recurrent neural network, and AutoKeras generated model. DNN and dropout were chosen specifically since there was a possibility of the model overfitting the dataset due to class imbalance. AutoKeras is a tool that identifies the optimal model architecture for a given dataset. Since it aligned with the objective to find the best deep learning model [11], AutoKeras was used to identify other possible architectures that may perform well. The deep learning classification models used were constructed using the Tensorflow framework [1].
3.4.1 Deep neural network
The deep neural network model used consisted of three dense layers, with the activation function ReLU (Fig. 4).

Visualisation of DNN model used for multi-class classification post SMOTE prior to feature selection
3.4.2 Convolutional neural network
The convolutional neural network model used consisted of a 1D convolution layer, Max pooling operation followed by flattening the input (Fig. 5).

Visualisation of CNN model used for multi-class classification post SMOTE prior to feature selection
3.4.3 Modified DNN − DNN + dropout model
This model is a modified version of the DNN discussed previously, with each dense layer followed by a dropout layer. Below is the plot of the modified DNN model (Fig. 6).

Visualisation of modified DNN model used for multi-class classification post SMOTE prior to feature selection
3.4.4 Recurrent neural network
This model made use of the simple RNN layer, a fully connected RNN where the output is to be fed back to input (Fig. 7).

Visualisation of modified DNN model used for multi-class classification post SMOTE prior to feature selection
3.4.5 AutoKeras
AutoKeras [19] is a publicly available library designed to facilitate automated machine learning (AutoML) processes specifically tailored for deep learning models. It leverages Keras models, implemented through the TensorFlow tf.keras API, to conduct the search.
3.5 Dataset
Gene expression data for invasive ductal carcinoma was extracted from 2 publicly available sources, TCGA (RNA-Seq) and METABRIC (microarray) (Tables 1 and 2).
4 Results
4.1 Performance analysis metrics
The following metrics were used to evaluate the performance of the models on the given multi-class classification and binary classification problems.
For the multi-class classification problem, accuracy measure and Cohen Kappa score [34] were used. For binary classification, accuracy, sensitivity, specificity, Matthew correlation-coefficient, and area under the ROC curve were used.
4.2 Feature selection
Post feature selection using Pipelines 1, 2, and 3, the relevant features were extracted and are given in Figs. 8, 9, and 10, respectively.

List of 88 features extracted by Pipeline 1 (mRMR and CFS)

List of 18 features extracted by Pipeline 2 (mRMR, mutual information, and CFS)

List of 100 features extracted by Pipeline 3 (mRMR and SVM-RFE)
4.3 Class balancing
To counter the issue of class imbalance as described in the previous section, two class balancing techniques were explored — SMOTE [15] and SMOTE followed by random undersampling, which were applied on the training set. The dataset was split into training and test sets in an 80:20 split (Table 3).
4.4 Deep learning classification models
The deep learning models being considered, as used by the authors of [2], are deep neural network (DNN), convolutional neural network (CNN), modified DNN: DNN + dropout, RNN, and Auto-Keras.
4.5 Results and inferences
4.5.1 Performance prior to feature selection
Prior to the application of feature selection, the results of the models applied on the dataset with 16,055 features, post SMOTE and post SMOTE, and random undersampling are described as in Tables 4 and 5.
It can be seen that there is not a significant difference in performance between the two class balancing methods chosen.
The accuracy across the models is in the range of 40–55%, with the AutoKeras generated model exhibiting highest accuracy post SMOTE and post SMOTE and random undersampling. The highest CKS is shown by the CNN model in both class balancing techniques, with the accuracy being comparable to the highest as well.
4.5.2 Feature selection using Pipeline 1 (mRMR and CFS)
The top 500 features were selected based on the mRMR technique, which was further reduced to 88 features through CFS. This is an 82.4% reduction in the number of features. In Tables 6 and 7, the results of the classification of each model using the two class balancing techniques post the application of feature selection using Pipeline 1 have been tabulated.
Here again, there is no significant difference in performance between the two class balancing methods chosen. There is a marked improvement in CKS scores across the models post feature selection. The accuracy remains in the same range. Therefore, it can be understood that feature selection does play a key role in the decision boundaries between the classes in this multi-class classification problem. The highest accuracy is shown by the DNN model in both class balancing techniques, and the highest CKS is shown by the AutoKeras model in the case of SMOTE, and the CNN model in the case of SMOTE and random undersampling.
4.5.3 Feature selection using Pipeline 2 (mRMR, MI, and CFS)
The top 500 features were selected using the mRMR and mutual information methods each. The intersection of these top features was found, which was a subset of 41 features. CFS was applied on this subset, which yielded 18 features. This is a reduction of 96.4% in the number of features. Tables 8 and 9 show the tabulation of the results of the classification of each model using the two class balancing techniques post the application of feature selection using Pipeline 2.
Yet again, there is no significant difference between the performance of the two class balancing methods; though accuracy wise, only SMOTE performs marginally better.
The overall performance based on CKS is worse than that of Pipeline 1. This can be attributed to the possibility that too few features were retained, which influenced the classification decision boundaries leading to poor classification performance.
Pipelines 1 and 2 relied on a combination of filter methods to construct a hybrid feature selection model. In the next pipeline, a wrapper method, SVM-RFE was implemented and its performance evaluated.
4.5.4 Feature selection using Pipeline 3 (mRMR and SVM-RFE)
In this pipeline, the top 500 features were identified using mRMR. Recursive feature elimination (RFE) was applied on this subset, retaining the top 100 features. Tables 10 and 11 show the performance of the models in classification post the application of class balancing and feature selection using Pipeline 3.
There is an increase in the accuracy across the models as compared to Pipeline 2, with DNN with SMOTE and random undersampling performing marginally better than the other models.
Importantly, while the other pipelines failed to correctly classify the stage 4 samples, Pipeline 3 was able to classify a Stage 4 sample correctly, in the DNN, CNN, and DNN + Dropout models as seen in the confusion matrices in Table 10.
4.5.5 Inference from multiclass classification results
The results of the multiclass classification from Tables 4, 5, 6, 7, 8, 9, 10 and 11 were based on the test set which is a 20% split of the combined TCGA and METABRIC data. The best overall results are seen from the DNN Model in Pipeline 3, with a CKS of 0.303 and an accuracy of 53.1%. Pipeline 3 DNN, CNN, and DNN + Dropout models were able to classify a Stage 4 sample correctly. Additionally, SMOTE along with undersampling did not improve the models as expected, with most results being within the same range as models that used only SMOTE.
4.5.6 Binary classification
The above results evaluated the classification of the samples into 4 classes. Stages 1 and 2 can be combined into a single early stage, and stages 3 and 4 into late stage, and this can be approached as a binary classification problem. The results of the same, post feature selection using Pipelines 1 and 3, are as follows in Tables 12, 13, 14 and 15.
The accuracy scores for binary classification are found to be significantly higher for all the models than the corresponding scores for multiclass classification. Across the models, it can be seen that the classification of early stage (stages 1 and 2) is quite good, but late-stage classification performance is poor. As in the multiclass results, mRMR followed by SVM-RFE seems to have performed best. The DNN, CNN, and DNN + dropout models in this Pipeline 3 with SMOTE and random undersampling all showed very similar performance. While DNN showed the best test accuracy, the DNN + dropout model obtained the best CKS of 0.280 and accuracy of 81%.
While all 3 models are able to classify early stage samples correctly, DNN + dropout was able to classify the most late stage samples correctly, which has been a major pain point across the analysis.
We can conclude that DNN + dropout in Pipeline 3 had the best overall performance, as it is able to strike the best balance of correct predictions for both late stage and early stage samples.
4.5.7 Inferences
Analysing the results from Tables 4, 5, 6, 7, 8, 9, 10, 1112, 13, 14 and 15, it is evident that feature selection has improved the performance of the classification system. Additionally, Pipeline 3 (mRMR followed by SVM-RFE) performed best of the 3 feature selection pipelines for both multiclass and binary classification. Overall, it can be seen that the models were able to classify stages 1 and 2 better than the later stages.
All the models were able to distinguish between stages 1 and 4 well. However, most were unable to correctly classify stage 4 samples. This could be attributed to the unclear decision boundaries between classes. This inference is supported by Fig. 11. Due to the high dimensionality inherent in our dataset, the visualisation of decision boundaries between classes is difficult. Hence, the authors have chosen to employ principal component analysis (PCA) as a means to facilitate visualisation. By examining the spatial arrangement of the projected data points, valuable insights into the separability of distinct classes can be obtained.

Plot of PCA on the original dataset
Figure 11 is a graphical representation of the PCA conducted on the original dataset. As evident from the plot, the classes demonstrate overlapping regions in the reduced-dimensional space. This observation suggests that accurately defining the decision boundaries between these classes poses a greater challenge. Owing to the unclear decision boundaries, it was possible for all models to differentiate between stages 1 and 4, but the majority struggled to correctly classify stage 4 samples.
The high training accuracy of the models alludes to possible over-fitting.
In the binary classification system, the accuracy is much higher than in the multiclass problem, with it being able to classify early stage samples well. However, late stage classification can be improved.
4.6 Comparison with existing research work
4.6.1 Binary classification for invasive ductal carcinoma cancer from GEO database
In the work done by the authors of [7], an external test set consisting of a microarray dataset, obtained from GEO with accession ID GSE61304 containing 56 samples of IDC, was used. The results of the models described earlier on this test set are mentioned in Tables 16 and 17.
The metrics used are accuracy, sensitivity, specificity, Mathew’s correlation coefficient (MCC), and area under the ROC curve [12, 13].
The model with the best performance in [27] was a Naive Bayes model that attained highest MCC of 0.27. While the deep learning models evaluated here do not perform as well as Naive Bayes, they perform just as well if not better than the other machine learning methods in [27]. Refinements in the feature selection process and the construction of the deep learning models may make them surpass the performance of their machine learning counterparts.
4.6.2 Multiclass classification on neuroblastoma cancer data from GEO database
Comparison of AUC scores on IDC dataset with relevant research in literature
The area under the ROC curve metric was calculated for multi-class classification, as used by the authors in [23]. Specifically, the macro-average AUC and One-versus-Rest (OVR) AUC values were computed, and have been compiled as in Table 21. The CNN used with the third feature selection pipeline had the highest macro-average AUC of 0.678 (Table 18).
The ROC curves for each classification model for feature selection using Pipeline 1 are depicted in Fig. 12.

ROC curves for each model post feature selection using Pipeline 1
The ROC curves for each classification model for feature selection using Pipeline 2 are depicted in Fig. 13.

ROC curves for each model post feature selection using Pipeline 2
The ROC curves for each classification model for feature selection using Pipeline 3 are depicted in Fig. 14.

ROC curves for each model post feature selection using Pipeline 3
The ROC curves obtained by the authors of [23] on neuroblastoma cancer [26] using a DNN is shown in Fig. 15.

Test set ROC curve from [8]
The model proposed by the authors of [23] had a marginally higher macro-average AUC of 0.71. The higher value is most likely due to the characteristics of the cancer (neuroblastoma) as well as not suffering from the problem of severe class imbalance. This is evident in the one-vs-rest AUC values as well with stage 4 having the highest value of 0.85, and is also represented most in the dataset with 124 samples out of 280 total.
However, the values obtained by [23] are comparable to the ones seen in Fig. 9 with the RNN stage 4 one-vs-rest AUC being 0.79, and the other values being in the similar ranges.
Comparing classification performance of hybrid feature selection pipeline on neuroblastoma dataset
In addition to comparing the AUC values obtained by the models on the IDC dataset, a feature selection pipeline was also implemented on the neuroblastoma dataset used by the authors in [23] (original data provided by [26], GEO accession GSE85047) (Table 19). Pipeline 3 was selected for this purpose since it had been performing the best as seen in earlier sections (Table 20).
The results obtained from implementing Pipeline 3 on the neuroblastoma data show good classification performance (Table 21). The CNN model obtained the highest overall macro-average AUC of 0.872 when class balancing using SMOTE was performed. In the same pipeline, DNN model showed slightly higher accuracy (0.643) and CKS (0.600), and a macro-average AUC score of 0.8. While using SMOTE + random undersampling, CNN performed the best with a macro-average AUC of 0.840. The ROC curves for each classification model for feature selection using Pipeline 3 post SMOTE and SMOTE + random undersampling are depicted in Figs. 16 and 17, respectively.

ROC curves for each model post feature selection using Pipeline 3 post SMOTE on the neuroblastoma dataset

ROC curves for each model post feature selection using Pipeline 3 post SMOTE and random undersampling on the neuroblastoma dataset
The macro-average values of 0.872 and 0.840 are significantly higher than the values obtained by the authors of [23], who obtained a macro-average score of 0.71, as mentioned in Fig. 15. This is an improvement of 22.81%. Looking more closely at the class-wise one-vs-rest AUC values, pipeline 3 again outperforms with the highest value of 0.95 for stage 2 in CNN with SMOTE. The same CNN classification model also achieved higher stage 4 one-vs-rest AUC of 0.91 compared to the authors’ 0.85, shown in Fig. 15.
It can be concluded that the class-balancing and feature selection pipelines described in this work significantly improve the multi-class classification performance of cancer staging.
5 Discussion
In order to perform the multi-class classification of invasive ductal carcinoma cancer into stages 1, 2, 3, and 4, class balancing methods, feature selection techniques, and deep learning methods were explored. In the past literature survey, multiple problems were identified. Firstly, the issue of small size of datasets was encountered, to counter which data from two different datasets were combined, and normalised and pre-processed the data accordingly. Additional samples were not the only benefit of combining the TCGA and METABRIC [6] datasets. Since they are 2 different types of gene expression data (RNA-Seq and microarray respectively), our model can be used on test samples for either type. This is extremely important since most of the earlier studies would have used microarray, but more recent studies would be using RNA-Seq as it continues to rise in popularity. To counter the class imbalance of the dataset, SMOTE and SMOTE followed by random undersampling were both implemented. It was seen that there was no significant difference in the performance of the two class balancing methods. Due to the high-dimensional nature of the dataset, feature selection was a key step. Three different pipelines of hybrid feature selection techniques were used — mRMR followed by CFS, mRMR, mutual information followed by CFS, and mRMR followed by SVM-RFE. All 3 pipelines had a positive effect, improving performance compared to models run on the full feature set. Additionally, class balancing using SMOTE and random undersampling, and feature selection using mRMR followed by SVM-RFE (Pipeline 3) performed the best for both multiclass classifications using a deep neural network classification model (0.303 CKS, 53.1%ACC) and for binary classification using a modified deep neural network classification model (0.280 CKS, 81.0% ACC).
On comparing with the existing work in [23], Pipeline 3 resulted in high classification performance on neuroblastoma data. The CNN model obtained the highest overall macro-average AUC of 0.872 when class balancing using SMOTE was performed. It achieved higher stage 4 OVR AUC of 0.91 compared to the previous work which resulted in 0.85. There was an improvement of 22.81% with respect to macro-average AUC while comparing with results of previous work in [23]. The DNN model showed slightly higher accuracy (0.643) and CKS (0.600), and a macro-average AUC score of 0.8.
6 Conclusion
The multi-class classification of stages of gene-expression cancer data using deep learning was attempted. Three different pipelines of hybrid feature selection techniques along with SMOTE were used which improved performance compared to models run on the full feature set. mRMR followed by SVM-RFE (Pipeline 3), and a deep neural network classification model performed the best for both multiclass classification (0.303 CKS, 53.1%ACC) and for binary classification (0.280 CKS, 81.0% ACC). The results and analysis reveal that feature selection techniques play a vital role in gene-expression data-based classification, and the proposed hybrid feature selection pipeline improves classification performance.
The limitation of this work was the lack of high accuracy obtained, particularly in the classification of stage 4. Most models based on medical data are highly sensitive to the dataset, and this was no different. Due to the nature of the dataset, the decision boundaries between the classes were not very clear, resulting in poor multi-class classification. As mentioned above, due to the nature of the dataset, study reproducibility will be an issue. However, the results show that there is scope for the same. Accuracy cannot be completely relied on, which is why Cohen-Kappa score was used as a metric. Other metrics can be looked at to better represent the results and performance of the models. A closer look at the deep learning models and hyperparameter tuning would be beneficial. Comparison of the types of SMOTE such as G-SMOTE and M-SMOTE can be performed. The feature selection techniques did not take into account the biological significance of the genes. This could be incorporated into the feature selection stage.
References
Abadi M, Agarwal A, Barham P, Brevdo E, Chen Z, Citro C, Zheng X (2015) TensorFlow: Large-scale machine learning on heterogeneous systems. Retrieved July 31, 2023, from https://www.tensorflow.org/
Ahmed O, Brifcani A (2019, April) Gene expression classification based on deep learning. In 2019 4th Scientific International Conference Najaf (SICN). IEEE, pp 145–149
American Cancer Society (2021, June 28) Stages of breast cancer: Understand breast cancer staging. Retrieved October 25, 2021, from https://www.cancer.org/cancer/breast-cancer/understanding-a-breast-cancer-diagnosis/stages-of-breast-cancer.html
Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, Yefanov A, Lee H, Zhang N, Robertson CL, Serova N, Davis S, Soboleva A (2013) NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res 41:D991–D995
Castillo D, Gálvez JM, Herrera LJ, Román BS, Rojas F, Rojas I (2017) Integration of RNA-Seq data with heterogeneous microarray data for breast cancer profiling. BMC Bioinformatics 18(1):506
cBioPortal for Cancer Genomics (2016) Breast cancer (METABRIC, Nature 2012 & Nat Commune 2016). Retrieved May 25, 2022, from http://www.cbioportal.org/study/summary?id=brca/_metabric
Daoud M, Mayo M (2019) A survey of neural network-based cancer prediction models from microarray data. Artif Intell Med 97:204–214
Dertat A (2017, October 9) Applied deep learning — part 1: Artificial neural networks. Medium. Retrieved October 25, 2021, from https://towardsdatascience.com/applied-deep-learning-part-1-artificial-neural-networks-d7834f67a4f6
Ding C, Peng H (2005) Minimum redundancy feature selection from microarray gene expression data. J Bioinform Comput Biol 3(02):185–205
Fathi H, AlSalman H, Gumaei A, Manhrawy II, Hussien AG, El-Kafrawy P (2021) An efficient cancer classification model using microarray and high-dimensional data. Comput Intell Neurosci 2021
Goodfellow I, Bengio Y, Courville A (2016) Deep learning. Retrieved October 25, 2021, from https://www.deeplearningbook.org
Google Developers (2020, Feb 11) Classification: Precision and recall | Machine learning crash course. https://developers.google.com/machine-learning/crash-course/classification/precision-and-recall
Google Developers (n.d.) Classification: ROC curve and AUC. Retrieved May 25, 2022, from https://developers.google.com/machine-learning/crash-course/classification/roc-and-auc
Google (n.d.) Google Colab. Google Colaboratory. Retrieved May 25, 2022, from https://research.google.com/colaboratory/faq.html
Gosain A, Sardana S (2017) Handling class imbalance problem using oversampling techniques: A review. 2017 International Conference on Advances in Computing, Communications and Informatics (ICACCI)
Griffith M, Walker J, Spies N, Ainscough B, Griffith O (2015) Informatics for RNA sequencing: a Web resource for analysis on the cloud. Plos Comput Biol 11(8):e1004393
Hambali MA, Oladele TO, Adewole KS (2020) Microarray cancer feature selection: Review, challenges and research directions. Int J Cogn Comput Eng 1:78–97
IBM Cloud Education (2020) What is deep learning? IBM. Retrieved October 25, 2021, from https://www.ibm.com/cloud/learn/deep-learning
Jin H, Chollet F, Song Q, Hu X (2023) AutoKeras: an AutoML library for deep learning. J Mach Learn Res 6:1–6
Liang H, Zhou G, Lv L et al (2021) KRAS expression is a prognostic indicator and associated with immune infiltration in breast cancer. Breast Cancer 28:379–386. https://doi.org/10.1007/s12282-020-01170-4
Lin Z, Ou-Yang L (2023) Inferring gene regulatory networks from single-cell gene expression data via deep multi-view contrastive learning. Brief Bioinforma 24(1):bbac586. https://doi.org/10.1093/bib/bbac586
Mignone P, Pio G, D’Elia D, Ceci M (2020) Exploiting transfer learning for the reconstruction of the human gene regulatory network. Bioinformatics 36(5):1553–1561. https://doi.org/10.1093/bioinformatics/btz781
Park A, Nam S (2019) Deep learning for stage prediction in neuroblastoma using gene expression data. Genom Inform 17(3)
Pedregosa F, Varoquaux G, Gramfort A, Michel V, Thirion B, Grisel O, Duchesnay É (2011) Scikit-learn: Machine learning in python. J Mach Learn Res 12:2825–2830
Pereira B, Chin SF, Rueda O et al (2016) The somatic mutation profiles of 2,433 breast cancers refine their genomic and transcriptomic landscapes. Nat Commun 7:11479. https://doi.org/10.1038/ncomms11479
Rajbhandari P, Lopez G, Capdevila C, Salvatori B et al (2018May) Cross-cohort analysis identifies a TEAD4-MYCN positive feedback loop as the core regulatory element of high-risk neuroblastoma. Cancer Discov 8(5):582–599
Roy S, Kumar R, Mittal V, Gupta D (2020) Classification models for invasive ductal carcinoma progression, based on gene expression data-trained supervised machine learning. Sci Rep 10(1):1–15
Scitable by Nature Education (2014) Gene Expression Is Analyzed by Tracking RNA. Retrieved May 25, 2022, from https://www.nature.com/scitable/topicpage/gene-expression-is-analyzed-by-tracking-rna-6525038/
Sun L, Kong X, Xu J, Zhai R, Zhang S (2019) A hybrid gene selection method based on ReliefF and ant colony optimization algorithm for tumor classification. Sci Rep 9(1):1–14
Suzuki E, Sugimoto M, Kawaguchi K et al (2019) Gene expression profile of peripheral blood mononuclear cells may contribute to the identification and immunological classification of breast cancer patients. Breast Cancer 26:282–289. https://doi.org/10.1007/s12282-018-0920-2
The Cancer Genome Atlas Program (n.d.) National Cancer Institute. Retrieved May 25, 2022, from https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga
UICC (2022) UICC and the TNM classification of malignant tumours. UICC. Retrieved May 25, 2022, from https://www.uicc.org/who-we-are/about-uicc/uicc-and-tnm-classification-malignant-tumours
Urbanowicz RJ, Meeker M, La Cava W, Olson RS, Moore JH (2018) Relief-based feature selection: introduction and review. J Biomed Inform 85:189–203
Viera AJ, Garrett JM (2005) Understanding interobserver agreement: the kappa statistic. Fam Med 37(5):360–363
World Health Organization (2021) Breast cancer. World Health Organization. Retrieved October 25, 2021, from https://www.who.int/news-room/fact-sheets/detail/breast-cancer
Yao F, Zhang C, Du W, Liu C, Xu Y (2015) Identification of gene-expression signatures and protein markers for breast cancer grading and staging. Plos One 10(9):e0138213
Yuan F, Lu L, Zou Q (2020) Analysis of gene expression profiles of lung cancer subtypes with machine learning algorithms. Biochimica et Biophysica Acta (BBA)-Mol Basis Dis 1866(8):165822
Yang ZJ, Yu Y, Chi JR et al (2018) The combined pN stage and breast cancer subtypes in breast cancer: a better discriminator of outcome can be used to refine the 8th AJCC staging manual. Breast Cancer 25:315–324. https://doi.org/10.1007/s12282-018-0833-0
Zhong L, Meng Q, Chen Y, Du L, Wu P (2021) A laminar augmented cascading flexible neural forest model for classification of cancer subtypes based on gene expression data. BMC Bioinformatics 22(1):1–17. https://doi.org/10.1186/s12859-021-04391-2
Author information
Authors and Affiliations
Contributions
Akash Kishore: literature survey, collected data set, data preparation and implementation. Lokeswari Y Venkataramana: evaluating the implementation, updating the manuscript, and reviewing the work. D Venkata Vara Prasad: reviewed the paper, suggestions on using different variations of dataset. Akshaya Mohan: literature survey, implementation, writing the manuscript. Bhavya Jha: literature survey, implementation, prepared figures and tables.
Corresponding author
Ethics declarations
Ethics approval
This article does not contain any studies with human participants or animals performed by any of the authors. All the authors have agreed to publish this manuscript. The manuscript is not submitted to any other journal or not under consideration of any journal.
Informed consent
Informed consent is not necessary as this article does not involve human or animal participants.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kishore, A., Venkataramana, L., Prasad, D.V. et al. Enhancing the prediction of IDC breast cancer staging from gene expression profiles using hybrid feature selection methods and deep learning architecture. Med Biol Eng Comput 61, 2895–2919 (2023). https://doi.org/10.1007/s11517-023-02892-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11517-023-02892-1