
Abstract
Perilla oil is one of the best sources of plant-based omega-3 fatty acids while its low oxidative stability triggers deteriorative changes of flavour and thereby it’s reduced domestic usage. The present study was aimed at the formation of one-layered and double-layered nanoliposomes for encapsulating perilla seed oil using chitosan, poly-L-lysine, sodium alginate and genipin. Moreover, physical and oxidative stability of developed nanoliposomes and their in vitro release behaviour were investigated. Formation of coated nanoliposomes was confirmed by FT-IR and TEM analysis, where they showed a satisfactory range of size (200–502 nm) and encapsulation efficiency (82–91%). Indeed, chitosan as primary coating and genipin as a GRAS cross-linker could improve physical and oxidative stability of the developed nanoliposomes and all coated nanoliposomes could benefit stability under gastric and intestinal conditions.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
There is an increasing demand for nutritive and healthy foods in the market and this fact has led the food industry to focus their research on products of this nature. Perilla (Perilla frutescens, Lamiaceae) is an edible plant native to East Asian countries while its leaves are sometimes used as a medicinal herb, and also as a substitute for basil leaves in Korean-style western foods. Perilla seeds contain approximately 30–45% oil and contribute a good supply of ALA (50–64% of its total fatty acids) [1, 2].The high content of n-3 fatty acid present in this oil allows the attribution of functional food, which means that besides the nutritional functions, its consumption may have beneficial effects on health. However, consumption of the excess amount of ALA-rich oils is limited by their poor storage stability as well as susceptibility to autoxidation and thermal oxidation [3,4,5].
A variety of approaches have been developed to overcome the limitations of the use of oils rich in n-3 fatty acids and to allow their therapeutic applications, such as complexation with phospholipids as well as encapsulation in liposomes, biodegradable microsphere, hydrogels, polymeric nanoparticles and lipid- based nanoparticles [6,7,8].
Liposome, as a bilayer lipid vesicle surrounding an aqueous core, is one of the most broadly applied delivery system used to encapsulate, preserve and controlled release of bioactive compounds in food, agricultural and pharmaceutical industries [9]. The heating method as a green technique for the fabrication of liposome that avoids hazardous chemicals and solvents have been developed for the preparation of food-grade liposomes. In particular, the Mozafari method has been used to prepare liposomes in a single step within less than 1 h, using a single apparatus, and in the absence of potentially toxic solvents [10].
However, liposomes are susceptible to oxidation over storage, as well as hydrolysis under low pH and enzymatic conditions as being the major obstacles in their uses [9]. In order to improve the oxidative stability of liposomes during long-term storage and stability in the gastrointestinal tract, diverse coatings have been previously developed using an electrostatic layer-by-layer (LBL) technique [9, 10]. In this regard, coating of carboxy-fluorescein (CF)-loaded liposomes using poly-L-lysine (PLL) to increase the CF retention inside the vesicles [11], medium-chain fatty acid nano-liposomes (NLs) using chitosan (CS) and sodium alginate (SA), in order to improve physical and in vitro digestion stability [12], liposomes loaded with flavonoid quercetin using whey protein isolate (WPI) to improve physical, storage and digestive stability [13], liposomes containing vitamin C using CS and SA in order to increase environmental stress stability [14], and coating the liposome with cold water fish skin gelatin, in order to increase the physical stability [15] have been successfully examined and reported.
Cross-linking interactions are continuously formed and destroyed providing a gel-like structure and hence dynamic stability of the polymer. As long as the correct coating polymers are combined, such interactions can contribute to the desired characteristics of stability and optimal drug delivery.
The cross-linking technique, also, has been successfully used for the sustained release of the drug from the nanoliposome [16]. Glutaraldehyde as a cross-linker leads to improvement of the mechanical properties, stability and controlled release of nanoparticles, but its high toxicity may limit the applications of the final product. Therefore, the use of non-toxic crosslinking agents has emerged as an alternative [17].
Genipin (C11H14O5) is a hydrolytic product of geniposide obtained from the Gardenia jasminoides Ellis fruit, a kind of traditional Chinese medicine, through enzymatic hydrolysis by ß-glucosidase. As a naturally occurring biocompatible cross-linking agent, GP has been documented to be around 10,000 times less cytotoxic compared with glutaraldehyde. GP permits the establishment of both intramolecular and intermolecular covalent bonds with primary amines (e.g., the side chain of lysine residues), and improves the mechanical strength, swelling, and drug-release properties of protein to a greater extent [18].
In spite of applying the above-mentioned coating polymers in liposomal systems for delivery purposes of drugs and nutraceuticals, to the best of our knowledge, fabrication of perilla oil-loaded NL by the green heating method, then coating the NLs by CS, PLL, SA have never been studied before. In addition, no attention has been paid to apply a non-toxic cross-linker and its impact on the physicochemical stability, which is related to its shelf-life, and its release behaviour.
The objective of this work was to explore the possibility of formation of colloidal protective NLs by the heating method as a green technique for encapsulating perilla seed oil, as well as one-layer and double-layer coating of NLs using CS, PLL and SA in companion with GP as cross- linker. Efficacy of the coating and cross-linker protocols was investigated by monitoring the physical stability of perilla oil-loaded NLs beside oxidative stability, by means of peroxide value, propanal, and hexanal formation. Furthermore, in vitro release behaviour of n-3 and n-6 PUFAs from developed NLs was studied.
Materials and Methods
Materials
Perilla seeds (Perilla frutescens, Lamiaceae family), verified by Seed and Plant Improvement Institute, were collected from a forest in Northern Province of Gilan in Iran (harvested on September 2016). L-alpha-lecithin from soybean oil (average molecular weight: 750 g/mol) was purchased from Acros Organics (Geel, Belgium). Chitosan (#448869, viscosity <50 mPa.s, 50 kDa), Poly-L-lysine hydrobromide (#p0879, molecular weight:1000–5000 g/mol), sodium alginate (#A1112, low viscosity, 12 kDa), genipin (#G4796), methyl linoleate (#L1876, ≥99% (GC)), methyl linolenate (#62200, ≥98.5% (GC)), pepsin obtained from pig gastric mucosa (#10108057, enzyme activity 2500 units/mg protein), pancreatin from porcine pancreas (#P1750, 4 × U.S. Pharmacopeial (USP) specifications), bile extract porcine (B8631) and dialysis tubing (#D0655) were supplied by Sigma-Aldrich (Sigma Chemical Co., St. Louis, Germany). propanal (#822133), hexanal (#802672) and all other chemicals (NaCl, KCl, Na2HPO4.2H2O, K2HPO4, ferrous-II-heptahydrate, sodium hydroxide, ammonium thiocyanate and barium chloride) and solvents were of laboratory grade and purchased from Merck Chemical Co. (Darmstadt, Germany) and Sigma Chemical Co. (St. Louis, Germany).
Methods
Extraction of Oil
Just before oil extraction, perilla seeds were ground at ambient temperature for 60 s using a Moulinex grinder (Type DPA1, France). Subsequently, powders were subjected to n-hexane extraction (1:4 w/v% of seeds powder to n-hexane) while agitating in a dark place at ambient temperature for 48 h. Then, the solvent was evaporated at 35 °C, using a rotary evaporator (Büchi, Flawil, Switzerland). Finally, the solvent residue was removed under a stream of nitrogen gas before being stored at −20 °C.
Liposome Preparation
Liposome samples were prepared according to the procedure outlined by Rasti, Jinap, Mozafari and Yazid (2012), with slight modifications. Briefly, a mixture of soybean lecithin (preheated to 35 °C) and perilla oil (1:0.2 w/w% of lecithin to perilla oil) was hydrated by adding stock solution of glycerol in deionized water (final concentration 2 v/v%) and then stirred at 1000 rpm on a hot plate (IKA® C-MAG HS 10, Petaling Jaya, Malaysia) at 35 °C for 60 min. All the samples were prepared in a six-baffled glass vessel. As prepared liposomes were placed in nitrogen atmosphere at 25 °C for 1 h for the sake of annealing and stabilizing the prepared liposomes. In order to prepare NL, the liposome suspension was subjected to sonication (Bendeline HD 3200, Germany) above the phase transition temperature (TM around −20 °C) of liposome before annealing. Sonication was conducted using a frequency of 20 kHz at 90% [19].
Preparation of Coated NLs
Chitosan solutions were made by dissolving CS in 1% glacial acetic acid aqueous solution at concentration of 0.2–0.8 w/v%. Fresh solutions of PLL and SA were prepared in deionized water in concentrations of 0.01–0.2 and 0.2–0.4 w/v%, respectively. Then, all of the solutions were stirred (500 rpm) overnight at ambient temperature. Finally, CS, PLL and SA solutions adjusted to pH 4.5, 6 and 6, respectively.
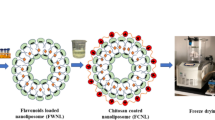
The first layer of coating was deposited by the addition of prepared NLs into coating solutions of CS (1:2 v/v% CS to NL) and PLL (1:2 v/v% PLL to NL) and then incubated at 25 °C for 1 h under gentle stirring (Fig. 1). The obtained solution was sonicated (20 kHz, 90%) for 5 min (18 s on and 30 s off) at 25 °C to achieve a well dispersed and stable solution.

Schematic diagrams of Preparation of coated nanoliposomes
Consequently, CS-NL and PLL-NL solutions were added drop-wise into the SA solution (1:2 v/v% CS-NL: SA and/or PLL-NL: SA) using the same method in an attempt to form the second layer of SA (Fig. 1). Then, the obtained SA-CS- and SA-PLL-coated NLs were sonicated.
The cross-linking reaction was carried out using GP solution (0.1 w/v%), where GP-CS-NL and GP-PLL-NL were formed by addition of CS-NL and PLL-NL solutions, respectively, into GP solution in ratio of 4:1 v/v% and then incubation at 30 °C for 1 h under gentle stirring (Fig. 1). Once the cross-linking reaction completed, the second layer of SA could be coated according to the abovementioned procedure.
Encapsulation Efficiency (EE) and Loading Contents (LC)
The percentage of the target compound incorporated into the NLs was measured through centrifugation of samples followed by the extraction of free PUFAs from the samples. Approximately 1 mL of the samples were centrifuged at 3500 g for 30 min at ambient temperature in order to sediment the liposomes (pellet) from the aqueous phase (supernatant) and then a volume of 5 mL of methanol-chloroform (50:50 v/v%) was added to the pellet. Then, the obtained solution was centrifuged at 1200 g for 10 min, followed by methyl esterification of chloroform phase after the solvent was removed under a stream of nitrogen [20]. Finally, the fatty acid profile was determined using GC equipped with a flame ionization detector (Agilent 6890 Series GC System, Agilent Technologies, Santa Clara, CA, USA) and helium as the carrier gas according to the method of AOAC (2002) [21]. The total content of n-3 and n-6 fatty acids was measured against authentic external standards of n-3 and n-6 fatty acids and applied in Eq. 1 and Eq. 2 to assess the encapsulation efficiency and loading contents, respectively.
where loaded (n-3 + n-6) content is the n-3 and/or n-6 PUFAs content entrapped within the NLs and total (n-3 + n-6) content is the both free and entrapped n-3 and/or n-6 PUFAs content in initial sample before centrifugation.
Size and ζ-potential analysis
The mean particle size and size distribution of the NLs were determined by dynamic light scattering (DLS, Zetasizer Nano ZS3600, Malvern Instruments Ltd., UK) at 633 nm and 25 °C using a quartz cell and a detection angle of 90°. Average size values were expressed as intensity mean size and the surface charge of samples was measured as ζ-potential. Measurement of the ζ-potential of the NLs was carried out using the same instrument by a combination of laser Doppler velocimetry and phase analysis light scattering. The samples were diluted 10 folds in their relevant media [13].
Morphological Analysis
Morphological analysis was conducted using transmission electron microscopy (TEM) on a Zeiss EM 10 (Zeiss, Oberkochen, Germany). First of all, the prepared sample solutions were diluted using deionized water in the mass ratio of sample: deionized water of 1:10 and subsequently were subjected to bath sonication (FUNGILAB S.A., UE06SFD, Barcelona, Spain) for 1 min. Then, a droplet of the samples was deposited on top of the 200 mesh copper grid. Afterward, the grid was stained with uranyl acetate solution (2%) for 2 min and after removing excess liquid with filter paper, allowed to dry at room temperature. Finally, the grid with the sample was examined by TEM at a voltage of 120 kV.
Fourier Transform Infrared (FT-IR) Analysis
Infra-red spectra of the samples were recorded over the range of 4000–400 cm−1 by the KBr tablet at a resolution of 0.09 cm−1 and a scan rate of 65 times per second on a FT-IR spectrophotometer (Thermo Nicolet, AVATAR, 370 FT-IR, USA).
Physical Stability
Both bare and coated NLs containing perilla oil were stored in sealed condition at two different temperatures of 4 °C and 45 °C for a storage period of up to 30 days. The average diameter, polydispersity index and ζ-potential (z) were determined to evaluate the physical stability of the samples.
Oxidative Stability
In order to examine the oxidative stability, the samples were hermetically sealed in a 20 mL glass vial and stored at 45 °C, to speed up the oxidation process, over a period of 30 days. Oxidative stability was measured by two distinct procedures of peroxide value determination using ferric thiocyanate [22] and headspace analysis through the measurement of propanal and hexanal volatile aldehydes as indicators of soy lecithin and perilla seed oil oxidation, respectively [8, 23].
In-Vitro Release Study
Selected formulations were analysed in vitro regarding their release profile under simulated gastric fluid (SGF) and simulated intestinal fluid (SIF) according to the procedure outlined by Minekus et al., (2014), with slight modifications. Briefly, 5 mL of each sample was taken in a dialysis bag with a cut off size of 10 kDa. The tube was immersed in 50 mL SGF and then the gastric-digested samples was mixed in 50 mL SIF. Both of release media incubated at 37 ± 0.5 °C under a continuous stirring speed of 100 rpm (Shaker-Incubator FSA, model ISH-251.5, Iran). The release media used was pepsin (0.32 w/v%) and 0.1 N HCl solution at pH 1.2 as SGF and pancreatin (1 w/v%) and 10 mM phosphate buffer saline (PBS) at pH 7.5 as SIF [24]. Release percentage of n-3 and n-6 PUFAs were monitored over 2 h and 8 h for SGF and SIF, respectively. Then, n-3 and n-6 PUFAs were extracted using n-Hexane and measured according to the method described previously under the section 2.2.4.
Statistical Analysis
Experimental results are expressed as the means of three independent experiments with standard deviation (mean ± standard deviation). Multiple comparisons of means (Tukey’s test) were used to assess the significance of differences between means in groups. Analysis of the variance (ANOVA) was performed on the experimental data using the SPSS software (SPSS Inc., Chicago, IL, USA, Version 21.0), and significance was tested at the 0.05 level of probability (p).
Results and Discussion
Characterization of Nanoliposomes
The particle size, polydispersity index, ζ-potential, encapsulation efficiency and loading contents of both coated and uncoated samples are presented in Table 1. Mean particle size of the liposomes significantly decreased from 254.5 nm to less than 200 nm (p < 0.05), after sonication treatment.
In the current study the mean diameter of empty NL was 109.45 nm, while increased significantly (p < 0.05) to 121.15 nm as perilla oil incorporated within formulation, which is in line with related research results [8, 16]. Moreover, PDI of liposomal samples was about 0.44 prior to sonication and substantially decreased (p < 0.05) after sonication to 0.23 in loaded NLs.
The concentrations of 0.4 w/v% for CS, 0.2 w/v% for PLL and 0.3 w/v% for SA were established to be the optimum, based on the response surface methodology (RSM) one factor design method and measurements of the average diameter, polydispersity index (PDI) and zeta potential (ζ-potential).
The mean particle size of NLs significantly increased (p < 0.05) after coating with the first layer of polymer (Table 1) which could be attributed to the construction of a thick layer on the surface of NL by electrostatic bonds and creation of bridges between polymer molecules [7, 12, 14, 25,26,27,28].
Regarding one-layer coated samples, the mean particle size of NLs coated with PLL (502.3 nm) was remarkably more (p < 0.05) than those coated with CS (241.5 nm). Considering ζ-potential values of one-layer coated samples (Table 1), PLL could create a thinner hydrodynamic layer and smaller electrostatic repulsion due to its low molar weight and plane spatial shape, which in turn would result in aggregation between NLs to overcome electrostatic attractions and yielding higher PDI as compared with CS [11, 29].
The introduction of GP as a cross-linking agent in the NL systems coated with either CS or PLL led to a decrease (p < 0.05) in the mean diameter to 200.5 nm and 472.5 nm, respectively, probably due to the compaction of the first layer of coating through cross-linking process [30].
The second layer of SA significantly increased (p < 0.05) the particle size of the SA-CS-NL to 335.5 nm compared with that of CS-NL (Table 1), which can be caused by the electrostatic bonds formed between \( {\mathrm{NH}}_3^{+} \) groups of CS and COO− groups of SA [14, 26], and also formation of bridges between SA molecules [31].
However, the second layer of SA significantly decreased (p < 0.05) the mean particle size of the SA-PLL-NL to 355.0 nm as compared with that of PLL-NL (Table 1), which can be ascribed by the formation of a thinner layer by PLL compared with CS which makes more compressed bonding with SA. Moreover, water trapped among PLL molecules could release and physical connections between PLL and SA could occur [27].
In this regard, Gibis et al. (2014) and Liu et al. (2016) suggested that CS as the first layer along with second layers of SA and pectin resulted in enlargement of anionic liposome size, though Haidar, Hamdy and Tabrizian (2008) reported a decrease in the liposomal size when SA and CS used as first and second layers, respectively [14, 26, 32]. However, our literature review found no reports on evaluating particle size of NLs coated with PLL and SA as first and second layers.
All the coated NLs showed a PDI lower than 0.3 (except for SA-PLL-NL) which was an indication of their size uniformity [16]. Higher PDI observed for SA-PLL-NL could be another indication of aggregation between NL particles which caused lowering the uniformity of the samples [33].
The ζ-potential is used to determine surface charge properties and stability of colloidal systems where values out of the range between −30 and + 30 mV indicate the suspending stability of the particles [19]. In the current study, ζ-potential of liposomal samples were − 38 mV before sonication while remarkably decreased (p < 0.05) following sonication (Table 1). This reduction of size caused inner layers to lead their polar head groups toward the outer layers and increased the absolute values of the negative charge [19]. In general, the negative charge of NLs may be attributed to the presence of phosphatidic acid in lecithin [30].
In CS-coated samples, ζ-potential reversed to positive values (Table 1) which was an indication of deposition of positively charged CS on the NL surface [16]. PLL could also decrease surface charge of liposome (p < 0.05), though it could not completely neutralize and reverse it to positive values (Table 1), which may be described by the formation of a discontinuous layer of coating on NL surface as mentioned earlier [19].
According to the results (Table 1), GP didn’t show a significant impact on surface charge and ζ-potential of samples. This observation could be explained in terms of GP potential which just reacts with the primary amino groups to form amide bonds. Published work by Elzoghby et al. (2013) has indicated that an increase in either GP concentration or cross-linking reaction time had no significant influence on ζ-potential values of samples [30].
Introduction of second layer of SA in the CS-coated NL changed the positive charge (42 mV in CS-NL) to a negative one (−35 mV in SA-CS-NL), whereas coating with PLL caused remarkable increase in absolute negative charge (−38 mV in SA-PLL-NL) as compared to those coated with just PLL (−18 mV in PLL-NL). The emerged negative charge could probably be correlated to the remained negative charge of functional groups of SA, following formation of electrostatic bonds between amines and carboxyl groups of the coating layers [14, 27].
Encapsulation efficiency and loading contents were determined based on the content of α-linolenic acid and linoleic acid. As can be seen from Table 1, all coatings in except PLL, significantly enhanced (p < 0.05) the encapsulation efficiency which could be attributed to the capability of three-dimensional polyelectrolyte network of coatings along and cross-linking which entrapped that portion of free oil that had not already been trapped in NL network [16]. As mentioned earlier, PLL coating was not capable of forming a continuous layer on the surface of NL and hence its encapsulation efficiency was lower than others [27].
Morphological Properties
As can be seen from TEM images in Fig. 2, a, prepared NLs were mostly as white uniform spherical particles with about 100 nm size. In CS-coated samples, NL was surrounded by a pale halo (CS coating) and its particle size verified that CS coating was deposited on the NL surface (Fig. 2, b).

Transmission electron micrographs of bare nanoliposome (NL) and nanoliposomes coated using chitosan (CS), sodium alginate (SA) and genipin (GP) at different amplifications
Samples coated using SA as the second layer were not uniform and spherical but showed a bigger size and smoother surface (Fig. 2, c) which was comparable to the observations of previous studies [12].
Incorporation of GP caused a drop in particle size and a darker halo in comparison with CS-coated samples (Fig. 2, d) which could be ascribed to the compression of CS in the presence of GP.
FT-IR Analysis
The subtle alteration in NL while coating with CS, PLL, SA, and cross-linking with GP monitored by analysing the changes in the FT-IR spectra. As depicted in Fig. 3, two absorption bands corresponding to C-H stretching (2925 cm−1 and 2853 cm−1), have remained unchanged in CS-coated NL as compared with bare NL. Absorption at of 3355 cm−1 was related to N-H and O-H stretching, which became broader in CS-coated sample in comparison with bare NL, indicating that NH functional group of CS was added to NL sample. NLs coated by CS showed characteristic absorption at 1415 cm−1 (Fig. 3), which represents OH bending vibration peak of CS. Moreover, absorption bands at 1051 cm−1 and 1167 cm−1 for NL, related to the symmetric and asymmetric stretching vibrations of PO2−, shifted to 1054 cm−1 and 1155 cm−1, respectively, after coating with CS. This feature suggested the sensitivity of PO2− to form hydrogen bond where such a bond formed between CS and NL. Observed absorption band of primary amine at 1572 cm−1 in the CS-coated sample confirmed the successful coating process of NLs [12].

FT-IR spectra of a) different coated materials, b) bare and coated nanoliposomes within 4000–400 cm−1
As shown in Fig. 3, absorbance observed at 1613 cm−1 in SA-CS-NL sample could represent COO− asymmetric stretching vibrations, indicating the presence of SA in the sample. The peak of SA at around 1126 cm−1 also altered to 1090 cm−1, which may be attributed to CO stretching vibrations of pyranose ring in SA. Furthermore, the absorption band at 1572 cm−1 that revealed the presence of primary amine group in CS-NL appeared to be shifted to 1578 cm−1 in SA-CS-NL sample, which was another indication of the bond formation between CS and SA [12].
GP is proven to have two characteristic peaks at 1681 cm−1 and 1622 cm−1, assigning to the tensile vibrations of carboxyl and C=C groups of olefin in GP, which were not observed in the IR spectra of GP-containing samples. On the other hand, the absorption band at 3355 cm−1 in CS-NL, which was attributed to the stretching vibrations of OH and NH2 functional groups, shifted to 3375 cm−1 in GP-CS-NL, suggesting a decrease in hydrogen bonds in favour of stronger OH or NH bonds. As can be seen in Fig. 3, amine group peak intensity at 1572 cm−1 and stretching vibrations of NH at 3355 cm−1 region increased in GP containing samples as compared to samples having no cross-linking agent (GP), due to the decrease in primary amine content.
It has been reported that addition of GP to CS has led to a reduction in the peak intensity of the primary amine group and also stretching vibration of N-H functional group after 3 h [34]. In addition, the spectral pattern of CS at 1469 and 1380 cm−1 in CS-NL shifted to 1466 and 1378 cm−1 in GP-CS-NL sample, respectively, indicating conformational change due to cross-linking with GP. Moreover, the bandwidth of stretching vibrations of CH at 2925 cm−1 decreased in GP containing sample (Fig. 3), revealing that the membrane fluidity decreased following the addition of GP and, consequently, sample stability improved [12]. All these observations confirmed the formation of cross-links between GP and CS in GP-CS-NL sample.
In SA-GP-CS-NL, the characteristic peaks of the CS primary amine at 1572 cm−1 and the carboxyl group of SA at 1623 cm−1 were not observed, suggesting that the electrostatic interaction between NH functional group of CS and carboxyl group of SA in SA-GP-CS-NL (Fig. 3).
Physical Stability
In order to investigate the physical stability, mean particle size, PDI and ζ-potential of developed samples were monitored over 30 days of storage at 4 °C and 45 °C. As represented in Fig. 4a and c mean particle size of samples (in exception of GP-CS-NL and GP-PLL-NL) and also ζ-potential of all samples did not show any significant changes (p > 0.05) after storing at 4 °C over 30 days. These observations along with PDI results (Fig. 4b) indicated stability of the developed samples at 4 °C during storage time which was in line with the results obtained by previous studies [8, 16, 35].


a mean particle size (nm), b polydispersity index and c) ζ-potential (mV) of nanoliposomes (NLs) coated with chitosan (CS), poly-L-lysine (PLL) and sodium alginate (SA) and cross-linked with genipin (GP)stored at 4 °C over 30 days; d mean particle size (nm), e) polydispersity index and f) zeta potential (mV) of samples stored at 45 °C over 30 days
However, presence of GP in both CS- and PLL-decorated NLs resulted in a significant reduction (p < 0.05) in mean particle size of samples over 30 days of storage at 4 °C (Fig. 4.a), which may be attributed to slow formation of cross-links between GP and primary amine groups over time, resulting in compaction of coatings and reduction of the NLs particle size [30, 36].
Mean particle size, polydispersity index and ζ-potential of samples stored at 45 °C over 30 days are depicted in Fig. 4d, e and f, respectively. As can be seen in Fig. 4d and e, mean particle size and PDI of both uncoated empty NL and perilla oil NL increased substantially (p < 0.05) over 30 days of storage at 45 °C, which could be due to aggregation of bare NLs. The ζ-potential of empty NL remained constant where the absolute value of the negative charge of perilla oil NL decreased (p < 0.05) over storage (Fig. 4f). This could be a consequence of leaking out and deposition of perilla oil on the liposome surface, which results in partial neutralization of NL surface charge [8].
Mean particle size of both CS- and PLL-coated NLs increased over storage time at 45 °C (Fig. 4d). Considering that PDI results of both the samples (Fig. 4e), In the case of CS coated NLs increase in particle size could be attributed to the cross-linking between CS chains of different NLs, while growth of particle size in PLL-coated NLs could be a result of the aggregation, due to its low zeta potential. Moreover, surface charge in CS-decorated NL remained constant whereas in PLL-decorated NL absolute surface charge increased significantly (p < 0.05) after 30 days of storage at 45 °C (Fig. 4f).
The mean particle size of GP-CS-NL tended to decrease significantly (p < 0.05) after 30 days of storage at 45 °C (Fig. 4d). Considering that no significant change was found in PDI (Fig. 4e), it can be concluded that CS three-dimensional network became more compact as a consequence of cross-linking between CS amine groups and GP over storage time. This can be further confirmed by the colour of GP-containing NLs, which getting more intense blue with passing time. However, the surface charge of GP-CS-NL samples did not show any significant change over time, as demonstrated in Fig. S1f (p > 0.05).
The mean particle size of GP-PLL-NL decreased meaningfully up to day 14 (p < 0.05) before a significant increase (p < 0.05) up to day 30 (Fig. S1d), where there was not any statistically significant difference (p > 0.05) in PDI up to day 14, and then increased remarkably (p < 0.05) up to the end of storage period (Fig. S1e). The observed behaviour could be attributed to the cross-linking effect of GP up to day 14, causing a reduction in mean particle size, which followed by aggregation of NLs resulting in particle size increment. The results were in good agreement with the results obtained for PDI (Fig. S1e) and surface charge (Fig. S1f). However, the lighter colour noticed in GP-PLL-NL samples may indicate the lower cross-linking degree in GP-PLL-NL compared with GP-CS-NL.
Results of mean particle size (Fig. 4d), PDI (Fig. 4e) and surface charge (Fig. 4f) of SA-CS-NL suggested the destruction of SA outer layers due to its temperature sensitivity, followed by cross-linking of CS chains and aggregation of NLs.
In SA-PLL-NL an overall increase in the mean particle size was observed (Fig. 4d and Fig. S1d). Results obtained for PDI (Fig. 4e and Fig. S1e) and surface charge which partially neutralized (Fig. 4f and Fig. S1f) could be an explanation of SA coating destruction followed by a profound increase in aggregation between the NLs from day 14 onward.
The observed decrease in SA-GP-CS-NL mean particle size up to day 7 (Fig. S1d) may be an indication of SA coating destruction and compaction of CS coating in the presence of GP which was further confirmed by the intensity of the blue colour of the sample. The mean particle size kept to remain constant up to day 21 (Fig. S1d), likely due to the integration of the coating as a result of its cross-linking in the presence of GP. A subsequent increase in mean particle size (Fig. S1d) and constant surface charge (Fig. S1f) of SA-GP-CS-NL from day 21 onward could be attributed to the fusion of the NLs which did not affect the surface charge.
Significant increase up to day 7 followed by decrease up to day 14 and then another increase up to the end of storage time in mean particle size of SA-GP-PLL-NL (Fig. 4d and Fig. S4d), maybe due to fusion of NLs, followed by destruction of SA coating and compaction of the coating layer and then aggregation of NLs, respectively, which is in high agreement with PDI (Fig. 4e and Fig. S1e) and surface charge (Fig. 4f and Fig. S1f) results. Moreover, the visual appearance of the SA-GP-PLL-NL indicated a bluer colour at the end of storage time though it was paler than GP-PLL-NL and SA-GP-CS-NL, probably due to less cross-linking in SA-GP-PLL-NL.
Altogether, it can be concluded that GP could effectively improve the physical stability of all developed NLs which was in line with results obtained by Muzzarelli (2009) [37].
Oxidative Stability
The primary oxidation products were determined through peroxide value analysis whereas secondary oxidation products were investigated based on the measurement of volatile propanal and hexanal as the oxidation indicators for linolenic acid and linoleic acid, respectively [38].
Analysis of fatty acid profiles showed that the linolenic and linoleic acid content of perilla oil was about 65% and 12% of the total fatty acid composition, respectively, whereas this content was about 5% and 60% in soy lecithin as NL wall-forming.
As illustrated in Fig. S2a, peroxide value of perilla oil at 45 °C tended to increase from the first week of the storage and raised its highest level in the third week before a subsequent decrease (p < 0.05) (Fig. S2a), suggesting a significantly higher rate of its degradation compared to its formation. Furthermore, concentration of hexanal (Fig. 5b and Fig. S2b) and propanal (Fig. 5c and Fig. S2c) showed an increase over time, where propanal amount was found to be remarkably more than hexanal content, which may be explained by higher linolenic acid content of perilla oil (p < 0.05).

Concentration of a) peroxide (meq/kg), b propanal (ng/ml) and c) hexanal (ng/ml) in nanoliposomes (NLs) coated with chitosan (CS), poly-L-lysine (PLL) and sodium alginate (SA) and cross-linked with genipin (GP) over 30 days of storage at 45 °C. The limit of detection (LOD) and limit of quantitation (LOQ) of propanal and hexanal were o.5 and 1.65 (ng/ml), respectively.
Investigation of oxidative stability of soy lecithin as wall material using empty NL as control, suggested an increase in the concentration of peroxide, propanal, and hexanal over time (p < 0.05), where the rate of increase improved substantially from the second week (Fig. 5a-c and Fig. S2a-c). The concentration of hexanal for empty NL was far more than propanal, which could be a result of its high content of linoleic acid.
Although concentration of peroxide, propanal, and hexanal increased significantly over storage time (p < 0.05) in oil-containing bare NL, peroxide and propanal content was substantially lower (p < 0.05) compared to perilla oil, stating the NL capability in improvement of perilla oil stability to some extent (Fig. 5a-c and Fig. S2a-c). However, the high concentration of hexanal in perilla oil-containing bare NL (Fig. 5b) suggested that the NL wall (soy lecithin) was likely to be significantly exposed to oxidation [8, 39].
In samples coated with first layer of either CS or PLL, hexanal and propanal content increased significantly (p < 0.05) with time over the study period (Fig. 5b and c), while peroxide value showed a significant increase from day 14 onward (Fig. 5a and Fig. S2a), being lower than perilla oil NL (p < 0.05).
However, the peroxide value of PLL-NL increased with a higher rate (more than 10 meq/kg on day 14) than that of CS-NL (about 10 meq/kg over 30 days). Both CS and PLL coatings displayed significant potential (p < 0.05) in terms of declining formation of hexanal and propanal compounds as compared to control (perilla oil NL), but CS coating performed more potent than PLL (Fig. 5b and c). These observations may be supported by the results of the ζ-potential, suggesting PLL deficiency in thoroughly coating the NL surface (section 3.1.), and also its lower physical stability (section 3.4.), which led to exposing of the NL surface to oxidation, as has been confirmed by the hexanal concentration (Fig. 5b). These findings may strength the results reached by other researchers in this field reporting effective capability of CS in oxidation retardation of nanoemulsion of n-3 fatty acids-rich oil in the presence of Fe2+ as pro-oxidant at 37 °C [40]. In addition, Caddeo et al. (2016) have found significant increase in stability of CS-coated liposomal samples stored at 20 and 40 °C in comparison with CS-tripolyphosphate (cross-linking agent) sample, which was attributed to a large positive ζ-potential effect of CS-coated sample and high electrostatic repulsion force in comparison with a small positive ζ-potential of CS-tripolyphosphate and only little repulsion force [16].
Coating of NLs using SA as secondary coating layer resulted in higher values (p < 0.05) of peroxide, propanal, and hexanal compared to one-layer coated samples, though the rate of increase was more pronounced in samples containing PLL than in those containing CS (Fig. 5a, b and c).
Significant increase in peroxide value (p < 0.05) for SA-CS-NL and SA-PLL-NL observed from days 7 and 14, respectively, in comparison with one-layer coated samples (Fig. S2a). This behavior may reflect SA destruction with time due to its sensitivity to heat which would likely contribute to impairment of first layer integrity.
Higher content of hexanal than propanal in SA-coated NLs (Fig. 5b and c) is another evidence of more oxidation in the NL surface compared with perilla oil entrapped within the liposome. However, oxidation rate was far lower (p < 0.05) in double-layered NLs than bare NL (Fig. 5a, b, and c), probably due to the protective effect of the remained coating on the liposome surface [14].
Regarding retardation of oxidation in both NL surface and perilla oil, the most effective coating showed to be GP-CS which led to a peroxide value of lower than 5 meq/kg after 30 days of storage whereas peroxide value reached about 15 meq/kg over 21 days in samples coated with GP-PLL (Fig. 5a and Fig. S2a). A possible explanation for these findings may be due to the compactness of the CS three-dimensional network in the presence of GP, which hinders the penetration of oxidative agents to the inner layers of NLs.
Peroxide value was found to be less than 10 meq/kg in SA-GP-CS-NL over 21 days of storage while it was just more than 15 meq/kg in SA-GP-PLL-NL in day 14 (Fig. S2a), which could be justified by the more efficient cross-linking of CS in the presence of GP and these findings are in agreement with the ζ-potential and physical stability results.
A similar study on PLL revealed lower oxidative stability of PLL than that of CS (p < 0.05), which could be due to the linear structure of the PLL and its deficiency in altering liposome surface charge to positive amounts. Accordingly, it can be concluded that the PLL coating was not suitable in terms of its inhibiting effect on the oxidation of NLs containing oil rich in n-3 fatty acids.
Therefore, further studies performed on the effect of CS (as primary coating), SA (as secondary coating) and GP (as cross-linker) on the release profile of n-3 and n-6 PUFAs in simulated gastrointestinal fluids.
Release Profile in Simulated Gastrointestinal Fluids
The NL’s ability to pass through the stomach and release the encapsulated perilla oil was investigated SGF and SIF. Figure 6 represents the release profile of linolenic and linoleic fatty acids from the NLs digested under SIF and SGF. As can be seen in Fig. 6, the highest release percentage in the SGF was observed for bare NL, representing the relative stability of the NL under gastric acidic conditions. Previous studies have also reported the stability of liposomal systems in low pH while passing through gastroenteric media [12, 41]. Furthermore, a reduction in the percentage of fatty acids released under SGF conditions was occurred with coating and cross-linking of NLs which was more prominent in GP-CS-NL and SA-GP-CS-NL samples, as fatty acids were not detectable in SGF (Fig. 6). Poor release in the gastric acidic condition is favorable for developing food products since this can result in a more bioavailability of bioactive compounds in intestinal juices [14].

Release profile for a) n-3, and b) n-6 fatty acids from bare nanoliposome (NL), nanoliposome-chitosan (CS-NL), nanoliposome-chitosan-genipin (GP-CS-NL) and nanoliposome-chitosan-genipin-sodium alginate (SA-GP-CS-NL) during digestion over 2 h and 8 h in simulated gastric (SGF) and intestinal (SIF) fluids, respectively. The limit of detection (LOD) and limit of quantitation (LOQ) of n-3 were 565.3 and 1713.0 (ng/μl), respectively. The limit of detection (LOD) and limit of quantitation (LOQ) of n-6 were 325.5 and 986.3 (ng/μl), respectively.
The release percentage of the fatty acids from the bare NL under SIF condition appeared to be substantially more than SGF (Fig. 6) which could be ascribed to the destruction of the NL membrane due to the activity of pancreatin enzyme [9, 42, 43]. In the monolayer-coated samples, the release percentage was again higher in SIF compared to SGF condition but release percentage was still lower than that of the bare NL (Fig. 6). However, the release percentage of bilayer-coated and GP cross-linked samples was slow and sustained under GIF conditions (Fig. 6), which may be attributed to the compressing three-dimensional network of CS and hindering penetration of pancreatin and subsequent hydrolysis of NL samples [35].
In general, digestive enzymes cause degradation of the liposomal phospholipids and the release of the encapsulated ingredients into the small intestine, whereas low pH or high pH in the whole GIT has little effect on the honesty of liposomes. Also, bile salts can increase the fluidity of the liposome membrane, resulting in a greater level of the enzymes adsorption and damage of the structural liposomes [10]. Therefore the digestion stability of liposomes can be increased by surface modification, through the formation of a barrier between enzymes and liposomal phospholipids [10]. Chiefly, SA (pKa values around 3.5) and CS (pKa values around 6.5) may have little or no negative charge under the SGF conditions, but have a high absolute charge in the SIF at the neutral pH. The digestive stability of the liposomes could be improved, when the liposomes surface modified by these biopolymers, because their properties in different GI fluids may alter the ability of enzymes to act on the surface of the lipids [10].
In this regard, Liu et al. (2013) investigated free fatty acids content and release profile of medium-chain fatty acids (MCFAs) in the bare liposome and those coated with CS and SA in SGF and SIF conditions [12]. They demonstrated that free fatty acids content was lower in the coated samples compared with uncoated samples. They reported low release percentage of 20.4 and 29.8% in SGF for both coated and uncoated samples, respectively, where less release percentage observed for coated ones was ascribed to SA potential in lowering pepsin activity. Moreover, the release percentage increased in intestinal conditions as compared with gastric conditions, for both coated (56.9%) and uncoated (79.8%) samples, but it kept again to be less in coated liposomes than that of uncoated ones. This observation was attributed to the spatial hindrance induced by the coatings which inhibited reaching pancreatic enzymes to liposome membrane and hence increased liposome stability under intestinal condition [12].
Release percentage of vitamin C in gastrointestinal simulated conditions for bare liposome, CS-coated liposome and SA-CS-coated liposome has already been investigated by Liu et al. (2016) and results have suggested the release percentage of 25, 27 and 14%, respectively, in gastric simulated conditions [14]. However, release percentage in intestinal condition for bare liposome raised to more than 80% and stayed unchanged for CS-coted (27%) and SA-CS-coated (14%) ones [14].
Conclusions
This study examined the development of NLs using different coating agents (CS, PLL, and SA) and GP as a cross-linker to overcome low oxidative stability of perilla oil and semi-permeability of NL membrane during storage and in vitro digestion. Formation of coated NLs was successfully achieved as shown by FT-IR and TEM analysis and showed to possess a satisfactory range of size (200–502 nm) and encapsulation efficiency (82–91%). Indeed, CS as primary coating and GP as a GRAS cross-linker could improve the physical and oxidative stability of both perilla oil and NL surface, though PLL as primary coating and SA as the secondary coating diminished the stability of NL system. All coated NLs could also benefit stability under gastrointestinal conditions through lowering the release percentage of n-3 and n-6 PUFAs. On that account, fabrication of NLs through heating method would be a promising and green technique from an industrial point of view for loading bioactive compounds into nanocarrier systems. The present study is anticipated to promote a better understanding of the advantages of combining NLs with the primary and secondary coatings and cross-linker, paving the way for the next in vitro and in vivo investigations to adjust release profile and modulate digestion rate of perilla oil.
References
T. Longvah, Y.G. Deosthale, P.U. Kumar, Food Chem. 70(1), 13–16 (2000)
S.S. Umesha, R. Sai Manohar, A.R. Indiramma, S. Akshitha, K. Akhilender Naidu, LWT-Food Sci. Technol. 62, 654–661 (2015)
R. Abuzaytoun, F. Shahidi, Am. Oil Chem. Soc. 83(10), 855–861 (2006)
W. Choo, J. Birch, J. Dufour, J. Food Compos. Anal. 20, 201–211 (2007)
K.B. Kim, Y.A. Nam, H. Sik Kim, A.W. Hayes, B.M. Lee, Food Chem. Toxicol. 70, 163–178 (2014)
D. McClements, Y. Li, Adv. Colloid Interf. Sci. 159, 213–228 (2010)
M. Hasan, N. Belhaj, H. Benachour, M. Barberi-Heyob, C.J.F. Kahn, E. Jabbari, M. Linder, E. Arab-Tehrany, Int. J. Pharm. 461(1–2), 519–528 (2014)
M. Frenzel, A. Steffen-Heins, Food Chem.185, 48–57 (2015a)
W. Liu, A. Ye, C. Liu, W. Liu, H. Singh, Food Res. Int. 48(2), 499–506 (2012)
W. Liu, A. Ye, F. Han, J. Han, Adv. Colloid Interf. Sci. 263, 52–67 (2019)
D. Volodkin, V. Ball, P. Schaaf, J.C. Voegel, H. Mohwald, Biochimica et Biophysica Acta (BBA)-Biomembranes 1768(2), 280–290 (2007)
W. Liu, J. Liu, W. Liu, T. Li, C. Liu, J. Agric. Food Chem. 61, 4133–4144 (2013)
M. Frenzel, A. Steffen-Heins, Food Chem. 173, 1090–1099 (2015b)
W. Liu, W. Liu, A. Ye, S. Peng, F. Wei, C. Liu, J. Han, Food Chem. 196, 396–404 (2016)
R. Einarsdottir, M. Gibis, B. Zeeb, K. Kristbergsson, J. Weiss, Food Biophysics 11(4), 417–428 (2016)
C. Caddeo, O. Diez-Sales, R. Pons, C. Carbone, G. Ennas, G. Puglisi, A.M. Fadda, M. Manconi, J. Colloid Interface Sci. 461, 69–78 (2016)
N. Shahgholian, G. Rajabzadeh, B. Malaekeh-Nikouei, Int. J. Biol. Macromol. 104, 788–798 (2017)
F. Song, L.M. Zhang, Ind. Eng. Chem. Res. 48(15), 7077–7083 (2009)
B. Rasti, S. Jinap, M.R. Mozafari, A.M. Yazid, Food Chem. 135(4), 2761–2770 (2012)
G. Lepage, C.C. Roy, J. Lipid Res. 27(1), 114–120 (1986)
AOAC, Official Methods of Anal. of AOAC Int. 17thedition. Maryland, USA (2002)
H. Stöckmann, K. Schwarz, Langmuir 15(19), 6142–6149 (1999)
Y. Serfert, S. Drusch, K. Schwarz, Food Chem. 113, 1106–1112 (2009)
M. Minekus, M. Alminger, P. Alvito, S. Ballance, T.O.R.S.T.E.N. Bohn, C. Bourlieu, C. Dufour, Food Funct. 5(6), 1113–1124 (2014)
H. Sasaki, K. Karasawa, K. Hironaka, K. Tahara, Y. Tozuka, H. Takeuchi, Eur. J. Pharm. Biopharm. 83(3), 364–369 (2013)
M. Gibis, B. Zeeb, J. Weiss, Food Hydrocoll.38, 28–39 (2014)
A.I. Gomaa, C. Martinent, R. Hammami, I. Fliss, M. Subirade, Front. Chem. 5, 103 (2017)
C. Laye, D.J. McClements, J. Weiss, J. Food Sci. 73(5), N7–N15 (2008)
O. Mertins, R. Dimova, Langmuir 27(9), 5506–5515 (2011)
A.O. Elzoghby, W.M. Samy, N.A. Elgindy, Pharm. Res. 30(2), 512–522 (2013)
A. Panya, M. Laguerre, J. Lecomte, P. Villeneuve, J. Weiss, D.J. McClements, E.D. Decker, J. Agri, Food Chem. 58, 5679–5684 (2010)
Z.S. Haidar, R.C. Hamdy, M. Tabrizian, Biomaterials 29(9), 1207–1215 (2008)
M. Danaei, M. Dehghankhold, S. Ataei, F. Hasanzadeh Davarani, R. Javanmard, A. Dokhani, S. Khorasani, M.R. Mozafari, J. Pharm. 10(2), 57 (2018)
M. Mekhail, K. Jahan, M. Tabrizian, Carbohydr. Polym. 108, 91–98 (2014)
H. Chen, W. Ouyang, C. Martoni, F. Afkhami, B. Lawuyi, T. Lim, S. Prakash, Int. J. Polymer Sci. 2010, 1–10 (2010)
T.M. Taylor, S. Gaysinsky, P.M. Davidson, B.D. Bruce, J. Weiss, Food Biophysics 2(1), 1–9 (2007)
R.A. Muzzarelli, Carbohydr. Polym. 77(1), 1–9 (2009)
M. Jimenez, H.S. Garcia, C.I. Beristain, J. Sci. Food Agric. 86(14), 2431–2437 (2006)
J. Heuvingh, S. Bonneau, Biophys. J. 97(11), 2904–2912 (2009)
L. Salvia-Trujillo, E.A. Decker, D.J. McClements, Food Hydrocoll. 52, 690–698 (2016)
J. Zhang, J. Han, A. Ye, W. Liu, M. Tian, Y. Lu, & M. P. Lou, Food Biophysics 1–13 (2019)
L.G. Hermida, M. Sabés-Xamaní, R. Barnadas-Rodríguez, J. Liposome Res. 19(3), 207–219 (2009)
J.S. Lee, H.W. Kim, D. Chung, H.G. Lee, Food Hydrocoll. 23(8), 2226–2233 (2009)
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Conflict of Interest
This study did not receive any specific grant from funding agencies in the public, commercial, or not-for-profit sectors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 60 kb)
Rights and permissions
About this article

Cite this article
Zamani-Ghaleshahi, A., Rajabzadeh, G., Ezzatpanah, H. et al. Biopolymer Coated Nanoliposome as Enhanced Carrier System of Perilla Oil. Food Biophysics 15, 273–287 (2020). https://doi.org/10.1007/s11483-019-09621-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11483-019-09621-y