
Abstract
Recent studies have demonstrated a close interaction between neuroinflammatory responses, increased production of inflammatory mediators, and neurodegeneration. Pathological findings in neurological diseases such as Alzheimer’s disease, Parkinson’s disease, and Huntington’s disease have shown common signs of neuroinflammation and neurodegeneration. Lupeol, a natural pentacyclic triterpene, has revealed a number of pharmacological properties including an anti-inflammatory activity. This study aimed to evaluate the effect of lupeol against lipopolysaccharide (LPS)-induced neuroinflammation in the cortex and hippocampus of adult mice. Our results showed that systemic administration of LPS induced glial cell production of proinflammatory cytokines, tumor necrosis factor (TNF)-α, inducible nitric oxide synthase (iNOS), and interleukin (IL)-1β, while co-treatment with lupeol significantly inhibited the LPS-induced activation of microglia and astrocytes, and decreased the LPS-induced generation of TNF-α, iNOS, and IL-1β. The intracellular mechanism involved in the LPS-induced activation of inflammatory responses includes phosphorylation of P38 mitogen-activated protein kinase (MAPK) and c-Jun N-terminal kinase (JNK), which was significantly inhibited by lupeol. We further elucidated that lupeol inhibited the LPS-induced activation of the mitochondrial apoptotic pathway and reversed the LPS-induced expression of apoptotic markers such as Bax, cytochrome C, caspase-9, and caspase-3. Taken together; our results suggest that lupeol inhibits LPS-induced microglial neuroinflammation via the P38-MAPK and JNK pathways and has therapeutic potential to treat various neuroinflammatory disorders.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
An inflammatory process is initiated as a first response of the resident immune system following invasion of a foreign particle, generating its signals via infection or irritation (Brown and Neher 2010). The immune system of the central nervous system (CNS) contains glial cells that include microglia and astrocytes, which are involved in the inflammatory process to defend the CNS from pathogens and maintain the normal integrity of neuronal cells (Skaper 2007). Several studies have demonstrated that prolonged inflammatory changes can exacerbate inflammatory diseases of the brain such as multiple sclerosis or meningitis (Aktas et al. 2007) or non-inflammatory brain diseases such as Alzheimer’s disease, Parkinson’s disease, or Huntington’s disease (Heneka and O’Banion 2007; Hirsch and Hunot 2009; Bjorkqvist et al. 2009). The neuroinflammatory process begins with the activation of glial cells, which generates a number of neurotoxic elements that include reactive oxygen species (ROS), nitric oxide species (NOS), cytokines, and other inflammatory mediators, all of which contribute to neurodegeneration (Skaper 2007; Di Filippo et al. 2010). Accordingly, Tansey et al. stated that the production of these inflammatory cytokines and chemokines leads to the induction of apoptosis in various subsets of brain cells, including oligodendrocytes and astrocytes (Tansey et al. 2009).
Lipopolysaccharide (LPS), a major component of the cell wall of Gram-negative bacteria, is known to be a potent stimulator of brain macrophages and activates the immune system (Block et al. 2007). Both in vitro and in vivo studies describe that LPS induction activates microglial cells, which lead to the generation of ROS, NOS, and a wide range of proinflammatory cytokines such as interleukin (IL)-1β, IL-6, tumor necrosis factor (TNF)-α, and interferons (IFNs) (Palsson-McDermott and O’Neill 2004; Chagnon et al. 2005; Block et al. 2007). LPS primarily attaches to the TLR4/CD14 receptor complex and initiates the inflammatory process via mitogen-activated protein kinase (MAPK) family proteins such as P38-MAPK, c-Jun N-terminal kinase (JNK), or the nuclear factor kappa B (NFκB) pathway (Kacimi et al. 2011). These pathways lead to the release of different inflammatory markers and the generation of oxidative stress, which ultimately leads to neurotoxicity and neurodegeneration (Shaulian and Karin 2002; Kacimi et al. 2011; Munoz et al. 2007). The downstream signal transduction of the P38 and JNK pathways has been described to activate a variety of transcription factors and generate different inflammatory mediators (Stambe et al. 2003). Furthermore, it has been described that JNK signaling induces activator protein (AP)-1-dependent BAX and caspase activation, which results in neuronal apoptosis (Putcha et al. 2003).
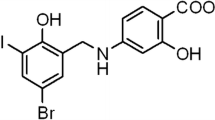
Lupeol, a biologically active dietary triterpenoid (Fig. 1), is found in many medicinal plants and different fruits such as olives, mangos, and strawberries (Saeed and Sabir 2002; Siddique and Saleem 2011; Papi Reddy et al. 2009). Several studies have demonstrated lupeol to exhibit a wide spectrum of pharmacological properties including anti-inflammatory, anti-cancer, anti-diabetic, anti-microbial, cardioprotective, and hepatoprotective activities (Ding et al. 2009; He et al. 2011; Siddique et al. 2011; Papi Reddy et al. 2009). Recent studies have shown that lupeol possesses anti-apoptotic properties, which include inhibition of the mitochondrial apoptotic pathway and ROS production in hepatocytes (Kumari and Kakkar 2012a, b). Moreover, Brimson et al. (2012) described a protective effect of lupeol against glutamate-induced neurotoxicity in HT22 mouse hippocampal cells.

Chemical structure of lupeol
The aim of our present study was to evaluate the anti-inflammatory effect of lupeol against LPS-evoked neuroinflammation in the cortex and hippocampus of adult mice. We analyzed the anti-apoptotic effect of lupeol by investigating the activities of different proteins in the mitochondrial apoptotic pathway. Moreover, LPS-induced neuroinflammation via the P38/JNK pathway and the inhibitory effect of lupeol were also evaluated.
Material and Methods
Reagents
LPS, lupeol, cresyl violet stain, and 3,3′-diaminobenzidine peroxidase (DAB) reagent were purchased from Sigma. The Vectastain ABC Elite kit was from Vector Laboratories (Burlingame, CA, USA) and Fluorojade B was purchased from Millipore. The primary antibodies p-JNK (sc-6254), NOS-2 (sc-651), TNF-α (sc-52746), IL-1β (sc-7884), Bax (sc-493), and cytochrome C (sc-8385) were purchased from Santa Cruz Biotechnology. Primary antibodies p-P38 (9211S), caspase-3 (9662), and caspase-9 (9502S) were from Cell Signaling Biotechnology. The secondary antibodies used in our experiments were goat anti-mouse IgG, goat anti-rabbit IgG, and rabbit anti-goat IgG, and were purchased from Santa Cruz Biotechnology.
Animals and Drug Treatment
LPS was dissolved in normal saline and injected intraperitoneally. An aqueous suspension of lupeol was made in 0.25 % (v/v) dimethyl; sulfoxide solution and administered intragastrically. C657LB male mice (n = 40, Gyeongsang National University Animal Breeding Center, Jinju, South Korea) weighing 25–30 g at the start of the treatment were housed in a temperature-controlled environment and maintained on a 12-h light/dark cycle (lights on at 6:00 am) with food ad libitum. All mice were randomly divided into four groups and treated for 7 days: control group (0.5 mL saline/day), LPS group (250 μg/kg/day), lupeol (50 mg/kg/day) + LPS (250 μg/kg/day) group, and lupeol group (50 mg/kg/day).
All experimental procedures were carried out in accordance with the rules established by the Animal Ethics Committee (IACUC) of the Division of Applied Life Sciences, Department of Biology, Gyeongsang National University, South Korea.
Western Blot Analysis
Western blot analysis was performed according to a previously defined procedure (Badshah et al. 2013). The mice were treated with respective drugs. Following the last day of treatment, mice were sacrificed and the cortex and hippocampi were collected. Tissue was homogenized in pro-prep extraction solution (iNtRON Biotechnology) and proteins samples were collected after centrifugation. Equivalent amounts of protein were loaded onto a 4–18 % (w/v) SDS-PAGE gel under reducing conditions and transferred onto a polyvinylidene difluoride membrane. The membranes were blocked with skim milk for 1 h to reduce non-specific binding, and probed with either rabbit-derived p-JNK, p-P38, Bax, caspase-9, caspase-3, NOS-2, and IL-1β, goat-derived cytochrome C, or mouse derived TNF-α antibodies. Membranes were incubated with goat-derived horseradish peroxidase-conjugated anti-rabbit IgG, anti-goat IgG, or anti-mouse IgG secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The immunoblots were visualized using Ez West Lumi western blotting detection reagent (Atto Corporation Tokyo). The x-ray films were scanned and optical densities of the bands were measured using computer-based Sigma Gel software (Jandel Scientific, San Rafeal, CA, USA).
Tissue Sample Preparation
For morphological analysis, the cortical and hippocampal tissue samples were prepared from different experimental groups. Mice were anesthetized and transcardially perfused with normal saline and 4 % (w/v) paraformaldehyde followed by removal of the whole brain. Brains were fixed with paraformaldehyde for 3 days followed by immersion in 20 % (w/v) sucrose phosphate buffer for 3 days. Brain samples were frozen with O.C.T. compound (A.O. Co., USA) and 12- to 14-μm-thick coronal sections were cut using a cryostat (Leica cryostat CM 3050, Germany). Coronal sections were collected on gelatin-coated slides at −20 °C and stored −70 °C.
Immunofluorescence Assays
Immunofluorescence assays were performed to identify microglial activation and the release of inflammatory mediators according to previously described procedures (Shah et al. 2014). Slides were dried at room temperature, washed twice with 0.01 M PBS for 5 min, and incubated with proteinase K for 5 min. Sections were next washed with PBS and blocked with normal serum (Vector Laboratories, CA) diluted 1:20 in PBS for 50 min. Primary antibodies (p-JNK, TNF-α, GFAP, and Iba-1) diluted 1:100 in PBS containing 2 % (v/v) normal serum and 0.1 % (v/v) Triton X-100 were applied at 4 °C overnight. The slides were rinsed twice with PBS and FITC or TRITC (anti-rabbit, anti-goat, or anti-mouse) secondary antibodies diluted 1:50 in PBS were applied at room temperature for 90 min. Tissue slides were washed twice with PBS and 4′,6-diamidino-2-phenylindole (DAPI) nucleus counterstaining solution was applied for 10 min. Slides were rinsed with PBS and glass cover slips were mounted on glass slides with fluorescent mounting medium (Dako 53023). Images were captured using a confocal laser scanning microscope (FV 1000MPE, Olympus, Japan) and integrated density was evaluated using Image J software.
Immunohistochemistry
Immunohistochemistry for caspase-3 activity in cortical and hippocampal tissue was performed as previously described (Badshah et al. 2014). Tissue sections prepared from control and experimental groups were air dried overnight, washed with PBS, and incubated with proteinase K for 5 min. Tissue was washed with PBS and peroxidase blocking solution (methanol containing 3 % (v/v) hydrogen peroxide) was applied for 10 min. Tissue slides were washed twice with PBS for 5 min, and then incubated for 60 min in blocking solution (5 % (v/v) normal goat serum and 0.3 % (v/v) Triton X-100). The caspase-3 (rabbit 1:100 in blocking solution) primary antibody was applied overnight, slides were washed three times with PBS, and the secondary antibody (goat anti-rabbit biotinylated, 1:500 in PBS) was applied at room temperature in the dark for 90 min. Tissue samples were washed with PBS and treated with ABC reagents for 60 min. Tissue sections were rinsed with PBS and immersed in a solution of PBS and DAB with some drops of hydrogen peroxide until the tissue sections changed to a light brownish color. Tissue slides were washed with PBS, immersed in xylene for 5 min, and glass cover slips were mounted on glass slides with mounting medium. Images were captured with a fluorescent light microscope.
Fluorojade-B Staining
Fluorojade B (FJB) staining was performed according to the manufacturer’s protocol (Cat# AG310, Millipore, USA). Tissue sections from the cortex and hippocampus of treated groups were air dried overnight. Sections were washed twice with PBS for 10 min and immersed in a solution of 1 % (w/v) sodium hydroxide and 80 % (v/v) ethanol for 5 min. Slides were then washed with 70 % (v/v) ethanol for 2 min followed by distilled water. The slides were then immersed in a solution of 0.06 % (w/v) potassium permanganate for 10 min, rinsed with distilled water, and transferred to a solution of 0.1 % (v/v) acetic acid and 0.01 % (v/v) FJB. After 20 min, the slides were washed three times with distilled water and allowed to dry at warm temperature. Glass cover slips were mounted using DPX non-fluorescent mounting medium and images from cortical and hippocampal sections were captured with a confocal laser scanning microscope (FV 100, Olympus).
Cresyl Violet Staining
Cresyl violet staining was performed according to a previously described procedure (Ali et al. 2015). Following overnight drying, the tissue slides were washed three times with 0.01 M PBS for 5 min. Tissue sections were stained with staining solution containing 0.5 % (w/v) cresyl violet acetate (Sigma) dissolved in distilled water with a few drops of glacial acetic acid. After 10 min, the slides were rinsed with distilled water and dehydrated with graded alcohol. Slides were dried and immersed in xylene for 5 min. Glass cover slips were mounted on slides and images were captured with a fluorescent light microscope.
Data Analysis
Data are presented as the mean ± standard error of mean (SEM). Data were analyzed by ANOVA followed by t-test. A level of p < 0.01 and p < 0.05 was considered to be significant. ‘*’ indicates a significant difference (p < 0.01) and ‘**’ indicates a significant difference (p < 0.05) with the control group. ‘#’ indicates a significant difference (p < 0.01) and ‘##’ indicates a significant difference (p < 0.05) with the LPS group.
Results
Lupeol Prevents LPS-Induced Activation of P38/JNK Pathways
Important members of the MAPK family of proteins involved stress-activated serine/threonine protein kinases such as P38-MAPK and c-JNK (Zarubin and Han 2005; Kyriakis and Ayruch 2012). The activation of these protein kinases has been implicated in oxidative stress and the activation of inflammatory mediators such as TNF-α, interleukins, and iNOS (Ajizan et al. 1999; Nakajima et al. 2004; El-Remessy et al. 2008; Han et al. 2001). To explore the effect of lupeol on the LPS-induced activation of the P38/JNK pathway, western blotting was performed on cortical and hippocampal samples of adult mice. Our results showed that LPS administration significantly elevated the level of phosphorylated p38 (p-P38) and phosphorylated JNK (p-JNK) compared with the control group. Lupeol co-treatment with LPS resulted in a significant reduction in p-P38 and p-JNK protein levels compared with the LPS-treated group (Fig. 2a). Moreover, morphological results of the cortex and hippocampal DG and CA1 regions also showed that p-JNK expression was significantly increased in the LPS-treated group compared with that of the control, while the LPS- and lupeol-treated group showed decreased expression of p-JNK compared with the LPS-treated group (Fig. 2b).

Lupeol prevents the LPS-induced activation of the P38/JNK pathway. a Representative western blots of p-P38 and p-JNK in the cortex and hippocampus of treated mice. The immunoblots were quantified using sigma gel software and the relative densities normalized with β-actin are shown. b Representative immunofluorescence photomicrographs of p-JNK-positive cells in the cortex and hippocampal regions of the mouse brain. Panels a–d represent the cortex, panels e–h represent the DG region of the hippocampus, while panels i–l represent the CA1 region of the hippocampus (magnification 10× objective field, scale bar cortex = 50 μm, scale bar hippocampus = 100 μm). Images are representative of at least three animals per group (n = 3). *p < 0.01 and **p < 0.05 with the control group; ##p < 0.05 with the LPS-treated group
Lupeol Inhibits the LPS-Induced Release of Inflammatory Markers
Activation of the MAPK family of proteins induces the phosphorylation of other protein kinases and the generation of cytotoxic factors and proinflammatory cytokines (Kyriasis and Avruch 2001; Kyriakis and Ayruch 2012). To evaluate the effect of LPS and lupeol on different inflammatory mediators, western blotting was performed to monitor the protein expression levels of TNF-α, NOS-2, and IL-1β. Our results showed that LPS administration significantly elevated the levels of these proteins in the cortex and hippocampus compared with the control group. Treatment with lupeol along with LPS significantly attenuated the LPS-induced increase in expression of TNF-α, NOS-2, and IL-1β compared with the LPS-treated group (Fig. 3a).

Lupeol inhibits the LPS-induced release of inflammatory markers. a Representative western blots for TNF-α, NOS-2, and IL-1β in the cortex and hippocampus of treated mice. The immunoblots were quantified using sigma gel software and the relative densities normalized with β-actin are shown. b Representative immunofluorescence photomicrographs of TNF-α-positive cells in the cortex and hippocampal regions of the mouse brain. Panels a–d represent the cortex while panels e–h represent the hippocampus region (magnification 10× objective field, scale bar = 100 μm). Images are representative of at least three animals per group (n = 3). *p < 0.01 and **p < 0.05 with the control group; #p < 0.01 and ##p < 0.05 with the LPS-treated group
Similar to the expression level of TNF-α, as detected by western blotting, immunofluorescence results showed that systemic administration of LPS increased the numbers of TNF-α-positive cells. Administration of lupeol along with LPS significantly attenuated the level of TNF-α-positive cells compared with the LPS-treated group in the cortex and hippocampus of adult mice (Fig. 3b).
Lupeol Attenuates the LPS-Induced Activation of Microglia and Astrocytes
Astrocytes and microglia are key cells in the neuroinflammatory and inflammatory neurodegenerative process, as they are the sources of released cytokines (Block et al. 2007; Maragakis and Rothstein 2006). Therefore, we investigated the protective effect of lupeol against astrocyte (GFAP reactive cells) and microglial activation (Iba-1 reactive cells). Immunofluorescence results revealed a significant increase in the number of GFAP and Iba-1 reactive cells in the cortex and hippocampus of LPS-injected mice compared with the control group. Treatment with lupeol along with LPS significantly decreased the number of GFAP and Iba-1 reactive cells in the cortex and hippocampus compared with LPS-treated group (Fig. 4).

Lupeol attenuates the LPS-induced activation of astrocytes and microglia. Representative photomicrographs of immunofluorescence staining for a astrocytes (GFAP-positive cells) and b microglia (Iba-1-positive cells) in LPS- and lupeol-treated groups. Images are representative of at least three animals per group. Panels a–c represent the cortex, panels d–f represent the CA1 region of the hippocampus, while panels g–i represent the DG region of the hippocampus (magnification 10× objective field, scale bar = 50 μm). **p < 0.05 with the control group; ##p < 0.05 with the LPS-treated group
Lupeol Attenuates the LPS-Induced Activation of the Mitochondrial Apoptotic Pathway
The intrinsic mitochondrial apoptotic pathway involves activation of the proapoptotic protein Bax, followed by cytochrome C release, and activation of caspase-9 and −3 (Debatin et al. 2002). To monitor the expression levels of these proteins, western blotting was performed in the cortical and hippocampal tissue of LPS- and lupeol-treated mice. Our results revealed that LPS administration induced the upregulation of Bax protein, which further enhanced cytochrome C release. Treatment with lupeol along with LPS downregulated the LPS-induced expression of Bax protein and resulted in a significant decrease in cytochrome C levels compared with the LPS-treated group (Fig. 5).

Lupeol inhibits LPS-induced Bax expression and cytochrome C release. Representative western blots of Bax and cytochrome C in the cortex and hippocampus of treated mice. The immunoblots were quantified using sigma gel software and the relative densities normalized with β-actin are shown. Immunoblots are representative of at least three animals per group (n = 3). *p < 0.01 and **p < 0.05 with the control group; #p < 0.01 and ##p < 0.05 with the LPS-treated group
Caspases are generally considered as the main mediator of apoptosis. Therefore, western blotting was performed to monitor the expression levels of caspase-9 and −3 following LPS and lupeol treatment. Our results showed that LPS treatment significantly increased the level of caspase-9 and caspase-3 compared with the control. Interestingly, co-treatment with lupeol and LPS inhibited the LPS-induced activation of caspases, and induced a significant decrease in the level of caspase-9 and −3 compared with the LPS treated group in the cortex and hippocampus of adult mice (Fig. 6a). In accordance with these results, immunohistochemistry results showed a significant increase in the expression of caspase-3 with LPS treatment, while lupeol administration along with LPS resulted in the decreased expression of caspase-3 compared with the LPS-treated group (Fig. 6b).

Lupeol inhibits the LPS-induced activation of caspase-9 and −3. a Representative western blots of caspase-9 and −3 in the cortex and hippocampus of treated mice. The immunoblots were quantified using sigma gel software and the relative densities normalized with β-actin are shown. b Representative immunohistochemical photomicrographs of caspase-3-positive cells in the cortex and hippocampal regions of the mouse brain. Panels a–d represent the cortex, while panels e–h represent the hippocampus. Images show caspase-3 stained brain tissue (magnification 10× objective field, scale bar = 500 μm). Data are the mean ± SEM of three independent experiments (n = 3). *p < 0.01 and **p < 0.05 with the control group; ##p < 0.05 with the LPS-treated group
Lupeol Inhibits LPS-Induced Neurodegeneration
FJB is a well-known marker of damaged or dead neuronal cells (Badshah et al. 2014; Schmued and Hopkins 2000). The FJB staining procedure was applied to the treated groups and the immunofluorescence results showed that systemic administration of LPS significantly elevated the level of FJB-positive neuronal cells (dead/damaged neurons) in the cortex and hippocampus relative to the control group. Interestingly, lupeol co-treatment with LPS significantly reduced neuronal damage compared with the LPS-treated group and retained neuronal viability (Fig. 7a).

Lupeol inhibits LPS-induced neurodegeneration. a Representative photomicrographs of FJB stained cortical and hippocampal tissue showing damaged or dead neuronal cells. Panels a–d represent the cortex, panels e–h represent the CA3 region of the hippocampus, while panels i–l represent the DG region of the hippocampus (magnification 10× objective field, scale bar = 50 μm). b Representative photomicrographs of cresyl violet stained cortical and hippocampal tissue showing damaged or dead neuronal cells. Panels a–d represent the cortex, panels e–h represent the CA3 region of the hippocampus, while panels i–l represent the DG region of the hippocampus (magnification 40× objective field, scale bar = 50 μm). Images are representative of at least three animals per group (n = 3). **p < 0.05 with the control group; ##p < 0.05 with the LPS-treated group
To determine the extent of neuronal viability after LPS and lupeol administration, cresyl violet staining was performed on cortical and hippocampal tissue slices of adult mice. In accordance with FJB results, cresyl violet staining results revealed that 1 week of LPS treatment significantly increased the number of apoptotic neurons as shown by damaged, fragmented, or shrunken neuronal cells (Fig. 7b; panels b, f, j). Administration of lupeol along with LPS significantly reduced the number of degenerated neurons compared with the LPS-treated group. Overall these results indicate that lupeol treatment effectively inhibits LPS-induced neurodegeneration in the cortex and hippocampus of adult mice.
Discussion
Recent studies have demonstrated that several mechanisms are involved in the pathogenicity of LPS that involve the JNK and P38-MAPK pathways in the production of inflammatory mediators (Xing et al. 2011; Wang et al. 2012). In the current study, we investigated the neuroprotective activity of an anti-inflammatory compound lupeol against LPS-induced neurotoxicity. Our result suggest that lupeol may inhibit the LPS-activated JNK and P38-MAPK pathways, which lead to depletion of inflammatory cytokines such as TNF-α, IL-1β, iNOS, and the apoptotic pathway.
Several lines of evidence have confirmed that systemic administration of LPS increases the production of inflammatory mediators including iNOS, COX2, and different cytokines such as TNF-α, IL-1β, and IL-6, which induce a number of neurobiological effects (Palsson-McDermott and O’Neill 2004; Parajuli et al. 2012). Neuroinflammation has been shown to induce neurodegeneration in different forms of brain disorders such as sepsis, Alzheimer’s disease, Parkinson’s disease, and multiple sclerosis (Gao and Hong 2008; Lull and Block 2010). Following systemic administration, LPS binds to Toll like receptor (TLR)-4 to activate glial cells that trigger the activation of inflammatory pathways such as P38-MAPK and JNK (Parajuli et al. 2012; Nakajima et al. 2004). Phosphorylation of P38 and JNK activate the pivotal transcription factor activator protein (AP)-1, which triggers different inflammatory genes such as IL-1, IL-2, CD40, TNF-α, and c-Jun (Kawai and Akira 2010; Shaulian and Karin 2002). Our results reveal that LPS administration induces the activation of the P38 and JNK pathways, which may increase the production of TNF-α, IL-1β, and iNOS, ultimately leading to cell death.
The P38 signaling cascade is known to contribute to the production of immune-related cytotoxic factors, proinflammatory cytokines, and neurodegenerative sequelae (Kyriakis and Ayruch 2012; Yasuda et al. 2011; Bendottil et al. 2006). Moreover, it has been determined that pharmacological inhibition of p38α-MAPK decreased the levels of the inflammatory cytokines TNF-α and IL-1β, and protected neuronal cells from synaptic protein loss and neurite degeneration (Xing et al. 2011; Munoz et al. 2007). The JNK signaling cascade acts on AP-1 to promote neuroinflammation and neurodegeneration or interferes with cell death-associated proteins such as the BCL-2 family of proteins to activate the mitochondrial apoptotic pathway (Bendottil et al. 2006; Putcha et al. 2003; Tournier et al. 2000). Accordingly Wang et al. showed that JNK inhibition reduced microglial activation, decreased the production of inflammatory mediators, and inhibited the apoptotic pathway (Wang et al. 2012). Although the exact neuroprotective mechanism of lupeol during LPS-mediated activation of the P38 and JNK pathways is not clear, our results indicate that lupeol decreases the levels of p-P38 and p-JNK, which may decrease the production of inflammatory mediators such as TNF-α, IL-1β, and iNOS.
Earlier reports have shown that lupeol exhibits a potent anti-inflammatory effect. Oral administration of lupeol suppresses the cytotoxic (CD8) and helper (CD4) T cells to inhibit the secretion of proinflammatory cytokines such as TNF-α, IFN-γ, IL-2, and IL-4 (Bani et al. 2006). In comparative studies, lupeol, or its derivatives, has shown more potency than classic non-steroidal anti-inflammatory drugs such as indomethacin in animal models of inflammation (Gupta et al. 2012; Davis et al. 1994; Akihisa et al. 1996). Attenuation of activated astrocytes and microglia is widely accepted as an important factor in the management of neuroinflammation. Astrocytes and microglia are supportive cells for neurons. Astrocytes regulate neuronal function via controlling synaptic transmission, whereas microglia maintain cellular homeostasis by performing immune surveillance (Haydon and Carmignoto 2006; Nimmerjahn et al. 2005). Various factors that influence the activation of glial cells in the CNS include the complement system, α-synuclein, and activated T cells. The activated T cells infiltrate through the blood brain barrier and release the proinflammatory cytokines TNF-α and IFN-γ, which interact with microglial cells to trigger the release of neuroinflammatory mediators that induce apoptosis and neurodegeneration (More et al. 2013; Gebicke-Haerter 2001). During inflammatory conditions, the morphologic characteristics of glial cells are also affected as astrocytes display a hypertrophic morphology and microglia display an amoeboid morphology (Huang et al. 2014). As stated elsewhere, LPS is a potent endotoxin that activates astrocytes and microglia both in vitro and in vivo (Palsson-McDermott and O’Neill 2004; Parajuli et al. 2012). Our morphological results in the cortex and hippocampus showed that lupeol effectively decreased the level of LPS-induced activated astrocytes and microglia (Fig. 4), indicating suppression of the inflammatory pathway.
LPS-induced neuroinflammation is known to activate the mitochondrial apoptotic pathway and induce neurodegeneration in the brain (Hotchkiss et al. 1999; Hattori et al. 2010). Moreover, studies in other cells such as myocardial cells, endothelial cells, and hepatocytes also showed evidence for LPS-induced apoptosis (Munshi et al. 2002; Chagnon et al. 2005; Cederbaum et al. 2012). Lupeol has previously been shown to inhibit excitotoxicity, mitochondrial depolarization, and the mitochondrial apoptotic pathway in hepatocytes (Kumari and Kakkar 2012a, b; Brimson et al. 2012). Our results have shown that lupeol administration downregulates the protein expression levels of the proapoptotic protein Bax, inhibits cytochrome C release, and prevents activation of the caspase cascade in the cortex and hippocampus of adult mice. Therefore, lupeol has a potent anti-apoptotic effect. Finally, we can conclude that lupeol has the potential to attenuate LPS-induced neuroinflammation and neurodegeneration. However, further research is needed to elucidate its exact mechanism of action and evaluate its neuroprotective role in various neurological disorders.
References
Ajizan SJ, English BK, Meals EA (1999) Specific inhibitors of p38 and extracellular signal-regulated kinase mitogen-activated protein kinase pathways block inducible nitric oxide synthase and tumor necrosis factor accumulation in murine macrophages stimulated with lipopolysaccharide and interferon-gamma. J Infect Dis 179:939–944
Akihisa T, Yasukawa K, Oinuma H, Kasahara Y, Yamanouchi S, Takido M, Kumaki K, Tamura T (1996) Triterpene alcohols from the flowers of compositae and anti-inflammatory effects. Phytochemistry 43:1255–1260
Aktas O, Ullrich O, Infante-Duarte C, Nitsch R, Zipp F (2007) Neuronal damage in brain inflammation. Arch Neurol 64:185–189
Ali T, Badshah H, Kim TH, Kim MO (2015) Melatonin attenuates D-galactose-induced memory impairment, neuroinflammation and neurodegeneration via RAGE/NF-K B/JNK signaling pathway in aging mouse model. J Pineal Res 58:71–85
Badshah H, Ullah I, Kim SE, Kim TH, Lee HY, Kim MO (2013) Anthocyanins attenuate body weight gain via modulating neuropeptide Y and GABAB1 receptor in rats’ hypothalamus. Neuropeptides 47:347–353
Badshah H, Kim TH, Kim MJ, Ahmad A, Ali T, Yoon GH, Naseer MI, Kim MO (2014) Apomorphine attenuates ethanol-induced neurodegeneration in the adult rat cortex. Neurochem Int 74:8–15
Bani S, Kaul A, Khan B, Ahmad SF, Suri KA, Gupta BD, Satti NK, Qazi GN (2006) Suppression of T lymphocyte activity by lupeol isolated from Crataeva religiosa. Phytother Res 20:279–287
Bendottil C, Tortarolol M, Borsellol T (2006) Stress activated protein kinases, JNK and p38, as new therapeutic approach for neurodegenerative diseases. Cent Nerv Syst Agents Med Chem 6:109
Bjorkqvist M, Wild EJ, Tabrizi SJ (2009) Harnessing immune alterations in neurodegenerative diseases. Neuron 64:21–24
Block ML, Zecca L, Hong JS (2007) Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci 8:57–69
Brimson JM, Brimson SJ, Brimson CA, Rakkhitawatthana V, Tencomnao T (2012) Rhinacanthus nasutus extracts prevent glutamate and amyloid-β neurotoxicity in HT-22 mouse hippocampal cells: Possible active compounds include lupeol, stigmasterol, and β-sitosterol. Int J Mol Sci 13:5074–5097
Brown GC, Neher JJ (2010) Inflammatory neurodegeneration and mechanisms of microglial killing of neurons. Mol Neurobiol 41:242–247
Cederbaum AI, Yang L, Wang X, Wu D (2012) CYP2E1 sensitizes the liver to LPS- and Tnf α-induced toxicity via elevated oxidative and nitrosative stress and activation of ASK-1 and JNK mitogen-activated kinases. Int J Hepatol 582790
Chagnon F, Metz CN, Bucala R, Lesur O (2005) Endotoxin-induced myocardial dysfunction. Circ Res 96:1095–1102
Davis RH, DiDonato JJ, Johnson RW, Stewart CB (1994) Aloe vera, hydrocortisone, and sterol influence on wound tensile strength and anti-inflammation. J Am Podiatr Med Assoc 84:614–621
Debatin KM, Poncet D, Kroemer G (2002) Chemotherapy: targeting the mitochondrial cell death pathway. Oncogene 21:8786–8803
Di Filippo M, Chiasserini D, Tozzi A, Picconi B, Calabresi P (2010) Mitochondria and the link between neuroinflammation and neurodegenertion. J Alzheimers Dis 20:369–379
Ding Y, Nguyen HT, Kim SI, Kim HW, Kim YH (2009) The regulation of inflammatory cytokine secretion in macrophage cell line by the chemical constituents of Rhus sylvestris. Chem Lett 19:3607–3610
El-Remessy AB, Tang Y, Zhy G, Matragoon S, Khalifa Y, Liu EK, Liu JY, Hanson E, Mian S, Fatteh N, Liou GI (2008) Neuroprotective effects of cannabidiol in endotoxin-induced uveitis: critical role of p38 MAPK activation. Mol Vis 14:2190–2203
Gao HM, Hong JS (2008) Why neurodegenerative diseases are progressive: uncontrolled inflammation drives disease progression. Trends Immunol 29:357–365
Gebicke-Haerter PJ (2001) Microglia in neurodegeneration: molecular aspects. Microsc Res Tech 54:47–58
Gupta R, Sharma AK, Sharma MC, Dobhal MP, Gupta RS (2012) Evaluation of antidiabetic and antioxidant potential of lupeol in experimental hyperglycemia. Nat Prod Res 26:1125–1129
Han Z, Boyle DL, Chang L, Bennett B, Karin M, Yang L, Manning AM, Firestein GS (2001) C-Jun N-terminal kinase is required for metalloproteinase expression and joint destruction in inflammatory arthritis. Clin Investig 108:73–81
Hattori Y, Takano K, Teramae H, Yamamoto S, Yokoo H, Matsuda N (2010) Inights into sepsis therapeutic design based on the apoptotic death pathway. J Pharmacol Sci 114:354–365
Haydon PG, Carmignoto G (2006) Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev 86:1009–1031
He Y, Liu F, Zhang L, Wu Y, Hu B, Zhang Y, Li Y, Liu H (2011) Growth inhibition and apoptosis induced by lupeol, a dietary triterpene, in human hepatocellular carcinoma cells. Biol Pharm Bull 34:517–522
Heneka MT, O’Banion MK (2007) Inflammatory processes in Alzheimer’s disease. J Neuroimmunol 184:69–91
Hirsch EC, Hunot S (2009) Neuroinflammation in Parkinson, s disease: a target for neuroprotection? Lancet Neurol 8:382–397
Hotchkiss RS, Swanson PE, Freeman BD, Tinsley KW, Cobb JP, Matuschak GM, Buchman TG, Karl IE (1999) Apoptotic cell death in patients with sepsis, shock, and multiple organ dysfunction. Crit Care Med 27:1230–1251
Huang YN, Lai CC, Lin JJ, Wang JY (2014) L-ascorbate attenuates the endotoxin-induced production of inflammatory mediators by inhibiting MAPK activation and NFκB translocation in cortical neurons/glia cocultures. PLoS ONE 9, e97276
Kacimi R, Giffard RG, Yenari MA (2011) Endotoxin-activated microglia injure brain derived endothelial cells via NF-κB. JAK-STAT and JNK stress kinase pathways. J Inflamm (Lond) 8:7
Kawai T, Akira S (2010) The role of pattern-recognition receptors in innate immunity: update on Toll-like receptors. Nat Immunol 11:373–384
Kumari A, Kakkar P (2012a) Lupeol prevents acetaminophen-induced in vivo hepatotoxicity by altering the Bax/Bcl-2 and oxidative stress-mediated mitochondrial signaling cascade. Life Sci 90:561–570
Kumari A, Kakkar P (2012b) Lupeol protects against acetaminophen-induced oxidative stress and cell death in rat primary hepatocytes. Food Chem Toxicol 50:1781–1789
Kyriakis JM, Ayruch J (2012) Mammalian MAPK signal transduction pathways activated by stress and inflammation: a 10-year update. Physiol Rev 92:689–737
Kyriasis JM, Avruch J (2001) Mammalian mitogen-activated protein kinase signal transduction pathways activated by stress and inflammation. Physiol Rev 81:807–869
Lull ME, Block ML (2010) Microglial activation and chronic neurodegeneration. Neurotherapeutics 7:354–365
Maragakis NJ, Rothstein JD (2006) Mechanism of disease: astrocytes in neurodegenerative disease. Nat Clin Pract Neurol 2:679–689
More SV, Kumar H, Kim IS, Song SY, Choi DK (2013) Cellular and molecular mediators of neuroinflammation in the pathogenesis of Parkinson’s disease. Mediators Inflamm doi: 10.115/2013/952375
Munoz L, Ranaivo HR, Rov SR, Hu W, Craft JM, McNamara LK, Chico LW, Van Eldik LJ, Watterso DM (2007) A novel P38MAPK inhibitor suppresses brain proinflammatory cytokine. J Neuroinflammation 4:21
Munshi N, Fernandis AZ, Cherla RP, Park IW, Ganju RK (2002) Lipopolysaccharide-induced apoptosis of endothelial cells and its inhibition by vascular endothelial growth factor. J Immunol 168:5860–5866
Nakajima K, Tohyama Y, Kohsaka S, Kurihara T (2004) Protien kinase C alpha requirement in the activation of p38 mitogen-activated protein kinase, which is linked to the induction of tumor necrosis factor alpha in lipopolysaccharide-stimulated microglia. Neurochem Int 44:205–214
Nimmerjahn A, Kirchhoff F, Helmchen F (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308:1314–1318
Palsson-McDermott EM, O’Neill LA (2004) Signal transduction by the lipopolysaccharide receptor, Toll-like receptor-4. Immunology 113:153–162
Papi Reddy K, Singh AB, Puri A, Srivastava AK, Narender T (2009) Synthesis of novel triterpenoid (lupeol) derivatives and their in vivo antihyperglycemic and antidyslipidemic activity. Bioorg Med Chem Lett 4463–6
Parajuli B, Sonobe Y, Kawanokuchi J, Doi Y, Noda M, Takeuchi H, Mizuno T, Suzumura A (2012) GM-CSF increase LPS-induced production of proinflammatory mediators via upregulation of TLR4 and CD14 in murine microglia. J Neuroinflammation 9:268
Putcha GV, Le S, Frank S, Besirli CG, Clark K, Chu B, Alix S, Youle RJ, LaMarche A, Maroney AC, Johnson EM Jr (2003) JNK-mediated BIM phosphorylation potentiates BAX-dependent apoptosis. Neuron 19:899–914
Saeed MA, Sabir AW (2002) Irritant potential of triterpenoids from Ficus carica leaves. Fitoterapia 73:417–420
Schmued LC, Hopkins KJ (2000) Fluoro-Jade B: a high affinity fluorescent marker for the localization of neuronal degeneration. Brain Res 874:123–130
Shah SA, Lee HY, Bressan RA, Yun DJ, Kim MO (2014) Novel osmotin attenuates glutamate-induced synaptic dysfunction and neurodegeneration via the JNK/PI3K/Akt pathway in postnatal rat brain. Cell Death Dis 5:1026
Shaulian E, Karin M (2002) AP-1 as a regulator of cell life and death. Nat Cell Biol 4:131–136
Siddique HR, Saleem M (2011) Beneficial health effects of lupeol triterpene: a review of preclinical studies. Life Sci 88:285–293
Siddique HR, Mishra SK, Karnes RJ, Saleem M (2011) Lupeol, a novel androgen receptor inhibitor: implications in prostate cancer therapy. Cancer Res 17:5379–5391
Skaper SD (2007) The brain as a target for inflammatory processes and neuroprotective strategies. Ann N Y Acad Sci 1122:23–34
Stambe C, Atkins RC, Hill PA, Nikolic-Paterson DJ (2003) Activation and cellular localization of the p38 and JNK MAPK pathways in rat crescentic glomerulonephritis. Kidney Int 64:2121–2132
Tansey M, Tran T, Lee JK (2009) Neuroinflammation in Parkinson’s disease. J Neuroimmune Pharmacol 4:419–429
Tournier C, Hess P, Yang DD, Xu J, Turner TK, Nimnual A, Bar-Sagi D, Jones SN, Flavell RA, Davis RJ (2000) Requirement of JNK for stress-induced activation of the cytochrome c-mediated death pathway. Science 288:870–874
Wang LW, Tu YF, Huang CC, Ho CJ (2012) JNK signaling is the shared pathway linking neuroinflammation, blood–brain barrier disruption, and oligodendroglial apoptosis in the white matter injury of the immature brain. J Neuroinflammation 9:175
Xing B, Bachstetter AD, Van Eldik LJ (2011) Microglial p38α MAPK is critical for LPS-induced neuron degeneration, through a mechanism involving TNFα. Mol Neurodegener 6:84
Yasuda S, Sugiura H, Tanaka H, Takigami S, Yamagata K (2011) p38 MAP kinase inhibitors as potential therapeutic drugs for neural diseases. Cent Nerv Syst Agents Med Chem 11:45–59
Zarubin T, Han J (2005) Activation and signaling of p38 MAP kinase pathway. Cell Res 15:11–18
Acknowledgments
This research was supported by the Commercializations Promotion Agency for R&D outcome (COMPA) and Pioneer Research Center Program through the National Research Foundation of Korea funded by the Ministry of Science, ICT & Future Planning (2012–0009521).
Conflict of Interest
The authors declare no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Additional information
Haroon Badshah and Tahir Ali contributed equally to this work.
Rights and permissions
About this article

Cite this article
Badshah, H., Ali, T., Rehman, Su. et al. Protective Effect of Lupeol Against Lipopolysaccharide-Induced Neuroinflammation via the p38/c-Jun N-Terminal Kinase Pathway in the Adult Mouse Brain. J Neuroimmune Pharmacol 11, 48–60 (2016). https://doi.org/10.1007/s11481-015-9623-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11481-015-9623-z