
Abstract
Infection, cancer and cardiovascular diseases are the major causes for morbidity and mortality in the United States according to the Center for Disease Control. The underlying etiology that contributes to the severity of these diseases is either hypoxia induced inflammation or inflammation resulting in hypoxia. Therefore, molecular mechanisms that regulate hypoxia-induced adaptive responses in cells are important areas of investigation. Oxygen availability is sensed by molecular switches which regulate synthesis and secretion of growth factors and inflammatory mediators. As a consequence, tissue microenvironment is altered by re-programming metabolic pathways, angiogenesis, vascular permeability, pH homeostasis to facilitate tissue remodeling. Hypoxia inducible factor (HIF) is the central mediator of hypoxic response. HIF regulates several hundred genes and vascular endothelial growth factor (VEGF) is one of the primary target genes. Understanding the regulation of HIF and its influence on inflammatory response offers unique opportunities for drug development to modulate inflammation and ischemia in pathological conditions.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Ischemic diseases are the major cause of morbidity and mortality worldwide. The spectrum of ischemic diseases is wide and includes both acute and chronic conditions. Ischemia can manifest as angina, acute coronary syndrome and chronic ischemic heart disease; as stroke in brain; as acute or chronic mesenteric ischemia in gastrointestinal system and as peripheral vascular diseases in limbs. Ischemic diseases share common risk factors, pathophysiology and etiology (Weber and Noels 2011; Noels and Weber 2011). It is well known that ischemia initiates an intense inflammatory response that is clinically relevant and worsens the ischemic injury (Braunwald 2013; Takahashi 2013). In stroke, cerebral ischemia is accompanied by infiltration of inflammatory cells, which is initiated by ischemia-induced expression of cytokines, adhesion molecules and other inflammatory mediators. Infiltration of inflammatory cells can lead to activation of microglia and astrocytes and persists for hours to days. Inflammation in ischemic tissues exacerbates tissue damage leading to worse neurological outcomes (Tuttolomondo et al. 2012; Iadecola and Alexander 2001; Zheng and Yenari 2004; Kim et al. 2013). The inflammatory changes also occur in ischemic myocardium and peripheral vascular diseases. Ischemic myocardium triggers activation of complement system leading to monocyte and neutrophil recruitment (Frangogiannis et al. 2002; Marchant et al. 2012). Atherogenesis, an important factor in the pathogenesis of cardiovascular and limb ischemia, is also linked to inflammation (Arnaud et al. 2011; Marsch et al. 2013). Furthermore, obesity is another clinical condition, in which hypoxia is associated with inflammation. The enlarging adipocytes experience an imbalance between demand and supply of oxygen leading to an increase in the secretion of inflammatory adipokines in the fat tissues. Chronic, low-grade inflammation in adipose tissues results in insulin resistance (Trayhurn 2013). As hypoxia induces inflammation, the reverse is also true in certain other diseases, wherein, inflammation causes hypoxia. Colitis (Colgan and Taylor 2010), acute lung injury, trauma and infection are some of the examples. Inflammation impairs tissue perfusion because of thrombosis. Vascular leak leads to edema, increased interstitial fluid pressure and compression of vessels (colitis), atelectasis of airways (acute lung injury, chronic obstructive pulmonary disease) and trauma (Eltzschig and Carmeliet 2011). Thus, cellular responses to hypoxia is central to the pathophysiology of major diseases (Fig. 1). Hypoxia inducible factor (HIF) is a pivotal transcription factor induced under hypoxia which transactivates target genes such as vascular endothelial growth factor (VEGF). In this review we will focus on the molecular regulation of HIF and VEGF signaling in inflammation and ischemia.

Hypoxia and inflammation associated diseases. a Summary of major pathological conditions arising from hypoxia/ischemia-mediated inflammation. b Shows how inflammation results in hypoxic conditions leading to pulmonary and gastrointestinal diseases
Hypoxia and Ischemia
Evolutionary adaptation to oxygen availability is well documented. However, dramatic variations in the distribution of oxygen concentration has been observed in different tissues and organs of higher mammals. The air we breathe has an oxygen concentration of about 21 % (pO2 = 160 mmHg) at sea level. Inhaled air passing through the trachea show a reduction in pO2 to 150 mmHg. At the alveoli the oxygen levels are about 110 mmHg. Oxygenated blood in arteries has pO2 range from 75 to 100 mmHg. Venous blood has a pO2 of 30–40 mmHg. Oxygen levels differ a lot depending on tissues and tolerance to changing levels of pO2. For example, brain pO2 levels are at 33.8 +/− 2.6 mmHg. In contrast, pO2 of kidney is 72 +/− 20 mmHg. Skeletal muscle pO2 is around 29.2 +/− 1.8. Brain tissues cannot tolerate lower pO2 whereas muscle can withstand changes in pO2 levels lower than the resting state. Interestingly, pO2 in developing fetus is much lower than the levels seen in maternal circulation. For example the umbilical vein blood has a pO2 of 20–30 mmHg and umbilical artery around 10–15 mmHg. These observations have changed the way mammalian cells are grown in vitro. Induced pluripotent stem cells (iPC) and embryonic stem cells maintain their pluripotency when cultured under hypoxic conditions. Relative pO2 again changes under pathological conditions such as inflammation, cancer, wound healing and tissue remodeling. Inflammation at skin will encounter a different oxygen level than inflammation in the gastrointestinal tract. Dermis and sub dermal tissues have a physiological pO2 of 24–35 mmHg whereas the intestine is exposed to higher levels of pO2, 57.6 +/− 2.3 mmHg. Hypoxia is defined as a condition in which oxygen supply to tissues or the whole organism is reduced. Ischemia refers to reduced blood flow or perfusion to a region of the body thereby lowering the relative oxygen levels. Each anatomical site in our body has different set point for oxygen requirements.
Inflammation creates hypoxic conditions at the local site due to increased metabolic activity outpacing the availability of oxygen. Furthermore, inflamed tissues often exhibit higher regional temperature, which can affect pO2 at the local site. Thus the hypoxic status of inflammation could be much severe than observed. Hypoxia at the inflammatory site can result in vascular breakdown, coagulation and blockage of blood flow. Tissue necrosis will ensue following prolonged loss of oxygen delivery. There is a strong cross talk between hypoxia and inflammation. Gene expression profile changes significantly when cells are exposed to hypoxia. Transcriptome of monocytes exposed to hypoxia (3–5 % O2) was much different than monocytes grown under normoxia (19.95 % O2). Hypoxia alters the repertoire of genes expressed by modulating transcription factors. Hypoxia induced inflammatory response and inflammation can cause hypoxic stress. HIF-1a has a direct effect on NFkB pathway and thereby links innate immunity, inflammation to ischemia. In IKKb knock out mice, Rius et al. found that NFkB is a transcriptional activator of HIF-1a (Rius et al. 2008). Deficiency of IKK beta resulted in abnormal expression of HIF-1 targets including VEGF. IkB alpha interacts with FIH. FIH is a negative regulator of HIF-1a by Asn hydroxylation. IkBa therefore can positively influence HIF-1 activity (Shin et al. 2009).
Cellular Responses to Hypoxia—Oxygen Sensing Molecular Switch
Cells adapt to hypoxia by recalibrating their metabolic needs and activating survival pathways. Availability of oxygen is directly linked to energy homeostasis. Lower oxygen levels compromises the function of mitochondria in generating cellular energy currency, ATP, through oxidative phosphorylation, which is the most efficient way of producing ATP from glucose. Cells activate three major pathways to survive the energy crisis created by hypoxia. The first priority for cells under hypoxic stress is to minimize energy demand by conservation. Low-priority, energy consuming processes such as endocytosis is shut down under hypoxia (Wang et al. 2009). Then, energy production by glycolysis is increased. In order to maintain energy balance, alternate sources of energy are also utilized. Finally, hypoxia forces cells to recycle macromolecules (macroautophagy) and organelles such as mitochondria (mitophagy) to make raw materials available for biosynthetic machinery (Bellot et al. 2009; Zhang et al. 2008).
Adaptation to nutrients and oxygen availability is an evolutionarily conserved process. From unicellular organisms to mammals, oxygen levels are continuously monitored by oxygen sensing machinery, which reprograms expression of several genes to adapt to changing conditions. Failure to do so will be catastrophic. In this review, we will focus on how mammalian cells respond to hypoxia with a complex regulatory system involving epigenetic and genetic pathways. Semenza and his colleagues discovered hypoxia inducible factor, HIF-1, while studying the regulation of erythropoietin (EPO) gene expression during hypoxic stress. Their pioneering studies identified cis-acting enhancer element in the promoter of EPO (Semenza and Wang 1992). Subsequent investigations revealed that HIF-1 binds to a consensus sequence, NCGTG, termed as hypoxia response element (HRE). HRE sites are present in the promoter of hundreds of genes, which are functionally related to hypoxic adaptation. For example, HIF induces expression of GLUT-1, glucose transporter that increases the intracellular levels of glycolytic substrate. At the same time, many of the enzymes needed for increased breakdown of sugar, hexokinase-1, glucose 6-phosphate isomerase, phophofructokinase, PFK1, the rate limiting enzyme of glycolysis, phosphoglycerate mutase-1 (PGAM1), pyruvate kinase type M-2 (PKM2), lactate dehydrogenase (LDHA) are increased by HIF-1-mediated transactivation. While increasing glycolysis, HIF-1 also blocks mitochondrial function and redirects the utilization of glutamine to citrate by upregulating isocitrate dehydrogenase-1 (IDH1) and aconitase1. This pathway is necessary to maintain fatty acid synthesis during hypoxia since, the production of acetyl-coA from pyruvate is prevented during hypoxia by the inhibition of pyruvate dehydrogenase enzyme by pyruvate dehydrogenase kinase1 (PDK1). Details of metabolic pathways regulated by HIF-1 under hypoxia is elegantly summarized in a recent review by Semenza (2013). Two recent studies have further confirmed the critical role of HIF-1 in glutamine utilization under hypoxia (Son et al. 2013; Gameiro et al. 2013). Reductive carboxylation is a preferred pathway to generate citrate and acetyl-coA from glutamine metabolism. Generally, cells use glutamate dehydrogenase 1 (GLUD1) to produce a-ketoglutarate. However, some malignant cells use a non-canonical to convert glutamine derived aspartate to oxaloacetate/a-ketoglutarate. This pathway was mediated by aspartate transaminase (GOT1) in a HIF-1 dependent manner. HIF-1 seems to be necessary and sufficient for reductive carboxylation, an important metabolic adaptation for the synthesis of fatty acids under hypoxia. Fatty acid precursors are needed for the synthesis of inflammatory mediators. In addition to maintaining energy homeostasis, HIF is also directly responsible for the increased production of EPO, vascular endothelial growth factor (VEGF), necessary for angiogenesis and nitric oxide synthase generating nitric oxide that is responsible for vasodilation and increased blood flow to ischemic tissues. In addition to metabolic and angiogenic regulation, HIF-1 is also responsible for the expression of inflammatory cytokines such as IL-33 in RA synovial fibroblasts. IL-33 binds to IL1Rl1 (ST2) and induce the production of Th-2 cytokines (Hu et al. 2013). IL-33 has dual role, intracellular and extracellular activation. NH-2 terminus has NLS motif and the full length IL-33 localizes to nucleus and regulates transcriptional activity (Carriere et al. 2007). Proteolysis allows extracellular presence of IL-33 and stimulates IL1RL1 receptors on Th-2 cells (Kurokawa et al. 2013). It may function as an ‘alarmin’ in directing immune/inflammatory response to the sites of tissue damage, similar to IL-1a and HMGB1 (Wahamaa et al. 2011). IL-33 is highly expressed in the heavy endothelial venule endothelial cells (HEV EC). HEV EC is directly linked to extravasation of lymphocytes from circulation into lymph nodes by diapedesis (Moussion et al. 2008). IL-33 may have an important role in sustained inflammation associated with diseases such as RA and Crohn’s disease. IL-33 plays a significant role in innate immunity as well (Yasuda et al. 2012). Thus HIF is a master regulator of hypoxic adaptation. The repertoire of genes modulated by HIF is being unraveled only recently.
Hypoxia Inducible Factor (HIF)
There are three members in the family of HIF, HIF-1, HIF-2 and HIF-3. HIF-1 is the founding member. HIF-2 was discovered later (Ema et al. 1997; Tian et al. 1997). HIFs are heterodimeric proteins consisting of an alpha subunit and a beta subunit. Alpha subunit is induced during hypoxia while the beta subunit is constitutively expressed. Beta subunit is called aryl hydrocarbon receptor nuclear translocator (ARNT). HIF belongs to the PER-ARNT-SIM (PAS) subfamily of basic helix-loop-helix (bHLH) family of transcription factors. Both alpha and beta subnits have similar structural domains. NH-2 domain contains a bHLH domain that mediates DNA binding. Central region has two PAS domains, PAS-A and PAS-B, which facilitate heterodimerization and a C-terminal region that regulates recruitment of co-activators and co-repressors (TAD, transactivation domain). TAD binds to co-activators such as CBP/p300, histone acetyltransferases/deacetylases and target gene specific factors such as Smad3 and C/EBP alpha. Most important functional domain of HIF-1, 2 alpha is the oxygen dependent degradation domain (ODDD), which determines the stability of the protein under different oxygen concentrations. HIF-3a is less studied and splice variants of HIF-3a have been suggested to play a dominant negative role in regulating HIF-1-mediated transactivation. HIFs are vital for developmental angiogenesis. Deletion of HIF-1 or HIF-2a leads to embryonic lethality at E10.5 stage due to defects in cardiovascular development (Iyer et al. 1998; Kotch et al. 1999; Tian et al. 1998). Conditional knock out mice were then generated to investigate the critical role of HIFs in developmental and pathological angiogenesis.
Regulation of HIF-1
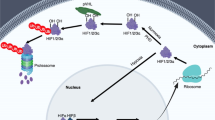
Intracellular HIF levels and its localization are dependent on oxygen levels. HIF-1a will be considered as a prototypical example in this review. In normoxia, HIF-1a undergoes post-transcriptional modifications in the ODDD region. Prolyl hydroxylases (PHD1,2,3) are key oxygen sensing molecular switches. PHDs are a family of 2-alphaketoglutarate-dependent dioxygenases. PHDs use ascorbic acid, iron and oxygen as cofactors in modifying prolyl residues in the presence of 2-alphaketoglutarate (oxoglutarate). PHDs hydroxylate two proline residues located at position 402 and 564. Hydroxylated HIF-1a is then recognized by von Hippel Lindau (VHL) protein and assembled into E3 ligase complex consisting of VHL, Cullin-2, Elongin B/C and ring box protein 1 (RBX1), VCBCR complex. Subsequent polyubiquitation of HIF-1a targets it to 26S proteasome for degradation (canonical pathway, Fig. 2). This process is very efficient and as a result very low levels of HIF-1a are maintained under normoxia. Under hypoxia however, PHDs cannot function efficiently due to lack of oxygen, a cofactor, and HIF-1a hydroxylation is attenuated. Thus, HIF-1a degradation is prevented in low oxygen concentration. HIF-1a then heterodimerize with HIF-1b and translocate to the nucleus. Though PHD-dependent hydroxylation is the predominant mechanism of HIF-1 α regulation, another post-translational modification may also play a role in HIF-1 α stability. A lysine residue at position 532 of ODD domain is acetylated by acetyl-transferase arrest defective-1 (ARD1) enzyme in an oxygen dependent manner. Acetylated HIF-1α binds to VCBCR complex efficiently and thereby results in enhanced degradation of HIF-1 α. Unlike PHD enzymes, oxygen levels do not directly affect the functional activity of ARD1 but ARD1 levels change under hypoxia by transcriptional regulation. Role of ARD1 in HIF-1 α regulation however remains controversial. Another important regulatory pathway that impinges upon the functional activity of HIF-1α is, hydroxylation of an asparagine residue at position 803. Factor inhibiting HIF-1, FIH, mediates the hydroxylation of Asn803 and prevents HIF-1 complex interaction with co-activators, CBP/P300. Inability to interact with co-activators attenuates the ability of HIF-1 to transactivate target genes. FIH activity is directly affected by oxygen, iron and oxoglutarate, which is very similar to the requirements for PHD enzymes. Additionally, HIF-1 α is also phosphorylated by mitogen-activated protein kinase (MAPK) p42/p44 and p38, which increases stability and translocation into the nucleus. Other post-translational modifications such as S-nitrosylation increases the transactivating function of HIF-1 and SUMOylation on the other hand represses HIF-1 function. Furthermore, chaperone proteins HSP70 and HSP90, which are stress-induced protective factors, can sequester HIF-1 α and prevent its degradation (non-canonical regulation). Chaperone-mediated autophagy seems to affect HIF-1 stability whereas macroautophagy did not influence HIF-1 stability (Hubbi et al. 2013a). In addition, sirtuins, NAD+-dependent histone deacetylases, influence stability of HIF. Sirt1 was found to differentially deacetylate HIF-2 α and not HIF-1 α (Dioum et al. 2009; Chen et al. 2012). However, another study found that Sirt1 could stabilize HIF-1 α (Lim et al. 2010). Thus role of Sirt1 in HIF-1 activity remains inconclusive and contextual. A recent study by Semenza’s group showed that Sirt7 physically interacts with HIF-1 α and HIF-2 α and negatively regulates their levels. Sirt7 mediated effects on HIF were found to be independent of the enzymatic activity (Hubbi et al. 2013b). Thus, complex regulatory pathways fine-tune the stability and function of HIF-1 under hypoxic stress.

a HIF-1 regulation. Schematic diagram shows canonical regulation of HIF-1α. In normoxia, HIF-1α is modified by prolyl-4-hydroxylases (PHD). PHD enzymes (PHD1, PHD2, PHD3) are Fe2+ and oxoglutarate dependent dioxygenases that use oxygen as a cofactor. PHD2 is involved in the regulation of developmental angiogenesis. PHD2 Gene deletion increases angiogenesis and erythropoiesis. Hydroxylation of proline residues in HIF-1α is recognized by VHL, tumor suppressor protein. Subsequent assembly of ubiquitin ligases results in polyubiquitination of HIF-1α and proteasomal degradation. HIF-2α is also regulated by a similar mechanism. Hypoxia and iron chelation can inhibit PHD activity and thereby prevents proteasomal degradation of HIF-1/2α. In addition, HIF-1α—mediated transactivation is modulated by hydroxylation of an Asn residue and its ability to recruit co-activators. Non-canonical regulation of HIF-1α involves chaperon-mediated sequestration, NFkB-depdendent signaling and microRNA network. b A representative confocal image showing HIF-2 nuclear translocation in hypoxia. A2780 ovarian cancer cells were exposed to either normoxia or hypoxia for 24 h. Indirect immunofluorescence using an antibody to HIF-2α shows nuclear accumulation of HIF-2α (red). Nuclei are stained with DAPI (blue)
HIF-2α and HIF-3α
HIF-2α is a structurally and functionally related transcription factor. HIF-2 α is also referred as EPAS1 and contains structural domain organizations similar to HIF-1α, Basic HLH, PAS and TAD domains. It shares about 48 % homology in primary sequence with HIF-1 α. HIF-2α is highly expressed in some tissues and cells such as lung vasculature and carotid body. HIF-1 α and HIF-2 α transactivate genes that are common to both as well as distinct groups of genes (Carroll and Ashcroft 2006; Sowter et al. 2003). For example expression of IL-8 and Myc in endothelial cells are dependent on HIF-2α and not HIF-1α (Florczyk et al. 2011). HIF-2 α is a major transcription factor expressed in endothelial cells and seems to play a major role in pathological angiogenesis (Skuli et al. 2012). HIF-2α is also the primary driver for erythropoietin expression. In some experimental conditions, loss of HIF-1 α is compensated by an increase in HIF-2α. Current evidence suggest tissue specific role for HIF-1α and HIF-2α in hypoxic responses. Unlike HIF-1α and HIF2-α, HIF-3α has not been extensively investigated. HIF-3α has multiple splice variants and expressed in tissue specific manner. Splice variants, HIF-3α2 and HIF-3α4 are shown to act as dominant-negative regulator of HIF-1α function (Maynard et al. 2005; Ando et al. 2013). As HIF-3α suppresses transcriptional activity of HIF, it is also called Inhibitory PAS domain protein, IPAS. HIF-3α has been found to interact with Bcl-xL, and may induce pro-apoptotic signaling (Torii et al. 2011). IPAS is transcriptionally upregulated by HIF-1 suggesting a negative feedback loop regulating HIF-1-mediated target gene expression (Makino et al. 2007).
HIF-1 Regulation by microRNA
MicroRNAs (miR) are short non-coding RNA 21–23 nucleotide long arising from either introns or exons. There are around one thousand miR characterized in human genome. MicroRNAs are transcribed as a longer (about 70 nt long) strand of RNA with unique stem loop structure (primary miR). Pri-mRNA is transported to the cytoplasm by exportin 5. Inside the cytoplasm they mature into pre-miR and then processed into a double stranded short segment of about 21–23 nucleotide by Dicer. Ago2 binding later selects either one of strand (3p or 5p) to base pair with target complementary sequence in 3′ untranslated region (3′UTR) of mRNA. The first 7 or 8 nucleotides of mature miR are termed ‘seed’ sequence. Binding of seed sequence at target sites in mRNA is necessary for regulating the function of transcripts. A complete match with target sequence results in degradation of target transcript while, an incomplete match affects translational initiation complex by interfering with Eukaryotic translation initiation factor 4G (eIF4G). Net result is inhibition of target protein expression. Each miR can target hundreds of targets and each target mRNA can have target sequence for multiple miR. Hypoxia in inflammatory sites can upregulate or down regulate classes of miR which in turn will have a reciprocal effect on target protein expression. Sequestering miR by competing transcripts can also modulate effects of miR. Our studies have established that under hypoxia, miR-424 is over expressed in endothelial cells. Increase in miR-424 was mediated by PU.1 transcription factor, which was regulated by C/EBP and RUNX2. miR-424 targeted CUL-2, the scaffolding protein which destabilized the E3 ubiquitin complex assembly and prevented degradation of HIF-1/HIF-2 a (Ghosh et al. 2010; Loscalzo 2010). Hypoxia also down regulates the levels of miR-199a-5P, which targets HIF-1α (Rane et al. 2009; Wang et al. 2011a). Reduction in miR-199a-5P contributes to increase in HIF-1 α transcripts and protein. Thus, a web of miR network fine-tunes the stability of HIF-1 under hypoxia. Furthermore, increased HIF-1 α under hypoxia transactivates the expression of miR-210 by binding to HRE elements present in the promoter of miR-210 (Cicchillitti et al. 2012; Kulshreshtha et al. 2008; McCormick et al. 2013). miR-210 is involved in suppressing mitochondrial function by targeting the iron sulfur complex assembly (ISCU), which constitutes the core of electron transport complex I-IV (Chan et al. 2009; Favaro et al. 2010). miR-210 further helps in the stabilization of HIF-1 by suppressing the expression of glyceroldehyde 3-phosphate dehydrogenase like 1 enzyme (Kelly et al. 2011; Peng et al. 2012). Functional activity of HIF-1 is strengthened by down regulation of FIH by miR-31 (Peng et al. 2012). Similarly, a recent study has established that miR-183 helps in stabilizing HIF-1 by targeting isocitrate dehydrogenase 2 (IDH2) (Tanaka et al. 2013). Thus, HIF-1 levels and its function are regulated by a group of microRNAs which fine-tunes hypoxic adaptation ranging from inflammation (Gonsalves and Kalra 2010), angiogenesis (Pulkkinen et al. 2008; Taguchi et al. 2008) and tissue remodeling (Wei et al. 2013; Zampetaki et al. 2013). Biological consequence of altering miR in diseased tissues has become a reality recently. Locked nucleic acid (LNA) strategy has been developed to bind and neutralize activity of specific miR under clinical settings. LNA-miR-92a (Hinkel et al. 2013) exerted cell-protective, proangiogenic, and anti-inflammatory effects in pig models. Similarly, miR-15 was found to protect against cardiac ischemic injury (Hullinger et al. 2012). These preclinical studies are encouraging and in fact LNA-based miR knock down methods are currently under Phase I/II trials. Clinical success of these methods however depends on effective delivery of LNA-reagents into affected target tissues. Pharmacological stability, tissue penetration, transport across cell membranes, escaping from nucleases and finally specific neutralization of target miR are some of the issues currently being investigated (Chistiakov et al. 2012; van den Boorn et al. 2013; Ramakrishnan 2011). Recent advances in the development of nanocarriers and functionalized nanopartlcles have helped in improving microRNA-mediated targeted therapies. These strategies are currently evaluated to knock down miRs (antagomiRs) targeting HIF-1a and VEGF to induce collateral vessel growth in ischemic tissues and to control inflammatory response.
VEGF and VEGF Receptors
VEGF is a major transcriptional target for HIF-1. Signaling through VEGF receptors have been recently reviewed (Jeltsch et al. 2013; Shibuya 2011). Three major ligand-receptor systems are upregulated under hypoxic stress, VEGF-VEGFR, Angiopoietins-Tie-1/Tie-2 and Delta-Notch. These signaling pathways help in the recovery from hypoxia-induced tissue damage and inflammation. Coordinated output from these signaling systems determines angiogenesis, blood flow, tissue perfusion, extravasation of inflammatory cells, tissue remodeling and repair. VEGF family of genes contains four members from mammals, one virus-encoded protein (VEGF-E) and a VEGF-like protein found in snake venom, svVEGF. The founding member of this family of pro-angiogenic growth factor is VEGF-A. Historically, Senger et al., in 1983 identified a protein which was ten thousand-times more potent than histamine to induce vascular permeability and named it as vascular permeability factor, VPF (Senger et al. 1983). VEGF was described by Leung et al. from Genentech in 1989 (Leung et al. 1989). Sequencing studies confirmed that VPF and VEGF are the same protein. Placenta derived growth factor, PlGF, VEGF-A, VEGF-B, VEGF-C and VEGF-D are induced under hypoxic stress in mammalian cells. VEGF-A is a homodimeric protein. Several splice variants of VEGF-A (121,145,165, 183, 189 and 206 amino acid residues) are generated by alternate splicing. VEGF-A121 lacks a heparin-binding domain (Cohen et al. 1995). Additional splicing of exon 8 at a proximal or distal site results in an anti-angiogenic variant, VEGF(xxx) family of molecules, which can compete with proangiogenic VEGF-A isoforms (Pritchard-Jones et al. 2007). VEGF-A165 and other longer isoforms have varying ability to interact with heparin sulfate proteoglycans (HSPG) and components of the extracellular matrix. VEGF-A189 and VEGF-A206 in particular contain a stretch of basic amino acid residues, thereby exhibiting stronger affinity to negatively charged HSPG. VEGF-A165 has a lower affinity to HSPGs. VEGF-sequestrated at the extracellular milieu can be conditionally released by matrix metalloproteinases which can be activated under different physiological conditions (Ferrara 2010; Miller et al. 2013). VEGF-A is a critical mediator of vasculogenesis during development. Deletion of even a single copy (VEGF-A +/−) is lethal and embryos died at E10-E11 stage indicating the importance of VEGF-A gene dosage during development (Carmeliet et al. 1996; Ferrara et al. 1996). Conditional deletion of VEGF-A in adult mice affects motor neuron development and ophthalmic complications including blindness (Kurihara et al. 2012; Oosthuyse et al. 2001). Consequently, VEGF-A has been found to have acute neuropotective effects in ischemic brain (Sun et al. 2003). Intraventricular delivery of VEGF stimulated and protected nascent neurons led to a reduction in infarct size. VEGF has a positive influence on Th2 inflammatory response in the lung. VEGF treatment decreased miR-1 levels in the lung. miR-1 targets Mpl. VEGF treatment therefore increased the levels of Mpl and P-selectin in lung endothelium. Attenuation of this pathway by intranasal delivery of miR-1 reduced lung inflammation in experimental models (Takyar et al. 2013). Therefore, VEGF is not only a versatile proangiogenic growth factor, but also limits disease progression in the brain and contributes to inflammation in the lung. Tissue specific role of VEGF can therefore be modulated for therapeutic purposes.
VEGF-B arises from a distinct gene located in chromosome 11. PlGF is encoded by PFG gene which is located in chromosome 14. Both PlGF and VEGF-B bind to VEGFR-1 and coreceptor, Neuropilin-1 (NRP-1) in promoting angiogenesis (Carmeliet et al. 2001). Neuropilin is a transmembrane glycoprotein originally identified as a coreceptor for members of the semaphorin family of secreted polypeptides (Pellet-Many et al. 2008). Neuropilins have a very short cytoplasmic domain and no kinase activity is associated with NRP. NRP functions as co-receptors to semaphorin receptors, plexins, and regulates axonal guidance during development. Recent studies suggest that NRP locks semaphorins to plexins in forming a ternary signaling complex (Janssen et al. 2012). The isoforms of different members of the VEGF family bind to NRP1 and NRP2, with distinct affinities. Exon 7 of VEGF encodes the key domain essential for the NRP binding. The VEGFR2 forms a complex with NRP1, which is important for VEGFR2 signaling and function in endothelial cells. The VEGF-A 121 isoform lacks exon 7 and is therefore unable to bind to NRP1. NRP2 has been found to complex with the receptor for VEGF-C, VEGF-D and VEGF receptor 3, also called fms-related tyrosine kinase receptor 4. Recent studies suggest that VEGF-B has a major role in regulating fatty acid transport across endothelium to tissues (Hagberg et al. 2010, 2012, 2013). VEGF-B expression pattern tightly correlated with nuclear encoded mitochondrial proteins constituting electron transport complex. VEGF-B knockout mice showed reduced fatty acid transport across endothelial cells to heart, muscle and brown adipose tissue but increased transport of fatty acids into white adipose tissues. These studies suggest a link between VEGF-B expression and mitochondrial lipid utilization and could have causal relationship with many pathological conditions. For example, peripheral tissue fat deposition contributes to pathobiology of Type 2 diabetes. When VEGF-B null mice were crossed into db/db diabetic mice, the offsprings showed marked improvement in diabetic phenotype. The double transgenic mice showed reduced fat deposition in peripheral tissues, increased glucose uptake in muscles, restored insulin sensitivity and glucose tolerance. These provocative findings suggest that VEGF-B is a novel pharmacological target for therapeutic development against type 2 diabetes and associated cardiovascular problems. Furthermore, absence and neutralization of VEGF-B may aggravate distal neuropathy observed in cancer patients during chemotherapy. VEGF-B is found to protect sensory neurons (Dhondt et al. 2011). VEGF-C and VEGF-D bind and activate VEGFR-3 receptor expressed in lymphatic vascular endothelium. VEGF-C is expressed during fetal development and in adult tissues while VEGF-D is not detectable in embryonic tissues but found during postnatal development. Physiological importance of VEGF-C is exemplified by the observation that VEGF-C knockout is embryonically lethal while VEGF-D knockout showed no lethality. Loss of VEGF-C leads to fluid accumulation due to impairment of lymphatic drainage (Karkkainen et al. 2004; Alitalo and Carmeliet 2002).
VEGF Receptor Signaling
Three major classes of VEGF receptors (VEGFR) are expressed in vascular and extravascular tissues. VEGFR are receptor tyrosine kinases containing an extracellular ligand binding domain, singly membrane spanning and an intracellular domain with a split kinase domain containing a long (60–70 amino acid residues) kinase insert. They are structurally related to PDGFR but contain seven immunoglobulin like (Ig) domains. Another distinction between PDGFR and VEGFR is that the latter lack the YxxM motif of autophosphorylation found in the former. Thus the signaling pathways of VEGFR differ from PDGFR albeit they are structurally related. VEGFR-1 is a high affinity (1–10 pM) receptor for VEGF-A but the binding does not result in strong kinase activation. VEGFR-1 has 10-fold weaker kinase activity when compared to VEGFR-2 (Sawano et al. 1996). Thus VEGFR-1 is generally considered as a non-signaling receptor but has an important biological role in regulating VEGF-mediated signaling through VEGFR-2. VEGF-A binding to VEGFR-2 results in autophosphorylation of Y1175 and recruits PLCg to the phosphorylated site. Binding of SH2 domain of PLCg then activates PKCb leading to stimulation of MAPK pathway (Sawano et al. 1997; Takahashi et al. 1999, 2001). This signaling pathway is essential for development since transgenic knock in mice having a mutation of Y1175 to F1175 led to embryonic lethality at E9.0 lacking blood vessel development. Similar phenotype was observed in total knockout of VEGFR-2. In addition to Y1175, another tyrosine residue at position 951 is also phosphorylated upon ligand binding. Y951 phosphorylation seems to regulate cell migration induced by VEGF-A (Sakurai et al. 2005). A general scheme of VEGF-VEGFR signaling system is shown in Fig. 3.

Schematic diagram shows VEGF family of growth factors and their receptors. Alternate splicing results in multiple splice variants of VEGF (not shown). Larger variants of VEGF have a heparin binding domain and are sequestered by heparin sulfate proteoglycans (HSPG). Sufatases and proteases secreted into the tissue microenvironment can release HSPG-bound VEGF. Shorter forms of VEGF are secreted and act on distant targets. VEGF binds to multiple VEGFRs. Neuropilin-1 and neuropilin-2 function as co-receptors for VEGF. Protease-mediated release of soluble VEGFR1 (sVEGFR or sFlt-1) can regulate VEGF-mediated signaling by sequestering the ligand. Furthermore, sFlt can heterodimerize with VEGFR2 and inhibit signaling by blocking autophosphorylation
VEGFA binding to VEGFR-1 does not lead to autophosphorylation of Y1169 site, which is analogous to Y1175 of VEGFR-2 (Sawano et al. 1997; Wang et al. 2011b). VEGFR-1 stimulation does not generate proangiogenic response in endothelial cells. However, VEGFR-1 has other important physiological role. VEGFR-1 is highly expressed in macrophages implicating its unique role in inflammatory response. VEGFR-1 activation in macrophages activates PI3-AKT pathway in a RACK1 dependent manner and stimulates macrophage cell migration. Thus VEGFA secreted at local inflammatory site can facilitate macrophage migration into target tissues and may help in tissue remodeling. Importance of VEGFR-1 in vascular development is demonstrated in knockout mouse models. VEGFR-1 deletion is embryonically lethal at E8.5 with over production of endothelial cells and complete lack of organized vasculature (Fong et al. 1995). This phenotype again reiterates the potential negative regulation of vasculogenesis by VEGFR-1. In a subsequent study, the kinase domain of VEGFR1 was deleted. Interestingly, kinase deleted VEGFR1 knockout mice developed normally (Hiratsuka et al. 1998). These studies suggest that ligand-binding domain of VEGFR-1 is important for orderly development of vasculature but not its kinase dependent signaling. However, VEGFR-1 mediated PI3-AKT activation in extravascular cells such as macrophages may still is important in postnatal development and inflammation mediated pathological angiogenesis (Hiratsuka et al. 1998; Murakami et al. 2006). For example, kinase deleted VEGFR-1 mice show reduced tumor angiogenesis due to impaired functions of tumor-associated macrophages.
VEGFR-3 is preferentially expressed in lymphatic endothelium, discontinuous or fenestrated endothelium of choroid plexus and high endothelial venules indicating an important role in lymphocyte trafficking (Kaipainen et al. 1995). VEGF-C binding activates PKC and Ras signaling pathways leading to lymphatic endothelial cell proliferation and migration. VEGFR-3 gene deletion leads to embryonic death at E9.5 (Dumont et al. 1998). However, deletion of its ligands VEGF-C and VEGF-D are viable (Haiko et al. 2008). It is likely therefore that VEGFR-3 is stimulated by alternate ligands present in the extracellular matrix. One such candidate has been identified to be Collagen and calcium-binding EGF domains protein-1 (CCBE1). Thus VEGFR-mediated signaling differs during development and depends on cellular and regional context (Hogan et al. 2009; Hagerling et al. 2013).
VEGF in Pathological Conditions
Inflammatory Cells
Inflammation refers to physical and physiological changes observed at the site of injury or infection. The route word for inflammation in Latin is ‘inflammo’ meaning ‘ignite’. Increased redness, heat, pain, and swelling are associated with inflamed tissues. Redness is caused by vasodilation accompanied by vascular stasis causing reduced movement of blood cells. Vasodialation is casued by bradykinin, histamine and VEGF released at the site of tissue damage and nitric oxide generated by vascular endothelium. Stasis is a culmination of changes occurring at the vasculature. Histamine and VEGF induce vascular leak at the site of inflammation leading to exudation of plasma. Vascular permeability increases cell density inside the vessels and interstitial pressure outside the blood vessels. Interstitial pressure can collapse capillaries and increase thrombotic events. Resulting sluggish blood flow causes stasis, which inadvertently helps the innate immune cells to extravasate into tissues. Endothelial cells respond to inflammatory signal by upregulating cell adhesion molecules such as ICAM, P-selectin and E-selectin. E-selectin, CD62, is a cell adhesion molecule selectively upregulated in endothelial cells upon cytokine stimulation. E-selectin has a lectin binding domains at the N and C termini, EGF like domain in the middle along with six-suchi domain repeats (CCR, complement control protein). E-selectin binds to sialyl-Lewisx carbohydrate antigen present on neutrophils, monocytes, eosinophils, T-cells and NK-cells. ESL-1 is described as a counter-receptor for E-selectin and is expressed on inflammatory cells. Expression of E-selectin is induced by P-selectin, IL-1b and TNF-α by transcriptional upregulation (Somers et al. 2000; Huang and Eniola-Adefeso 2012; Zou et al. 2005; Nimrichter et al. 2008). P-selectin, CD62P, another cell adhesion molecule that is expressed on endothelial cells and platelets is an early responder to inflammatory stimuli (McEver et al. 1989). P-selectin is stored in vesicles and are quickly expressed on cell surface by vesicle fusion (degranulation). Platelets and endothelial cells respond to thrombin, type II collagen, ATP and respond by expressing P-selectin on cell surface. Similar to E-selectin, P-selectin binds to PSGL-1 ligand expressed on inflammatory cells (Cleator et al. 2006; Chen and Geng 2006; Woltmann et al. 2000). Expression of E and P-selectins are modulated by sheer stress of blood flow and cytokines. Hypoxia alone is not sufficient for E-selctin upregulation but it is superinduced in the presence of cytokines such as TNF-α or LPS.
Intercellular adhesion molecule, ICAM1, and VCAM are other important mediators of inflammatory cell attachment to endothelial cell under hypoxic conditions. ICAM1 (CD54) is a glycoprotein expressed on endothelial cells, macrophages and lymphocytes when stimulated with TNF- a or IL-1 a. ICAM binds to LFA-1, an integrin (a L, CD11a; b2, CD18) expressed on lymphocytes and macrophage adhesion ligand-1,MAC-1, (Yang et al. 2005; Heiska et al. 1998). ICAM1 can also be induced by RANTES secreted by macrophages and granulocytes. ICAM1 is highjacked by rhinovirus (Bella et al. 1998) and malaria parasite infected erythrocytes for transendothelial cell migration (Chakravorty and Craig 2005). Recent studies suggest that ICAM1 upregulation during hypoxic stress is independent of HIF-1 but dependent on NFkB-mediated transactivation (Winning et al. 2010). Even though ICAM1 expression both on macrophages and endothelial cells were independent of HIF-1 but mediated by a novel pathway regulated by prolyl-hydroxylases (PHD) during hypoxia. In a recent study, the role of PHD isoforms on inflammatory response was elucidated in gene knockout models (Kiss et al. 2012). These studies showed that PHD3 is very important for controlled innate immune response during sepsis. PHD3 deficiency (PHD3−/−) aggravated innate immune response leading to early organ failure and death in a HIF-1 and NFkB dependent manner. VCAM-1 (CD106) is an another endothelial cell adhesion molecule upregulated during inflammation and hypoxia (Barreiro et al. 2002). VCAM-1 binds to VLA-1 antigen (alpha4 beta1 and alpha4 beta7 integrins) expressed on lymphocytes. VCAM-1 also interact with erzin and Moesin, which act as intermediates of cell membrane (ICAM-2) to actin bundles thereby regulating cell attachment and migration.
Blood borne monocytes and lymphocytes are recruited to inflammatory sites by a complex signaling cascade that involves a plethora of cytokine and chemokines. Circulating monocytes for example attach to cell adhesion molecules over expressed on endothelial cells under hypoxic/inflammatory stimuli (rolling model). Subsequently, monocytes extravasate to reach tissues by a processes called diapesis, squeezing between endothelial cells without affecting barrier functions. Outside the vasculature monocytes differentiate into tissue macrophage or dendritic cells in mediating innate or adaptive immune response respectively. VEGF, CXCL12, endothelial monocyte activating peptide-II (EMAPII) and Angiopoietin-2 (Ang2) mediate extravasation of monocytes under hypoxia. Upon extravasation, human monocyte derived macrophages (hMDM) adapt to hypoxic environment quickly by differentially expressing genes such as FGF, VEGFR and hypoxia-inducible proinflammatory cytokines such as IL-1 beta, TNF-alpha, and acute phase protein, IL-6 (Bosco et al. 2008). A comprehensive list of chemokines elaborated under hypoxia is summarized in Tables 1 and 2. In addition to chemokines, chemokine receptors are also modulated in monocyte-derived macrophages at hypoxic environment. Of particular interest is that hypoxia inhibits migration of macrophages by inhibiting cell motility promoting factor, and inducing the expression of GRO family of chemokines such as CXCL2 and CXCL3 which are negative regulators of macrophage migration (Smith et al. 2005). Thus, hypoxia traps monocyte-derived macrophages after their extravasation without affecting their ability to differentiate into immature dendritic cells (iDC). Immature dendritic cells are highly migratory and reach the draining lymph nodes. iDC differentiate to mature CD (mDC), which are retained at the site of inflammation. Hypoxia at the site of inflammation therefore modulates chemokine and chemokine receptor expression in inflammatory cells. One of the final outcomes of these changes is recruitment of monocytes to local site of tissue damage, differentiation of monocytes to tissue macrophage, retention of macrophages at hypoxic site and generation of highly motile immature dendritic cells and less motile mature dendritic cells. These changes are achieved by altering transciptome of monocyte/macrophage by hypoxia. For example, nuclear factor-kappa b (NFkB), the central transcription factor mediating inflammatory response is intricately involved in hypoxia-induced expression of monocyte chemoattractant protein-1 (MCP-1). VEGF induces MCP-1 through the activation of NFkB pathway (Marumo et al. 1999). MCP-1, in turn, can induce VEGF thereby establishing a feed-forward loop in human aortic endothelial cells (Hong et al. 2005). Inflammatory cytokine, IL-1b, induced NFkB -cyclooxygenase-2 axis was found to stabilize HIF-1 and as a consequence induced VEGF (Jung et al. 2003). Similarly, TNF-a-induced TLR4- NFkB pathway intersects with HIF-1a mediated inflammatory response as well (Tewari et al. 2012). These studies reiterate cross talk between inflammatory cytokines and hypoxia-induced responses in recruiting and regulating innate immune cells.
VEGF is effective in treating ischemia-induced damage of neuronal tissues (Greenberg and Jin 2013). VEGF administration induced not only angiogenesis but also protected neurons from cell death and neurogenesis in preclinical models of focal cerebral ischemia (Sun et al. 2003). VEGF gene therapy using AAV has a significant impact on reducing neuronal damage following stroke (Greenberg and Jin 2013). VEGF reduced inflammatory cytokine levels in the brain following ischemic stroke and attenuated immune cell infiltration (Herz et al. 2012). An alternate approach is to transplant CNS stem cells, which secrete significant amounts of VEGF. Cell-derived VEGF was necessary to recover from ischemic injury. VEGF was necessary to establish BBB and suppress inflammation in addition to neovascularization. Such an approach has been successful in promoting angiogenesis and tissue remodeling at the damaged site (Horie et al. 2011). VEGF therapy increases pericyte covering, vessel normalization and improved blood flow in experimental animals (Zechariah et al. 2013a). Same group of researchers have again showed that VEGF mediated signaling and recovery following stroke was attenuated in hyperlipidemia or in ApoE−/− genetic background (Zechariah et al. 2013b). This raises the potential limitation of VEGF centric therapeutic strategies to improve angiogenesis and blood flow to limit stroke-induced tissue damage (Table 3).
Inflammation Associated with Solid Tumors
Various in-situ hybridization studies have shown increased VEGF mRNA levels in various solid tumors including carcinomas of lung, gastrointestinal tract, breast, endometrium, urogenital tract and intracranial tumors (Ferrara 2004). The expression is particularly found to be correlated to hypoxia in the tumor cells. Although, tumor cells are the major source of VEGF, stromal cells have been identified to be an important source as well (Fukumura et al. 1998). Tumor stroma is made of fibroblasts, inflammatory cells, adipocytes and endothelial cells. This unique microenvironment resembles tissues under persistent inflammation in many respects. Tumor hypoxia and inflammation are prognostic in several types of cancers. Higher levels of VEGF secreted by tumor cells and stromal components helps in further mobilization of mesenchymal stem cells and precursors of inflammatory cells from the bone marrow. VEGF induces tumor angiogenesis. Consequently, VEGF inhibitors are effective in inhibiting tumor growth and metastasis. Blocking VEGF and its receptors has been much more effective than monotherapy. Gerber et al. showed that chimeric receptor containing first three Ig-like domains of VEGFR-1, common to both human and mouse, resulted in nearly complete suppression of tumor growth in a mouse model of human rhabdomyosacroma (Gerber et al. 2000). VEGF inhibitors combined with chemotherapy and radiotherapy resulted in better tumor suppression compared to either therapy alone (Lee et al. 2000; Jain 2001). Klement et al. found that combining a monoclonal neutralizing antibody (DC101) targeting the flk-1/KDR (VEGFR-2) with low dose vinblastine chemotherapy enhanced the anti-vascular effects (Klement et al. 2000). Currently, many VEGF inhibitors are in various phases of clinical development (antibodies to VEGF, VEGF-R, VEGF-neutralizing aptamers and inhibitors of receptor tyrosine kinases). One of the strategies to inhibit VEGF-VEGFR signaling is a humanized antibody against VEGF, Avastin (Bevacizumab) which has been approved by FDA for clinical use.
Hematological Malignancies
Similar to the solid tumors, VEGF is also expressed in a variety of hematological malignancies like T and B-cell lymphomas, acute and chronic myeloid leukemia, multiple myeloma and Burkitt’s lymphoma (Gerber and Ferrara 2003). VEGFR-1 and VEGFR-2 are also detected in some leukemia cell lines, however VEGFR-2 is found more frequently compared to VEGFR-1 (Dias et al. 2000). Additionally, a neutralizing monoclonal antibody IMC-1C11, specific to human VEGFR-2 inhibited proliferation of xenotransplanted human leukemia cells and increased survival in the mouse models (Smolich et al. 2001). Based on the promising results in pre-clinical trials, many of the VEGF inhibitors are currently being tested in the clinical trials of hematological malignancies.
Female Reproductive Tract
Polycystic ovarian syndrome (PCOS) is an important cause of female infertility characterized by hirsutism, obesity, polycystic ovaries and menstrual irregularities. Hyperplasia of stroma and theca contributes to the excessive androgen production responsible for the symptoms of PCOS. Stromal angiogenesis is an important feature of the polycystic ovaries. VEGF levels are elevated in PCOS patients, compared to healthy females (Peitsidis and Agrawal 2010). VEGF mRNA levels were also elevated in the cyst walls. An increased expression of VEGF is associated with increased ovarian stromal blood flow, which is believed to disrupt the ovarian autoregulatory mechanisms, thereby leading to uninhibited growth of all cohort follicles. These findings reflect that VEGF may be an important player in the pathogenesis of PCOS. Another important condition characterized by pathologic angiogenesis is endometriosis. Endometriosis is a condition, in which endometrial implants are found outside the uterine cavity. These ectopic implants develop vessels that enable them to survive and grow. Increased VEGF levels are observed in the peritoneal cavity of endometriosis patients (McLaren et al. 1996). Two VEGF inhibitors, a soluble truncated receptor (decoy) and an antibody to VEGF were recently studied in the mouse models and were found to result in significant reduction in the endometrial implant size (Kerbel and Folkman 2002; Hull et al. 2003). VEGF is implicated in pre-eclampsia, a serious obstetrics complication leading to hypertension, proteinuria, and glomerular endotheliosis. Increased circulating levels of soluble VEGFR1 (sFlt1) is observed in pre-eclampsia patients (Maynard et al. 2003). This study demonstrated that endothelial dysfunction was a result of decreased VEGF and PlGF as a consequence of increased levels of sFlt1. Exogenous administration of VEGF and PlGF reversed pre-eclampsia in model systems. Current studies are in progress to evaluate the use of VEGF therapy in pre-eclampsia patients.
Intraocular Neovascularization
The conditions associated with intraocular ischemia include diabetes, central retinal vein occlusion, prematurity and wet-type of age-related macular degeneration. These conditions subsequently lead to intraocular neovascularization which is associated with grave consequences including vitreous hemorrhages, retinal detachment and blindness (Chang et al. 2012; Ciulla and Rosenfeld 2009). Earlier studies have shown increase in VEGF in aqueous and vitreous humor associated with intraocular ischemia, contributing to the neovascular disorders. Various pre-clinical and clinical studies are currently investigating the effects of VEGF inhibitors in these conditions (Andreoli and Miller 2007). The VISION trial first showed that pegaptanib, an anti-VEGF agent was able to prevent vision loss in neovascular age related macular degeneration. Fab fragment of a humanized anti-VEGF antibody, Lucentis (Ranibizumab) is now clinically used to treat wet, age-related macular degeneration.
VEGF Therapies to Rescue Ischemic Tissues
The ability of VEGF to regulate pathologic angiogenesis in ischemic conditions, led to prospects in vascular occlusive diseases, in which restoration of blood flow is life saving. VEGF targeted therapies have been tried in myocardial infarction, peripheral limb ischemia and stroke (Gupta et al. 2009). An increase in vascularity was observed with adenoviral and plasmid liposome mediated delivery of VEGF165 in limb ischemia patients (Makinen et al. 2002). A double blind randomized controlled trial including diabetic patients with peripheral arterial disease also showed a significant improvement in symptoms in patients treated with intramuscular VEGF165(Kusumanto et al. 2006). Patients treated with VEGF had fewer amputations and more skin ulcer healing compared to the placebo group. RAVE, a double-blind trial assessing the intramuscular adenoviral gene transfer of VEGF121 however showed no improvement in the symptoms, in patients with limb ischemia (Rajagopalan et al. 2003). This raises important concerns regarding the difference in efficacy of the various isoforms of VEGF and the importance of consistent method to measure primary endpoint of improved perfusion in different trials.
A phase II KAT trial, assessing the efficacy of VEGF165 gene therapy in patients with coronary artery disease also found improvement in myocardial perfusion, but without any change in rate of restenosis (Hedman et al. 2003). Another trial, Euroinject One, showed significant improvement in the ventricular function in patients treated by intramyocardial VEGF165 plasmid gene transfer (Kastrup et al. 2005). However, the results in myocardial infarction have been disappointing in terms of improvement in myocardial stress perfusion and angina class. In an effort to improve these outcomes, Ripa et al. conducted a pilot study of combined VEGF165 gene therapy and stem cell mobilization (Ripa et al. 2006). There was still no improvement in primary end point of myocardial stress perfusion probably because of inadequate homing. Future trials should include co-transfer of plasmid encoding SDF-1, a homing factor, timely administration of G-CSF and a consistent method for perfusion assessment. Overall, current evidence suggests numerous factors that could result in lack of benefit in some efficacy end points. Several laboratories are currently investigating these factors and the effects of targeted VEGF therapies in various ischemic diseases.
Concluding Remarks
Hypoxia-induced changes at the transcriptome and inflammation-induced cytokine/chemokine responses modulate endothelial cells and innate immune cells. As cancer, cardiovascular and infectious diseases are modulated by hypoxia and inflammation, it provides an opportunity to develop novel drugs to intervene this co-dependent signaling pathways. Hypoxia inducible factor, HIF, is an evolutionarily conserved key transcription factor that regulates vascular biology, angiogenesis, metabolic reprogamming and inflammation. HIF is regulated at the cellular level by canonical pathway involving proteasomal degradation and non-canonical regulation involving HSP-70, NFkB and microRNA. HIF regulates recruitment of inflammatory cells, erythropoiesis, tissue remodeling, and pH homeostasis. Furthermore, HIF-mediated metabolic adaptation creates a niche for stem cells to thrive in a hypoxic microenvironment. One of the primary target genes regulated by HIF is VEGF-A. VEGF-family of growth factors is necessary for developmental angiogenesis, collateral vessel growth in peripheral arterial disease, stroke, and myocardial infraction. Inducing local production of VEGF is therapeutically useful in limiting hypoxia/ischemia-induced tissue damage. Conversely, pathological angiogenesis can be inhibited by strategies to neutralize VEGF or inhibit its signaling pathways. VEGF family of growth factors bind to three distinct receptors. Pleotropism in receptor binding and differences in tissue distribution of receptor/co-receptor offers therapeutic strategies to modulate VEGF signaling. VEGF-induced changes in inflammatory signals and its positive influence on neuroprotection provide a road map for the future development of VEGF-based therapies.
References
Alitalo K, Carmeliet P (2002) Molecular mechanisms of lymphangiogenesis in health and disease. Cancer Cell 1(3):219–227
Ando H et al (2013) A hypoxia-inducible factor (HIF)-3alpha splicing variant, HIF-3alpha4 impairs angiogenesis in hypervascular malignant meningiomas with epigenetically silenced HIF-3alpha4. Biochem Biophys Res Commun 433(1):139–144
Andreoli CM, Miller JW (2007) Anti-vascular endothelial growth factor therapy for ocular neovascular disease. Curr Opin Ophthalmol 18(6):502–508
Antoine M et al (2005) Upregulation of pleiotrophin expression in rat hepatic stellate cells by PDGF and hypoxia: implications for its role in experimental biliary liver fibrogenesis. Biochem Biophys Res Commun 337(4):1153–1164
Arnaud C, Poulain L, Levy P, Dematteis M (2011) Inflammation contributes to the atherogenic role of intermittent hypoxia in apolipoprotein-E knock out mice. Atherosclerosis 219(2):425–431
Barreiro O et al (2002) Dynamic interaction of VCAM-1 and ICAM-1 with moesin and ezrin in a novel endothelial docking structure for adherent leukocytes. J Cell Biol 157(7):1233–1245
Battaglia F et al (2008) Hypoxia transcriptionally induces macrophage-inflammatory protein-3alpha/CCL-20 in primary human mononuclear phagocytes through nuclear factor (NF)-kappaB. J Leukoc Biol 83(3):648–662
Bella J, Kolatkar PR, Marlor CW, Greve JM, Rossmann MG (1998) The structure of the two amino-terminal domains of human ICAM-1 suggests how it functions as a rhinovirus receptor and as an LFA-1 integrin ligand. Proc Natl Acad Sci U S A 95(8):4140–4145
Bellot G et al (2009) Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol 29(10):2570–2581
Bosco MC et al (2004) Hypoxia selectively inhibits monocyte chemoattractant protein-1 production by macrophages. J Immunol 172(3):1681–1690
Bosco MC et al (2006) Hypoxia modifies the transcriptome of primary human monocytes: modulation of novel immune-related genes and identification of CC-chemokine ligand 20 as a new hypoxia-inducible gene. J Immunol 177(3):1941–1955
Bosco MC et al (2008) Monocytes and dendritic cells in a hypoxic environment: Spotlights on chemotaxis and migration. Immunobiology 213(9–10):733–749
Braunwald E (2013) Cardiovascular science: opportunities for translating research into improved care. J Clin Invest 123(1):6–10
Carmeliet P et al (1996) Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 380(6573):435–439
Carmeliet P et al (2001) Synergism between vascular endothelial growth factor and placental growth factor contributes to angiogenesis and plasma extravasation in pathological conditions. Nat Med 7(5):575–583
Carriere V et al (2007) IL-33, the IL-1-like cytokine ligand for ST2 receptor, is a chromatin-associated nuclear factor in vivo. Proc Natl Acad Sci U S A 104(1):282–287
Carroll VA, Ashcroft M (2006) Role of hypoxia-inducible factor (HIF)-1alpha versus HIF-2alpha in the regulation of HIF target genes in response to hypoxia, insulin-like growth factor-I, or loss of von Hippel-Lindau function: implications for targeting the HIF pathway. Cancer Res 66(12):6264–6270
Chakravorty SJ, Craig A (2005) The role of ICAM-1 in plasmodium falciparum cytoadherence. Eur J Cell Biol 84(1):15–27
Chan SY et al (2009) MicroRNA-210 controls mitochondrial metabolism during hypoxia by repressing the iron-sulfur cluster assembly proteins ISCU1/2. Cell Metab 10(4):273–284
Chang JH et al (2012) Corneal neovascularization: an anti-VEGF therapy review. Surv Ophthalmol 57(5):415–429
Chen WY, Chang MS (2009) IL-20 is regulated by hypoxia-inducible factor and up-regulated after experimental ischemic stroke. J Immunol 182(8):5003–5012
Chen M, Geng JG (2006) P-selectin mediates adhesion of leukocytes, platelets, and cancer cells in inflammation, thrombosis, and cancer growth and metastasis. Arc Immunol Ther Exp 54(2):75–84
Chen R et al (2012) The acetylase/deacetylase couple CREB-binding protein/Sirtuin 1 controls hypoxia-inducible factor 2 signaling. J Biol Chem 287(36):30800–30811
Chistiakov DA, Sobenin IA, Orekhov AN (2012) Strategies to deliver microRNAs as potential therapeutics in the treatment of cardiovascular pathology. Drug Deliv 19(8):392–405
Cicchillitti L et al (2012) Hypoxia-inducible factor 1-alpha induces miR-210 in normoxic differentiating myoblasts. J Biol Chem 287(53):44761–44771
Ciulla TA, Rosenfeld PJ (2009) Anti-vascular endothelial growth factor therapy for neovascular ocular diseases other than age-related macular degeneration. Curr Opin Ophthalmol 20(3):166–174
Cleator JH, Zhu WQ, Vaughan DE, Hamm HE (2006) Differential regulation of endothelial exocytosis of P-selectin and von Willebrand factor by protease-activated receptors and cAMP. Blood 107(7):2736–2744
Cohen T et al (1995) VEGF121, a vascular endothelial growth factor (VEGF) isoform lacking heparin binding ability, requires cell-surface heparan sulfates for efficient binding to the VEGF receptors of human melanoma cells. J Biol Chem 270(19):11322–11326
Colgan SP, Taylor CT (2010) Hypoxia: an alarm signal during intestinal inflammation. Nat Rev Gastroenterol Hepatol 7(5):281–287
Dhondt J et al (2011) Neuronal FLT1 receptor and its selective ligand VEGF-B protect against retrograde degeneration of sensory neurons. FASEB J Off Publ Fed Am Soc Exp Biol 25(5):1461–1473
Dias S et al (2000) Autocrine stimulation of VEGFR-2 activates human leukemic cell growth and migration. J Clin Invest 106(4):511–521
Dioum EM et al (2009) Regulation of hypoxia-inducible factor 2alpha signaling by the stress-responsive deacetylase sirtuin 1. Science 324(5932):1289–1293
Dumont DJ et al (1998) Cardiovascular failure in mouse embryos deficient in VEGF receptor-3. Science 282(5390):946–949
Egger M et al (2007) Hypoxia up-regulates the angiogenic cytokine secretoneurin via an HIF-1alpha- and basic FGF-dependent pathway in muscle cells. FASEB J 21(11):2906–2917
Eltzschig HK, Carmeliet P (2011) Hypoxia and inflammation. N Engl J Med 364(7):656–665
Ema M et al (1997) A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1alpha regulates the VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci U S A 94(9):4273–4278
Falanga V et al (1991) Hypoxia upregulates the synthesis of TGF-beta 1 by human dermal fibroblasts. J Invest Dermatol 97(4):634–637
Favaro E et al (2010) MicroRNA-210 regulates mitochondrial free radical response to hypoxia and krebs cycle in cancer cells by targeting iron sulfur cluster protein ISCU. PloS ONE 5(4):e10345
Ferrara N (2004) Vascular endothelial growth factor: basic science and clinical progress. Endocr Rev 25(4):581–611
Ferrara N (2010) Binding to the extracellular matrix and proteolytic processing: two key mechanisms regulating vascular endothelial growth factor action. Mol Biol Cell 21(5):687–690
Ferrara N et al (1996) Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 380(6573):439–442
Florczyk U et al (2011) Opposite effects of HIF-1alpha and HIF-2alpha on the regulation of IL-8 expression in endothelial cells. Free Radic Biol Med 51(10):1882–1892
Fong GH, Rossant J, Gertsenstein M, Breitman ML (1995) Role of the Flt-1 receptor tyrosine kinase in regulating the assembly of vascular endothelium. Nature 376(6535):66–70
Frangogiannis NG, Smith CW, Entman ML (2002) The inflammatory response in myocardial infarction. Cardiovasc Res 53(1):31–47
Fu H, Luo F, Yang L, Wu W, Liu X (2010) Hypoxia stimulates the expression of macrophage migration inhibitory factor in human vascular smooth muscle cells via HIF-1alpha dependent pathway. BMC Cell Biol 11:66
Fukumura D et al (1998) Tumor induction of VEGF promoter activity in stromal cells. Cell 94(6):715–725
Gameiro PA et al (2013) In vivo HIF-mediated reductive carboxylation is regulated by citrate levels and sensitizes VHL-deficient cells to glutamine deprivation. Cell Metab 17(3):372–385
Ganat Y, Soni S, Chacon M, Schwartz ML, Vaccarino FM (2002) Chronic hypoxia up-regulates fibroblast growth factor ligands in the perinatal brain and induces fibroblast growth factor-responsive radial glial cells in the sub-ependymal zone. Neuroscience 112(4):977–991
Gerber HP, Ferrara N (2003) The role of VEGF in normal and neoplastic hematopoiesis. J Mol Med (Berl Germany) 81(1):20–31
Gerber HP, Kowalski J, Sherman D, Eberhard DA, Ferrara N (2000) Complete inhibition of rhabdomyosarcoma xenograft growth and neovascularization requires blockade of both tumor and host vascular endothelial growth factor. Cancer Res 60(22):6253–6258
Ghezzi P et al (1991) Hypoxia increases production of interleukin-1 and tumor necrosis factor by human mononuclear cells. Cytokine 3(3):189–194
Ghosh G et al (2010) Hypoxia-induced microRNA-424 expression in human endothelial cells regulates HIF-alpha isoforms and promotes angiogenesis. J Clin Invest 120(11):4141–4154
Gonsalves CS, Kalra VK (2010) Hypoxia-mediated expression of 5-lipoxygenase-activating protein involves HIF-1alpha and NF-kappaB and microRNAs 135a and 199a-5p. J Immunol 184(7):3878–3888
Greenberg DA, Jin K (2013) Vascular endothelial growth factors (VEGFs) and stroke. Cell Mol Life Sci 70(10):1753–1761
Gupta R, Tongers J, Losordo DW (2009) Human studies of angiogenic gene therapy. Circ Res 105(8):724–736
Hagberg CE et al (2010) Vascular endothelial growth factor B controls endothelial fatty acid uptake. Nature 464(7290):917–921
Hagberg CE et al (2012) Targeting VEGF-B as a novel treatment for insulin resistance and type 2 diabetes. Nature 490(7420):426–430
Hagberg C, Mehlem A, Falkevall A, Muhl L, Eriksson U (2013) Endothelial fatty acid transport: role of vascular endothelial growth factor B. Physiology 28(2):125–134
Hagerling R et al (2013) A novel multistep mechanism for initial lymphangiogenesis in mouse embryos based on ultramicroscopy. EMBO J 32(5):629–644
Haiko P et al (2008) Deletion of vascular endothelial growth factor C (VEGF-C) and VEGF-D is not equivalent to VEGF receptor 3 deletion in mouse embryos. Mol Cell Biol 28(15):4843–4850
Hara S et al (2006) Hypoxia enhances c-Met/HGF receptor expression and signaling by activating HIF-1alpha in human salivary gland cancer cells. Oral Oncol 42(6):593–598
Hedman M et al (2003) Safety and feasibility of catheter-based local intracoronary vascular endothelial growth factor gene transfer in the prevention of postangioplasty and in-stent restenosis and in the treatment of chronic myocardial ischemia: phase II results of the Kuopio Angiogenesis Trial (KAT). Circulation 107(21):2677–2683
Hedtjarn M et al (2002) Interleukin-18 involvement in hypoxic-ischemic brain injury. J Neurosci 22(14):5910–5919
Heiska L et al (1998) Association of ezrin with intercellular adhesion molecule-1 and −2 (ICAM-1 and ICAM-2). Regulation by phosphatidylinositol 4, 5-bisphosphate. J Biol Chem 273(34):21893–21900
Herz J et al (2012) Intracerebroventricularly delivered VEGF promotes contralesional corticorubral plasticity after focal cerebral ischemia via mechanisms involving anti-inflammatory actions. Neurobiol Dis 45(3):1077–1085
Hinkel R et al (2013) Inhibition of MicroRNA-92a protects against ischemia/reperfusion injury in a large-animal model. Circulation 128(10):1066–1075
Hirani N et al (2001) The regulation of interleukin-8 by hypoxia in human macrophages–a potential role in the pathogenesis of the acute respiratory distress syndrome (ARDS). Mol Med 7(10):685–697
Hiratsuka S, Minowa O, Kuno J, Noda T, Shibuya M (1998) Flt-1 lacking the tyrosine kinase domain is sufficient for normal development and angiogenesis in mice. Proc Natl Acad Sci U S A 95(16):9349–9354
Hitchon C et al (2002) Hypoxia-induced production of stromal cell-derived factor 1 (CXCL12) and vascular endothelial growth factor by synovial fibroblasts. Arthritis Rheum 46(10):2587–2597
Hogan BM et al (2009) Ccbe1 is required for embryonic lymphangiogenesis and venous sprouting. Nat Genet 41(4):396–398
Hong KH, Ryu J, Han KH (2005) Monocyte chemoattractant protein-1-induced angiogenesis is mediated by vascular endothelial growth factor-A. Blood 105(4):1405–1407
Horie N et al (2011) Transplanted stem cell-secreted VEGF effects post-stroke recovery, inflammation, and vascular repair. Stem Cells. doi:10.1002/stem.584
Hu F et al (2013) Hypoxia-inducible factor-1alpha and interleukin 33 form a regulatory circuit to perpetuate the inflammation in rheumatoid arthritis. PloS ONE 8(8):e72650
Huang RB, Eniola-Adefeso O (2012) Shear stress modulation of IL-1beta-induced E-selectin expression in human endothelial cells. PloS ONE 7(2):e31874
Hubbi ME et al (2013a) Chaperone-mediated autophagy targets hypoxia-inducible factor-1alpha (HIF-1alpha) for lysosomal degradation. J Biol Chem 288(15):10703–10714
Hubbi ME, Hu H, Kshitiz GDM, Semenza GL (2013b) Sirtuin-7 inhibits the activity of hypoxia-inducible factors. J Biol Chem 288(29):20768–20775
Hull ML et al (2003) Antiangiogenic agents are effective inhibitors of endometriosis. J Clin Endocrinol Metab 88(6):2889–2899
Hullinger TG et al (2012) Inhibition of miR-15 protects against cardiac ischemic injury. Circ Res 110(1):71–81
Iadecola C, Alexander M (2001) Cerebral ischemia and inflammation. Curr Opin Neurol 14(1):89–94
Iyer NV et al (1998) Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes Dev 12(2):149–162
Jain RK (2001) Normalizing tumor vasculature with anti-angiogenic therapy: a new paradigm for combination therapy. Nat Med 7(9):987–989
Janssen BJ et al (2012) Neuropilins lock secreted semaphorins onto plexins in a ternary signaling complex. Nat Struct Mol Biol 19(12):1293–1299
Jeltsch M, Leppanen VM, Saharinen P, Alitalo K (2013) Receptor tyrosine kinase-mediated angiogenesis. Cold Spring Harb Perspect Biol 5(9). doi:10.1101/cshperspect.a009183
Jeong HJ et al (2005) Hypoxia-induced IL-6 production is associated with activation of MAP kinase, HIF-1, and NF-kappaB on HEI-OC1 cells. Hear Res 207(1–2):59–67
Jung YJ, Isaacs JS, Lee S, Trepel J, Neckers L (2003) IL-1beta-mediated up-regulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J Off Publ Fed Am Soc Exp Biol 17(14):2115–2117
Kaipainen A et al (1995) Expression of the fms-like tyrosine kinase 4 gene becomes restricted to lymphatic endothelium during development. Proc Natl Acad Sci U S A 92(8):3566–3570
Karakurum M et al (1994) Hypoxic induction of interleukin-8 gene expression in human endothelial cells. J Clin Invest 93(4):1564–1570
Karkkainen MJ et al (2004) Vascular endothelial growth factor C is required for sprouting of the first lymphatic vessels from embryonic veins. Nat Immunol 5(1):74–80
Kastrup J et al (2005) Direct intramyocardial plasmid vascular endothelial growth factor-A165 gene therapy in patients with stable severe angina pectoris A randomized double-blind placebo-controlled study: the Euroinject One trial. J Am Coll Cardiol 45(7):982–988
Kelly TJ, Souza AL, Clish CB, Puigserver P (2011) A hypoxia-induced positive feedback loop promotes hypoxia-inducible factor 1alpha stability through miR-210 suppression of glycerol-3-phosphate dehydrogenase 1-like. Mol Cell Biol 31(13):2696–2706
Kerbel R, Folkman J (2002) Clinical translation of angiogenesis inhibitors. Nat Rev Cancer 2(10):727–739
Kim J et al (2008) Hypoxia-induced IL-18 increases hypoxia-inducible factor-1alpha expression through a Rac1-dependent NF-kappaB pathway. Mol Biol Cell 19(2):433–444
Kim JY, Kim N, Zheng Z, Lee JE, Yenari MA (2013) The 70 kDa heat shock protein protects against experimental traumatic brain injury. Neurobiol Dis 58:289–295
Kiss J et al (2012) Loss of the oxygen sensor PHD3 enhances the innate immune response to abdominal sepsis. J Immunol 189(4):1955–1965
Klement G et al (2000) Continuous low-dose therapy with vinblastine and VEGF receptor-2 antibody induces sustained tumor regression without overt toxicity. J Clin Invest 105(8):R15–R24
Kotch LE, Iyer NV, Laughner E, Semenza GL (1999) Defective vascularization of HIF-1alpha-null embryos is not associated with VEGF deficiency but with mesenchymal cell death. Dev Biol 209(2):254–267
Kulshreshtha R, Davuluri RV, Calin GA, Ivan M (2008) A microRNA component of the hypoxic response. Cell Death Differ 15(4):667–671
Kurihara T, Westenskow PD, Bravo S, Aguilar E, Friedlander M (2012) Targeted deletion of Vegfa in adult mice induces vision loss. J Clin Invest 122(11):4213–4217
Kurokawa M et al (2013) Interleukin-33-activated dendritic cells induce the production of thymus and activation-regulated chemokine and macrophage-derived chemokine. Int Arch Allergy Immunol 161(Suppl 2):52–57
Kusumanto YH et al (2006) Treatment with intramuscular vascular endothelial growth factor gene compared with placebo for patients with diabetes mellitus and critical limb ischemia: a double-blind randomized trial. Hum Gene Ther 17(6):683–691
Lee CG et al (2000) Anti-Vascular endothelial growth factor treatment augments tumor radiation response under normoxic or hypoxic conditions. Cancer Res 60(19):5565–5570
Leung DW, Cachianes G, Kuang WJ, Goeddel DV, Ferrara N (1989) Vascular endothelial growth factor is a secreted angiogenic mitogen. Science 246(4935):1306–1309
Lim JH et al (2010) Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol Cell 38(6):864–878
Loscalzo J (2010) The cellular response to hypoxia: tuning the system with microRNAs. J Clin Invest 120(11):3815–3817
Makinen K et al (2002) Increased vascularity detected by digital subtraction angiography after VEGF gene transfer to human lower limb artery: a randomized, placebo-controlled, double-blinded phase II study. Mol Ther J Am Soc Gene Ther 6(1):127–133
Makino Y et al (2007) Transcriptional up-regulation of inhibitory PAS domain protein gene expression by hypoxia-inducible factor 1 (HIF-1): a negative feedback regulatory circuit in HIF-1-mediated signaling in hypoxic cells. J Biol Chem 282(19):14073–14082
Marchant DJ et al (2012) Inflammation in myocardial diseases. Circ Res 110(1):126–144
Marsch E, Sluimer JC, Daemen MJ (2013) Hypoxia in atherosclerosis and inflammation. Curr Opin Lipidol 24(5):393–400
Marumo T, Schini-Kerth VB, Busse R (1999) Vascular endothelial growth factor activates nuclear factor-kappaB and induces monocyte chemoattractant protein-1 in bovine retinal endothelial cells. Diabetes 48(5):1131–1137
Maynard SE et al (2003) Excess placental soluble fms-like tyrosine kinase 1 (sFlt1) may contribute to endothelial dysfunction, hypertension, and proteinuria in preeclampsia. J Clin Invest 111(5):649–658
Maynard MA et al (2005) Human HIF-3alpha4 is a dominant-negative regulator of HIF-1 and is down-regulated in renal cell carcinoma. FASEB J Off Publ Fed Am Soc Exp Biol 19(11):1396–1406
McCormick RI et al (2013) miR-210 is a target of hypoxia-inducible factors 1 and 2 in renal cancer, regulates ISCU and correlates with good prognosis. Br J Cancer 108(5):1133–1142
McEver RP, Beckstead JH, Moore KL, Marshall-Carlson L, Bainton DF (1989) GMP-140, a platelet alpha-granule membrane protein, is also synthesized by vascular endothelial cells and is localized in Weibel-Palade bodies. J Clin Invest 84(1):92–99
McLaren J et al (1996) Vascular endothelial growth factor is produced by peritoneal fluid macrophages in endometriosis and is regulated by ovarian steroids. J Clin Invest 98(2):482–489
Metinko AP, Kunkel SL, Standiford TJ, Strieter RM (1992) Anoxia-hyperoxia induces monocyte-derived interleukin-8. J Clin Invest 90(3):791–798
Miller JW, Le Couter J, Strauss EC, Ferrara N (2013) Vascular endothelial growth factor a in intraocular vascular disease. Ophthalmology 120(1):106–114
Moussion C, Ortega N, Girard JP (2008) The IL-1-like cytokine IL-33 is constitutively expressed in the nucleus of endothelial cells and epithelial cells in vivo: a novel ‘alarmin’? PloS ONE 3(10):e3331
Murakami M et al (2006) Signaling of vascular endothelial growth factor receptor-1 tyrosine kinase promotes rheumatoid arthritis through activation of monocytes/macrophages. Blood 108(6):1849–1856
Murdoch C, Muthana M, Lewis CE (2005) Hypoxia regulates macrophage functions in inflammation. J Immunol 175(10):6257–6263
Nimrichter L et al (2008) E-selectin receptors on human leukocytes. Blood 112(9):3744–3752
Noels H, Weber C (2011) Catching up with important players in atherosclerosis: type I interferons and neutrophils. Curr Opin Lipidol 22(2):144–145
Oosthuyse B et al (2001) Deletion of the hypoxia-response element in the vascular endothelial growth factor promoter causes motor neuron degeneration. Nat Genet 28(2):131–138
Peitsidis P, Agrawal R (2010) Role of vascular endothelial growth factor in women with PCO and PCOS: a systematic review. Reprod BioMed Online 20(4):444–452
Pellet-Many C, Frankel P, Jia H, Zachary I (2008) Neuropilins: structure, function and role in disease. Biochem J 411(2):211–226
Peng H et al (2012) MicroRNA-31 targets FIH-1 to positively regulate corneal epithelial glycogen metabolism. FASEB J 26(8):3140–3147
Pritchard-Jones RO et al (2007) Expression of VEGF(xxx)b, the inhibitory isoforms of VEGF, in malignant melanoma. Br J Cancer 97(2):223–230
Pulkkinen K, Malm T, Turunen M, Koistinaho J, Yla-Herttuala S (2008) Hypoxia induces microRNA miR-210 in vitro and in vivo ephrin-A3 and neuronal pentraxin 1 are potentially regulated by miR-210. FEBS Lett 582(16):2397–2401
Rajagopalan S et al (2003) Regional angiogenesis with vascular endothelial growth factor in peripheral arterial disease: a phase II randomized, double-blind, controlled study of adenoviral delivery of vascular endothelial growth factor 121 in patients with disabling intermittent claudication. Circulation 108(16):1933–1938
Ramakrishnan S (2011) Hydrogel-siRNA for cancer therapy. Cancer Biol Ther 11(9):849–851
Rane S et al (2009) Downregulation of miR-199a derepresses hypoxia-inducible factor-1alpha and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ Res 104(7):879–886
Ricciardi A et al (2008) Transcriptome of hypoxic immature dendritic cells: modulation of chemokine/receptor expression. Mol Cancer Res 6(2):175–185
Ripa RS et al (2006) Intramyocardial injection of vascular endothelial growth factor-A165 plasmid followed by granulocyte-colony stimulating factor to induce angiogenesis in patients with severe chronic ischaemic heart disease. Eur Heart J 27(15):1785–1792
Rius J et al (2008) NF-kappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 453(7196):807–811
Sakurai Y, Ohgimoto K, Kataoka Y, Yoshida N, Shibuya M (2005) Essential role of Flk-1 (VEGF receptor 2) tyrosine residue 1173 in vasculogenesis in mice. Proc Natl Acad Sci U S A 102(4):1076–1081
Sawano A, Takahashi T, Yamaguchi S, Aonuma M, Shibuya M (1996) Flt-1 but not KDR/Flk-1 tyrosine kinase is a receptor for placenta growth factor, which is related to vascular endothelial growth factor. Cell Growth Differ Mol Biol J Am Assoc Cancer Res 7(2):213–221
Sawano A, Takahashi T, Yamaguchi S, Shibuya M (1997) The phosphorylated 1169-tyrosine containing region of flt-1 kinase (VEGFR-1) is a major binding site for PLCgamma. Biochem Biophys Res Commun 238(2):487–491
Scannell G et al (1993) Hypoxia induces a human macrophage cell line to release tumor necrosis factor-alpha and its soluble receptors in vitro. J Surg Res 54(4):281–285
Semenza GL (2013) HIF-1 mediates metabolic responses to intratumoral hypoxia and oncogenic mutations. J Clin Invest 123(9):3664–3671
Semenza GL, Wang GL (1992) A nuclear factor induced by hypoxia via de novo protein synthesis binds to the human erythropoietin gene enhancer at a site required for transcriptional activation. Mol Cell Biol 12(12):5447–5454
Senger DR et al (1983) Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 219(4587):983–985
Shibuya M (2011) Vascular endothelial growth factor (VEGF) and its receptor (VEGFR) signaling in angiogenesis: a crucial target for anti- and pro-angiogenic therapies. Genes Cancer 2(12):1097–1105
Shin DH et al (2009) Inhibitor of nuclear factor-kappaB alpha derepresses hypoxia-inducible factor-1 during moderate hypoxia by sequestering factor inhibiting hypoxia-inducible factor from hypoxia-inducible factor 1alpha. FEBS J 276(13):3470–3480
Simon MP, Tournaire R, Pouyssegur J (2008) The angiopoietin-2 gene of endothelial cells is up-regulated in hypoxia by a HIF binding site located in its first intron and by the central factors GATA-2 and Ets-1. J Cell Physiol 217(3):809–818
Simons D et al (2011) Hypoxia-induced endothelial secretion of macrophage migration inhibitory factor and role in endothelial progenitor cell recruitment. J Cell Mol Med 15(3):668–678
Skuli N et al (2012) Endothelial HIF-2alpha regulates murine pathological angiogenesis and revascularization processes. J Clin Invest 122(4):1427–1443
Smith DF, Galkina E, Ley K, Huo Y (2005) GRO family chemokines are specialized for monocyte arrest from flow. Am J Physiol Heart Circ Physiol 289(5):H1976–H1984
Smolich BD et al (2001) The antiangiogenic protein kinase inhibitors SU5416 and SU6668 inhibit the SCF receptor (c-kit) in a human myeloid leukemia cell line and in acute myeloid leukemia blasts. Blood 97(5):1413–1421
Somers WS, Tang J, Shaw GD, Camphausen RT (2000) Insights into the molecular basis of leukocyte tethering and rolling revealed by structures of P- and E-selectin bound to SLe(X) and PSGL-1. Cell 103(3):467–479
Son J et al (2013) Glutamine supports pancreatic cancer growth through a KRAS-regulated metabolic pathway. Nature 496(7443):101–105
Sowter HM, Raval RR, Moore JW, Ratcliffe PJ, Harris AL (2003) Predominant role of hypoxia-inducible transcription factor (Hif)-1alpha versus Hif-2alpha in regulation of the transcriptional response to hypoxia. Cancer Res 63(19):6130–6134
Sun Y et al (2003) VEGF-induced neuroprotection, neurogenesis, and angiogenesis after focal cerebral ischemia. J Clin Invest 111(12):1843–1851
Taguchi A et al (2008) Identification of hypoxia-inducible factor-1 alpha as a novel target for miR-17-92 microRNA cluster. Cancer Res 68(14):5540–5545
Takahashi M (2013) Role of innate immune system in inflammation and cardiac remodeling after myocardial infarction. Curr Vasc Pharmacol (in press)
Takahashi T, Ueno H, Shibuya M (1999) VEGF activates protein kinase C-dependent, but Ras-independent Raf-MEK-MAP kinase pathway for DNA synthesis in primary endothelial cells. Oncogene 18(13):2221–2230
Takahashi T, Yamaguchi S, Chida K, Shibuya M (2001) A single autophosphorylation site on KDR/Flk-1 is essential for VEGF-A-dependent activation of PLC-gamma and DNA synthesis in vascular endothelial cells. EMBO J 20(11):2768–2778
Takyar S et al (2013) VEGF controls lung Th2 inflammation via the miR-1-Mpl (myeloproliferative leukemia virus oncogene)-P-selectin axis. J Exp Med 210(10):1993–2010
Tanaka H et al (2013) MicroRNA-183 upregulates HIF-1alpha by targeting isocitrate dehydrogenase 2 (IDH2) in glioma cells. J Neurooncol 111(3):273–283
ten Freyhaus H et al (2011) Hypoxia enhances platelet-derived growth factor signaling in the pulmonary vasculature by down-regulation of protein tyrosine phosphatases. Am J Respir Crit Care Med 183(8):1092–1102
Tewari R, Choudhury SR, Ghosh S, Mehta VS, Sen E (2012) Involvement of TNFalpha-induced TLR4-NF-kappaB and TLR4-HIF-1alpha feed-forward loops in the regulation of inflammatory responses in glioma. J Mol Med 90(1):67–80
Tian H, McKnight SL, Russell DW (1997) Endothelial PAS domain protein 1 (EPAS1), a transcription factor selectively expressed in endothelial cells. Genes Dev 11(1):72–82
Tian H, Hammer RE, Matsumoto AM, Russell DW, McKnight SL (1998) The hypoxia-responsive transcription factor EPAS1 is essential for catecholamine homeostasis and protection against heart failure during embryonic development. Genes Dev 12(21):3320–3324
Torii S et al (2011) Pro-apoptotic activity of inhibitory PAS domain protein (IPAS), a negative regulator of HIF-1, through binding to pro-survival Bcl-2 family proteins. Cell Death Differ 18(11):1711–1725
Trayhurn P (2013) Hypoxia and adipose tissue function and dysfunction in obesity. Physiol Rev 93(1):1–21
Tuttolomondo A et al (2012) Inflammation in ischemic stroke subtypes. Curr Pharm Des 18(28):4289–4310
van den Boorn JG, Dassler J, Coch C, Schlee M, Hartmann G (2013) Exosomes as nucleic acid nanocarriers. Adv Drug Deliv Rev 65(3):331–335
Wahamaa H et al (2011) High mobility group box protein 1 in complex with lipopolysaccharide or IL-1 promotes an increased inflammatory phenotype in synovial fibroblasts. Arthritis Res Ther 13(4):R136
Wang Y et al (2009) Regulation of endocytosis via the oxygen-sensing pathway. Nat Med 15(3):319–324
Wang C et al (2011a) Underexpressed microRNA-199b-5p targets hypoxia-inducible factor-1alpha in hepatocellular carcinoma and predicts prognosis of hepatocellular carcinoma patients. J Gastroenterol Hepatol 26(11):1630–1637
Wang F et al (2011b) RACK1 regulates VEGF/Flt1-mediated cell migration via activation of a PI3K/Akt pathway. J Biol Chem 286(11):9097–9106
Wang Y et al (2012) Hypoxia promotes ligand-independent EGF receptor signaling via hypoxia-inducible factor-mediated upregulation of caveolin-1. Proc Natl Acad Sci U S A 109(13):4892–4897
Weber C, Noels H (2011) Atherosclerosis: current pathogenesis and therapeutic options. Nat Med 17(11):1410–1422
Wei Y, Schober A, Weber C (2013) Pathogenic arterial remodeling: the good and bad of microRNAs. Am J Physiol Heart Circ Physiol 304(8):H1050–H1059
White JR et al (2004) Genetic amplification of the transcriptional response to hypoxia as a novel means of identifying regulators of angiogenesis. Genomics 83(1):1–8
Winning S, Splettstoesser F, Fandrey J, Frede S (2010) Acute hypoxia induces HIF-independent monocyte adhesion to endothelial cells through increased intercellular adhesion molecule-1 expression: the role of hypoxic inhibition of prolyl hydroxylase activity for the induction of NF-kappa B. J Immunol 185(3):1786–1793
Woltmann G, McNulty CA, Dewson G, Symon FA, Wardlaw AJ (2000) Interleukin-13 induces PSGL-1/P-selectin-dependent adhesion of eosinophils, but not neutrophils, to human umbilical vein endothelial cells under flow. Blood 95(10):3146–3152
Yang L et al (2005) ICAM-1 regulates neutrophil adhesion and transcellular migration of TNF-alpha-activated vascular endothelium under flow. Blood 106(2):584–592
Yasuda K et al (2012) Contribution of IL-33-activated type II innate lymphoid cells to pulmonary eosinophilia in intestinal nematode-infected mice. Proc Natl Acad Sci U S A 109(9):3451–3456
Zampetaki A, Dudek K, Mayr M (2013) Oxidative stress in atherosclerosis: the role of microRNAs in arterial remodeling. Free Radic Biol Med 64:69–77
Zechariah A et al (2013a) Vascular endothelial growth factor promotes pericyte coverage of brain capillaries, improves cerebral blood flow during subsequent focal cerebral ischemia, and preserves the metabolic penumbra. Stroke J Cereb Circ 44(6):1690–1697
Zechariah A et al (2013b) Hyperlipidemia attenuates vascular endothelial growth factor-induced angiogenesis, impairs cerebral blood flow, and disturbs stroke recovery via decreased pericyte coverage of brain endothelial cells. Arterioscler Thromb Vasc Biol 33(7):1561–1567
Zhang H et al (2008) Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 283(16):10892–10903
Zheng Z, Yenari MA (2004) Post-ischemic inflammation: molecular mechanisms and therapeutic implications. Neurol Res 26(8):884–892
Zou X et al (2005) PSGL-1 derived from human neutrophils is a high-efficiency ligand for endothelium-expressed E-selectin under flow. Am J Physiol Cell Physiol 289(2):C415–C424
Acknowledgments
This work was supported in part by the following grants, DA012104, DA033881, DA034582, DA031202 and Sparboe Endowment for Women’s Cancer Research.
Conflict of interest
There is no conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Ramakrishnan, S., Anand, V. & Roy, S. Vascular Endothelial Growth Factor Signaling in Hypoxia and Inflammation. J Neuroimmune Pharmacol 9, 142–160 (2014). https://doi.org/10.1007/s11481-014-9531-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11481-014-9531-7