
Abstract
Hypoxia is an important regulator of angiogenesis and it stimulates neovascularization from existing blood vessels. At the molecular level, it occurs mostly through transcriptional regulation of genes which contain a core consensus sequence called hypoxia response element (HRE) via hypoxia inducible factors (HIFs) action. The discovery of HIFs hydroxylases as a family of dioxygenases that regulate HIFs dependently on the oxygen availability have significantly improved our understanding of the mechanisms of hypoxia signaling. Moreover, a broad number of factors have been shown to influence HIF stability and their effects could be mediated via several possible mechanisms including nitrosylation, deacetylation, or oxidation. The induction of HIF leads to the complex regulation of the pro- and antiangiogenic factors and extensive research will be essential for thorough understanding of the role of hypoxia in disease development and will help to identify new therapeutic targets for treatment of such hypoxia-dependent disorders.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others


Keywords
1 Introduction
Oxygen is vital for living cells and it plays a fundamental role in their metabolism. An inadequate oxygen supply to tissues and cells causes hypoxia, a state which can restrict their functions. The ability to maintain oxygen homeostasis is essential to the survival of all vertebrate species. On the other hand, several pathological conditions like stroke (cerebral ischemia) and heart infarction (myocardial ischemia) may result from the detrimental effect of hypoxia. Moreover, hypoxia has been recognized as one of the fundamentally important features of tumor growth and metastasis.
The hypoxic response is primarily mediated by the family of hypoxia inducible transcription factors (HIFs). Their activation leads to transcription of HIFs target genes—nowadays more than 150 HIFs-dependent genes controlling different processes including energy metabolism, cell growth and apoptosis, or vasomotor control was identified (Fig. 8.1). Moreover, to counteract the undesirable effects of low oxygen concentration, the new blood vessels providing oxygen to the ischemic tissues are formed. The expression of pro-angiogenic factors like vascular endothelial growth factor (VEGF) or interleukin-8 (IL-8) was thought to be strongly increased in hypoxic conditions; however, recent studies underline the complexity of angiogenic response after HIFs induction. The complication of cellular response to low oxygen concentration may be also related to context specific effects of individual HIF isoforms. Latest research underlined that HIF-1 and HIF-2 possess both overlapping and unique target genes and may also trigger specific roles: HIF-1 predominantly drives the initial response to hypoxia (<24 h), whereas HIF-2 is responsible for chronic response (>24 h).

Selected HIF-dependent genes. HIFs transactivate genes involved in a number of adaptive processes in response to hypoxia including angiogenesis, energy metabolism, cell migration, proliferation, etc.
2 Stabilization of the Hypoxia Inducible Factor
2.1 The Hydroxylation of HIF-α as a Major Mechanism for Its Destabilization
The discovery of the hypoxia response element (HRE), the oxygen-regulated sequence, in the erythropoietin gene and the identification of the hypoxia inducible factor-1 (HIF-1) as a master transcription factor binding this sequence [1, 2] had started the intensive research in hypoxia field. Further studies performed in other than erythropoietin-producing cells have revealed that the same regulatory element is involved in the hypoxic regulation of gene expression indicating the presence of the general oxygen-sensing system in mammalian cells.
It is now well established that the regulatory sequence, called HRE, comprises of consensus-binding site (HBS, HIF-1-binding site) with a core motif (RCGTG) [3] and HIF-1 ancillary sequence (HAS), located eight to nine nt down- or upstream of HBS, which facilitates HIF-1-mediated transcription activation [4]. The existence of HRE was confirmed in hundreds of genes regulated in hypoxic conditions.
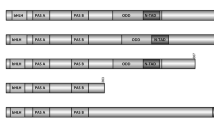
HRE is recognized by HIFs transcription factors, a heterodimers consisting of oxygen-labile α (HIF-α) and oxygen-independent β (HIF-β, also called aryl hydrocarbon receptor nuclear translocator, ARNT) subunits. Both α and β subunits are members of the bHLH/PAS (Basic Helix–Loop–Helix/PER-ARNT-SIM) domain family [5]. Three genes encoding distinct HIF-α isoforms exist in humans: HIF1A, encoding HIF-1α; EPAS1, encoding HIF-2α; and HIF3A, which is expressed as multiple HIF-3α splice variants [5–7]. Mainly HIF-1α and HIF-2α mediate the hypoxia-dependent signaling, whereas HIF-3α was shown to inhibit transcriptional activity of HIF-1α [8]. Such difference in isoform-specific effects may be the result of their structure: HIF-3α has high similarity to HIF-1α and HIF-2α in the bHLH and PAS domains but it lacks the C-terminal transactivation domain (C-TAD), which is required for transcriptional activation (Fig. 8.2).

The schematic structure of HIF-α isoforms. HIF-1α is closely related to HIF-2α, whereas the variant of HIF-3α, HIF-3α1 lacks C-terminal transactivation domain (C-TAD), which is required for transcriptional activation. However all α subunits are prone to oxygen-dependent degradation by the von Hippel–Lindau protein (pVHL) E3 ligase complex. The modifications of specific residues are highlighted above each protein, and the proteins that perform those modifications are shown (PHDs, FIH-1)
Only HIF-β subunit is constitutively expressed and not regulated by oxygen concentration. On the other hand, the expression of HIF-α subunits is strictly controlled at the protein level. This occurs, predominantly, through the hydroxylation of specific proline residues (Pro402 and Pro564 in HIF-1, Pro405 and Pro531 in HIF-2, Pro490 in HIF-3) within oxygen-dependent degradation domain (ODDD) present in α subunits [7, 9, 10]. The hydroxylation is performed by a class of enzymes called prolyl hydroxylases (PHDs). They require molecular O2, 2-oxoglutarate, iron ions (Fe2+), and ascorbic acid to be fully active, and they are inhibited in hypoxic conditions [11]. From three PHDs identified so far and shown to hydroxylate HIF-1α in vitro (PHD1, 2, and 3) [12], in vivo PHD2 isoform plays a major role in normoxic HIF-1α regulation [13, 14]. Hydroxylated HIF-α is recognized by the β-domain of von Hippel–Lindau tumor suppressor protein (pVHL) and is targeted for degradation by the 26S proteasome [15–17]. This process is preceded by the ubiquitination of the pVHLα domain by the elongin-C/elongin-B/cullin-2 E3-ubiquitin-ligase complex. The requirement of oxygen for PHDs functioning suggests that under hypoxic condition their activity is abrogated. In fact, when the oxygen level drops down, HIF-α subunit escapes recognition by the pVHL ubiquitin–ligase complex and proteasomal degradation and could be transported to the nucleus. There, after dimerization with HIF-1β [18], the active complex binds to HRE to induce transcription of numerous hypoxia-responsive genes (Fig. 8.3).

HIFs stabilization during hypoxia. Under normoxia, HIF-α is subjected to oxygen-dependent prolyl hydroxylation by PHDs leading to pVHL E3 ligase complex binding and ubiquitination and degradation by the 26S proteasome. Moreover, asparagine hydroxylation by FIH-1 inhibits binding of the HIFα coactivator p300/CBP. In hypoxic conditions, both PHDs and FIH-1 are not active and non-hydroxylated HIF-1α associates with HIFβ in nucleus. HIFα/HIFβ heterodimer binds to HRE sequence at the promoters of HIF-responsive genes and upon binding to the coactivators p300/CBP initiates their transcription
Importantly, there are also other mechanisms responsible for the controlling of the HIF-mediated transcription. To form an active HIF-α/β complex able to transactivate numerous genes, the binding of specific coactivators, like p300/CREB-binding protein (CBP) is necessary. Other than PHDs, oxygen-regulated hydroxylase-domain protein termed factor-inhibiting HIF-1 (FIH-1) regulates the binding of p300/CBP. FIH-1 modifies asparagine residue (Asn803 in human HIF-1α, Asn851 in HIF-2α) in the C-TAD in the presence of oxygen, what leads to blockage of recruitment of p300/CBP coactivator to HIF-α [19]. The regulation of HIF-α by FIH-1 leads to the inhibition of interactions between HIF-α and transcriptional coactivators via sterical hindrance caused by Asn hydroxylation. Similarly to PHDs, FIH-1 activity is inhibited under hypoxic conditions, allowing the binding of p300/CBP to HIF-1/2α, thus increasing HIF transactivation (Fig. 8.3).
2.2 Not Only Hydroxylation: ROS-Mediated HIFs Stabilization
In the past, reactive oxygen species (ROS) have been considered only as toxic by-products of metabolic processes. It is also believed now that abnormally high levels of ROS contribute to the oxidative stress-mediated damage leading to the development of many diseases. However, recent studies underline the importance of the low levels of ROS as a part of homeostatic signaling pathway. One of the ROS-regulated pathway is the HIF signaling.
A numerous studies have presented so-called ROS hypothesis and underlined the significance of ROS for HIF-1α stabilization [20–22]. It was proposed that hypoxia-mediated production of superoxide at complex III of the mitochondrial electron transport chain and its conversion to H2O2 by the superoxide dismutase (SOD) directly inhibits PHDs enzymes by oxidizing the essential nonheme-bound iron. However, recently Masson and colleagues have shown that PHDs have low sensitivity to inhibition by H2O2. In contrast, the other HIF-hydroxylated enzyme, FIH-1, was much more susceptible to H2O2-dependent inactivation [23]. These results suggest that molecular mechanisms responsible for the interaction between ROS and HIFs still have to be investigated.
On the other hand, there are other observations linking ROS and PHDs/HIF. Sirtuins are NAD-dependent deacetylases involved in metabolism, stress response, and longevity [24]. It was recently reported that the member of sirtuins family, SIRT3, destabilizes HIF-1α by inhibiting ROS production and keeping PHDs in the active state, thereby leading to HIF-1α degradation. In SIRT3 null cells, ROS level is increased and it contributed to increased HIF-1α stabilization/activity and finally to increased glycolysis and cellular growth [25]. Additionally, Bell et al. [26] proved that SIRT3 modulates the progression of tumors in ROS- and HIF-dependent way. The absence of SIRT3 led to increased ROS level and to activation of pro-tumorigenic HIF-1α in normoxia and its hyperactivation in hypoxia. After injection of HCT116 colorectal carcinoma cells with stable knockdown of SIRT3 to immunodeficient Nu/Nu mice, the growing tumors were bigger than tumors derived from control HCT116 cells. Additionally, when injected mice were subjected to potent antioxidant, N-acetylcysteine (NAC), the rate of tumor growth and its size was comparable to control conditions [26]. Moreover, there are other studies suggesting the effect of SIRT3 on ROS production. SIRT3 can directly target isocitrate dehydrogenase 2 (IDH2), a major source of NADPH in the mitochondria [27] and SOD2, activating mitochondrial ROS scavenging [28] and consequently influence cellular redox status and affect HIF signaling.
2.3 Not Only Hydroxylation: The Role of (De)acetylation in HIFs (De)stabilization
As mentioned above, SIRT3, a member of deacetylases family may regulate HIF stabilization through ROS- and PHD-dependent signaling. However, the action of SIRT3 and other members of sirtuins family may also rely on the direct deacetylation of HIFs protein (Fig. 8.4).

The regulation of angiogenic factors by sirtuins. SIRT1 and SIRT6 have direct, complex effect on several factors involved in the angiogenic response, as they inhibit pro-angiogenic HIF-1α, NFκB, as well as anti-angiogenic Foxo1 with concomitant upregulation of HIF-2α. SIRT3 indirectly, via inhibition of ROS, through several mechanisms including deacetylation of SOD2 and IDH2 proteins, activates PHDs leading to inhibition of HIF-1α
Interestingly, it was shown that specific members of sirtuins family may exert different effects on HIF transcription factors. Deacetylation of HIF-1α at Lys674 by SIRT1 led to blocking p300 recruitment and consequently this repressed HIF-1 target genes [29]. On the other hand, the activity of HIF-2α increases after deacetylation by the same nuclear SIRT1 [30]. Moreover, the expression of HIF-2α-dependent genes, namely, VEGF, SOD2, and erythropoietin was also induced in response to SIRT1 action [30]. SIRT1 was also able to downregulate PHD2 protein level through deacetylase activity [31]. Importantly, SIRT1 is regulated by hypoxia in HIF-1α- and HIF-2α-dependent manner what suggests both positive and negative feedback loops between SIRT1 and HIFs [32]. Moreover, SIRT6 was also shown to be a negative regulator of HIF-1α stability and protein as SIRT6-deficient cells exhibit increased HIF-α activity [33].
Additionally, the direct involvement of sirtuins in the controlling angiogenic response was underlined. SIRT1 was found to be highly expressed in the vasculature during blood vessel growth, where via deacetylation of the forkhead transcription factor Foxo1, a crucial negative regulator of blood vessel development inhibits its anti-angiogenic activity [34]. Moreover, the knockout of SIRT1 led to dysregulation of number of genes essential for vascular growth, maturation, and remodeling resulting in the defective blood vessel formation and blunted ischemia-induced neovascularization [34].
Sirtuins represent class III histone deacetylases (HDACs) enzymes and also other members of HDACs have been reported to interact with and influence HIF-1 activity. HDAC4 was found to modulate HIF-1α protein N-terminal lysine acetylation level, stability, and HIF-1 activity [35]. On the other hand, inhibitors of class I/II HDACs (HDACIs), like valproic acid or trichostatin A, have been shown to downregulate HIF signaling by either reducing functional HIF-1α levels or repressing HIF-α transactivation activity (reviewed in [36]).
The aberrant expression of HDACs was implicated in the cancer development and the extensive number of HDAC inhibitors have been shown to exert the antitumor effects. The molecular mechanism of the observed action may involve HIFs regulation, however, as deacetylases play a role in numerous processes like transcriptional regulation, epigenetic programming, chromosomal remodeling and they regulate plethora of factors, HIFs modification represents only one possibility.
3 The Cross Talk Between HIFs and NFκB: The Implications for Inflammatory Angiogenesis
Inflammatory reaction is not only characterized by the excessive expression of inflammatory cytokines like TNF-α or IL-1β but is also associated with reduced oxygen tension [37]. It was shown that various inflammatory stimuli like lipopolysaccharide (LPS) [38] or pro-inflammatory cytokines [39–41] are able to induce HIF system under normoxic conditions. Such situation may be especially important during tumor progression as inflammation can facilitate the development of cancer [42, 43]. The cross talk between HIFs and inflammatory machinery may lead to the upregulation of the expression of pro-angiogenic factors, resulting in the increased vascular leakage, tumor vascularization, and finally metastasis. Moreover, a growing body of evidence indicates the presence and activation of HIF signaling in various inflammatory diseases, like rheumatoid arthritis [44], asthma [45], or atherosclerosis [46].
The regulation of HIF-1α by inflammatory cytokines was shown to occur both at the transcriptional and translational level. Especially important seems to be the interaction of HIFs and nuclear factor κB (NFκB) as both transcription factors are induced by hypoxia and inflammation and influence each other. Importantly, not only hypoxia may regulate HIF-1α mRNA level through NFκB [38, 47] but also in normoxic conditions NFκB is a direct modulator of HIF-1α expression [48]. In fact, the functional NFκB response element is present in the HIF-1α promoter at −197/−188 base pairs upstream of the transcriptional start site [48]. First indication that an active NFκB-binding site is present in HIF-1α promoter was shown in 2006, when Frede and colleagues studied the effect of LPS on HIF-1α mRNA in human monocytes and macrophages [38]. Additionally, during hypoxic induction of HIF-1α in pulmonary artery smooth muscle cells, the binding of the NFκB subunits p50 and p65 to the HIF-1α promoter have been shown [47]. Finally, direct interaction (although with different affinity) of all NFκB family members—p65, RelB, c-Rel, p52, and p50 with HIF-1α gene/promoter at an NFκB consensus site has been reported, whereas a truncated version of the HIF-1α promoter construct which lacks the NFκB site was not activated by the NFκB subunits [48].
Of note, the regulation of HIF by NFκB is evolutionary conserved—NFκB-binding element was found to be conserved across different species [47]. Moreover, recent work by van Uden et al. showed that even in Drosophila, NFκB regulates HIF-1β (tango) and HIF-α (sima) levels and activity both in normoxia and hypoxia [49].
As discussed earlier, sirtuins have been shown to associate, deacetylate, and regulate the activity of HIF-1α. Similarly, NFκB acetylation status and transcriptional activity could be modified by sirtuins. Sirtuins, although with different mechanisms of action, act as a negative regulators of NFκB activity (reviewed in [50]) (Fig. 8.4).
4 Nitric Oxide: Important Regulator of Hypoxia-Induced Angiogenesis
As discussed above, the activation of HIF transcription factors has been increasingly implicated in inflammatory diseases. A common hallmark of inflammatory disorders is the increased synthesis of nitric oxide (NO) by induction and activation of inducible nitric oxide synthase (iNOS). The implication of NO in the regulation of angiogenic mediators as well as HIF transcription factor has been suggested in a great number of papers.
In the past, several studies have shown the discrepant data concerning the effect of NO on HIF-1α accumulation—both stabilization [51, 52] and destabilization [53, 54] have been reported. Such confusing results have been nicely explained by Mateo and colleagues who have shown that NO may exert biphasic effect on HIF-α stabilization, dependently on the concentration used. At low concentration, up to 400 nM diminishment in HIF-1α stability was observed, whereas higher NO concentrations (above 1 μM) increased HIF-1α stability [55].
Moreover, the discrepant data about NO-mediated effects could be the result of various NO donors used in the experiments. Different factors which release NO are used, mostly SNAP (S-nitroso-N-acetylpenicillamine), SIN-1 (S-morpholinosydnonimine), GSNO (S-nitrosoglutathione), and DETA (diethylenetriamine NONOate). Unfortunately, sodium nitroprusside (SNP), a complex compound which releases NO together with iron ions and cyanides, was also used in many studies. SNP was shown to inhibit HIF-1 accumulation in human glioblastoma A-172 cells, whereas in the same experiment SNAP or GSNO increased HIF-1α accumulation [56]. Similarly, in the human bladder cancer and in the human prostate cancer cell lines SNP decreased hypoxia-induced HIF-1α protein level [57]. As mentioned earlier, SNP, after releasing NO, is converted to ferrocyanide and ferricyanide. Cyanides, inhibitors of cytochrome c oxidase, are cytotoxic molecules as well as iron which could be released from SNP is a possible toxic factor generating highly reactive radicals, such as hydroxyl radicals via the Fenton reaction.
Importantly, NO-dependent accumulation of HIF-α might be a possible mechanism responsible for increase in HIF-dependent gene expression in tumors. Both increased NOS expression and NO level led to stabilization of HIF-1α protein in human oral squamous cell carcinoma [58]. Similarly, in prostate cancer, Nanni and colleagues have identified the nuclear co-localization between endothelial NOS, estrogen receptor β, HIF-1α, and HIF-2α and they found that these proteins cooperate to activate transcription of protumorigenic genes [59].
The induction of HIF by NO may lead to the increased transcription of angiogenic gene expression, and nowadays it is well proven that NO is an important mediator of angiogenesis. We [60, 61] and others [62] have shown that NO is significant regulator of VEGF signaling both in vitro and in vivo. In rat vascular smooth muscle cells, IL-1β-induced NO production, NO donors, or plasmid delivery of NOS led to the upregulation of VEGF synthesis, which was abolished when inhibitor of NOS, like L-NAME was added [60, 61]. In rat ischemic hindlimb injected with eNOS cDNA increase in peripheral blood flow in ischemic tissue, better vascularization (increased number of CD31-positive cells in muscles), and increased VEGF level have been detected [62].
Similarly to discrepant data about HIF-1 accumulation, also there are results showing inhibition of VEGF expression after SNP treatment [56, 63] again indicating the importance of the appropriate NO donor usage. In our hands, direct comparison of SNAP, SIN-1, DETA, GSNO, and SNP on VEGF production in rat or human vascular smooth muscle cells showed that all donors, but not SNP, potently upregulate VEGF release [61, 64]. These results strongly indicate that NO increases VEGF level, but SNP cannot be used as a NO donor.
In summary, it seems that different effects of NO on HIF-1 or VEGF level described in the literature may be the result of various methodological approaches including different NO donors and various concentrations used.
The mechanism responsible for NO-mediated HIF modification relies mostly on S-nitrosylation of critical cysteine residues of several key proteins involved in HIF regulation, including HIF-1α itself but not on cGMP involvement. For example, NO was shown to have inhibitory effect on PHDs 1–3 in vitro. In human embryonic kidney cells stimulated with exogenous NO donor (GSNO) dose dependently inhibition of PHD activity and reduction of HIF-1α hydroxylation leading to its stabilization was observed [65]. Moreover, SNAP was shown to inhibit FIH-1 leading to enhanced HIF-1α C-TAD activity. Additionally, NO may S-nitrosylate the VHL blocking its ubiquitination activity [66].
Interesting findings linking NO and mitochondrial respiratory chain were published recently [67]. The authors based on the information that NO could be produced in the mitochondria in the reaction catalyzed by cytochrome c oxidase (Cco/NO) [68]. To find if NO produced in mitochondria could be responsible for HIF-α stabilization, HEK 293.7 cells without NOS activity have been used. Interestingly, the cells used Cco/NO activity to generate NO and further for the stabilization of HIF-1α [67].
5 microRNA and Hypoxia-Induced Angiogenesis
Identified in 1993 as small, noncoding RNA molecules which bind to the 3′ UTRs of target mRNAs to negatively regulate gene expression [69], microRNAs have been shown to be implicated in many different processes including angiogenesis. To date, more than 1,500 human miRNAs have been reported (http://www.mirbase.org) and it is suggested that they could regulate more than one-third of the mRNAs produced.
In 2010, Chan and Loscalzo have introduced the term “hypoxamirs” to stress the existence of a number of hypoxia-regulated miRNAs [70]. Several HIF-related miRNAs have been identified till now, with the most prominent miR-210. Moreover, miR-20a, miR-20b, miR-199a, miR-424, miR-130a, miR-130b, and miR-155 have been shown to affect HIF expression, although it seems that only miR-20a (encoded by miR-17-92 cluster), miR-20b, and miR-199a directly target the 3′UTR of HIF-1α [31, 71, 72].
From well-described hypoxamirs, miR-210 was shown to be regulated by both HIF-1 and HIF-2 [73] and is able to target several angiogenic factors. Overexpression of miR-210 in human umbilical vein endothelial cells increases the expression of VEGF and its receptor, VEGF-R2 leading to increased renal angiogenesis [74]. The receptor tyrosine kinase ligand Ephrin-A3, an important regulator of endothelial cells survival, migration, and differentiation is another example of angiogenic gene regulated by miR-210 [75]. Moreover, Notch signaling pathway was suggested to be activated by miR-210 in a model of cerebral ischemia [76].
Importantly, the overexpression of miR-210 has been detected in a variety of cardiovascular diseases and solid tumors. Particularly, its high level characterizes the renal clear cell carcinomas (RCCs), in which also HIF is overexpressed as a consequence of pVHL inactivation. Its important role in tumor development may be related to the fact that in response to hypoxia this miRNA regulates a wide spectrum of genes involved not only in angiogenesis but also in the mitochondrial metabolism, DNA repair, or cell survival.
6 Hypoxia and Angiogenic Regulators
6.1 Regulation of VEGF by HIFs: A Classic Example of Pro-angiogenic Gene Regulation in Hypoxic Conditions
Angiogenesis is controlled through the equilibrium of pro- and anti-angiogenic factors. However, under special conditions, e.g., during tumor development, the level of pro-angiogenic factors increases and this facilitates the blood vessel formation.
HIFs regulate the expression of more than 150 genes, including several, involved in angiogenesis (Fig. 8.1), like VEGF, the major pro-angiogenic factor which is known to enhance proliferation, survival, and tube formation by endothelial cells.
The mechanism of VEGF regulation in hypoxic conditions is well known. It was shown already in 1995 that the promoter of the VEGF gene contains HRE and binding of HIF to this region increases the transcription of VEGF [77]. Moreover, the stability of VEGF mRNA, which is very low under normoxia is dramatically increased in hypoxia [78]. It is interesting, that whereas the average half-life of eukaryotic mRNAs is 10–12 h, the half-life of VEGF mRNA is less than 1 h, e.g., in PC12 rat pheochromocytoma cells it is about 40 min [79].
Post-transcriptional VEGF regulation may be controlled at least in part by RNA-binding proteins (RBPs) and miRNAs. The VEGF 3′UTR contains several important cis-acting elements including CA-rich element (CARE) or AU-rich element (ARE) and is regulated by its RBPs such as AUF1, tristetraprolin (TTP), heterogeneous nuclear ribonucleoprotein (hnRNP L), and HuR [80–83]. One of the recently discovered factor with strong binding preference for the VEGF mRNA 5′UTR is the DEAD-box RNA helicase DDX6 [84]. Some proteins like TTP and AUF destabilize VEGF mRNA in macrophages and tumor cells, respectively [81, 82]. In contrast, the hypoxia-induced inhibition of DDX6 positively affects VEGF expression [84].
The stabilization of labile VEGF mRNA relies largely on the activity of specific RNA-binding protein HuR, a member of the Elav family of proteins found in Drosophila. HuR binds to ARE in the VEGF 3′UTR, forming an RNA–protein complex in a hypoxia-inducible fashion [83]. Till now, the molecular details of how HuR regulates VEGF expression are not well understood. It was suggested that the mechanism of HuR action may be related to its protective effect against endonucleases as a 40 bp region adjacent to the HuR-binding site in the VEGF stability region has been identified to be susceptible to ribonucleases in the absence of HuR [85].
Data accumulated over the last years have highlighted the significant roles for miRNAs, small noncoding RNAs, for the regulation of VEGF signaling. Not only VEGF but also its receptors and components of the intracellular signaling pathway are controlled by different miRNAs, including miR-15, -16, -23, -27, -93, -200b, -221, or miR-424 (reviewed in [86]). Recently, numerous studies indicate also the cross talk between RBPs and miRNAs in the post-transcriptional regulation of gene expression, like the interplay between HuR and miRNAs which associate with the HuR-regulated mRNAs. Chang et al. indicated that HuR suppressed miR-200b expression and antagonizes its anti-angiogenic effects, leading to increase in VEGF level [87].
The transcriptional regulation of VEGF by HIFs can be modulated by other transcription factors. Interestingly, the complex regulation is achieved by the members of E2F transcription factors family. This family consists of eight members which may have the opposite effect on VEGF transcription. E2F7 and E2F8 were shown to stimulate angiogenesis via transcriptional induction of VEGF [88]. The key event in this regulation is the formation of an E2F7/8–HIF-1α transcriptional complex that directly binds and stimulates the activity of VEGF promoter. The positive regulation of VEGF transcription by E2F7/8 is quite surprising as these factors are classified as repressors, and activation of VEGF transcription is unique mechanism. However, the significance of these factors for angiogenic process is also provided by in vivo studies using E2F7−/−E2F8−/− mice. Deletion of the E2F7 and E2F8 genes causes serious vascular defects and lethality around embryonic day 10.5 [89]. In contrast, another member of E2F family, E2F1 negatively regulates hypoxia-induced VEGF expression [90]. In hypoxic conditions, E2F1 associates with p53 and downregulates VEGF expression exclusively, without affecting other hypoxia-inducible genes. Importantly, E2F1−/− mice display enhanced angiogenesis, endothelial cell proliferation, and reperfusion in a hindlimb ischemia model, resulting from enhanced VEGF expression [90].
Additional transcription factors play important role in the hypoxic regulation of VEGF-driven angiogenesis. Related transcription enhancer factor-1 (RTEF-1) and early growth response 1 (EGR-1) can both target VEGF to enhance angiogenesis [91, 92]. RTEF-1 was shown to regulate HIF-1α both in normoxic and hypoxic conditions and to increase capillary density and blood recovery after the hindlimb ischemia [93]. Not only VEGF (VEGF-A) is a target gene of RTEF-1. Several studies have shown that other genes involved in the regulation of vascular growth, like fibroblast growth factor receptor-1, FGFR-1 [94], or VEGF-B [95] are regulated by RTEF-1. Similarly, EGR-1 acts not only on VEGF. This transcription factor was suggested to be a master regulator of angiogenic, inflammatory, procoagulant, and permeability-related genes [92].
Finally, translation of VEGF mRNA is increased under low oxygen tension. It was shown that even in such unfavorable stress conditions like hypoxia, when cap-dependent initiation of translation is compromised, the synthesis of VEGF protein is maintained at high level. This is because VEGF mRNA contains the internal ribosomal entry site (IRES) in the 5′UTR sequence and when cap-dependent translation is suppressed under hypoxia this IRES-dependent translation predominates [96].
6.2 Complex Effect of Hypoxia on Angiogenesis Stimulators: Not Only Induction Is Observed
The hypoxia-induced upregulation of VEGF expression at mRNA and protein level was shown in the multiple cell lines, including cancer cells. However, although very important, VEGF is not the sole-acting angiogenesis factor. Among others, there are platelet-derived growth factor (PDGF), placental growth factor (PlGF), transforming growth factor-β (TGFβ), fibroblast growth factor (FGF), angiopoietins (Ang), matrix metalloproteinases (MMP), or interleukin-8 (IL-8) (Fig. 8.5).

Angiogenesis regulation—the balance between pro-and anti-angiogenic factors. The balance between pro- and antiangiogenic factors is important for the quiescent endothelial cells. Generally, the expression of proangiogenic factors increases whereas inhibitors of angiogenesis decreases in hypoxic conditions; however, recent reports suggest the complex regulation of factors regulating angiogenesis
A lot of studies have found the correlation between HIF expression and intratumoral microvascular density (reviewed in [97]). Since hypoxia is one of the major inducers of tumor vasculature, HIFs and VEGF were suggested to be the attractive targets for anticancer therapy. However, clinical data suggest that the benefit of anti-angiogenic therapies with anti-VEGF antibodies does not last long, as many patients encounter progression of cancers. This can be explained by the recurrence of tumor angiogenesis through the compensatory production of mediators other than VEGF, indicating for the complex regulation of angiogenic factors in response to hypoxia.
In relation to the above observations we demonstrated that 24 h incubation of human microvascular endothelial cell line (HMEC-1) in the atmosphere of 1 % O2 led to decrease of PlGF expression, the stimulator of blood vessel growth [98]. Moreover, we have also shown that the expression of another pro-angiogenic agent, IL-8 was downregulated by hypoxia/HIF-1 in endothelial cells [98, 99]. Importantly, in such conditions VEGF was induced, showing opposite effect of hypoxia conditions on the expression of different pro-angiogenic factors. Next we reported that in contrast to HIF-1, overexpression of HIF-2 isoform resulted in increased expression of IL-8 [100]. The mechanism of the opposite effect of HIF-1 and HIF-2 on IL-8 expression involves the recruitment of different transcription factors—IL-8 inhibition by HIF-1 is mediated via the down-regulation of the NF-E2-related factor 2 (Nrf2) transcription factor [99], whereas stimulatory effect of HIF-2 on IL-8 relies on the induction of Sp-1 [100]. Moreover, c-Myc transcription factor plays a crucial role in the regulation of HIF-1/HIF-2 mediated IL-8 expression. We have shown that HIF-1α not only downregulates the level of c-Myc but concomitantly it also increases the production of Mxi-1, c-Myc antagonist. On the other hand, HIF-2α increases c-Myc activity [100].
There are a growing number of studies underlying the complexity of the regulation of hypoxia-dependent angiogenesis regulators. Interestingly, in HIF-1-deficient colon cancer cells, the hypoxic induction of VEGF was only partially blocked, whereas NFκB-dependent increase in IL-8 expression was observed. What is more, HIF-1 inhibition did not influence vascularization of tumors—the microvessel density was identical in tumors that had wild-type or knock-down HIF-1α [101]. The unexpected and surprising results have been recently presented by Yu and Hales [102]. In lung cancer tumor, long-term exposure to hypoxia repressed the growth of tumor and decreased microvessel density in the tumor tissues. Importantly, the same conditions led to acceleration of tumor progression and microvasculature in the colon cancer. The mechanism responsible for the opposite effect of hypoxia on angiogenesis and tumorigenesis in different tumor types observed in this study was not investigated in details, however, the differences in the expression of Na+–K+ ATPase, a versatile signal transducer, between these two tumor types was underlined. The inhibition of the sodium pump expression was observed in lung cancer tumors but not in colon cancer tumors. It is possible that the basal and hypoxia-induced expression of pro-angiogenic mediators was different in these two models, but it was not checked in this study. In another work [103], in the transgenic polyomavirus middle T breast cancer mouse model, the exposure of mice to acute cyclic hypoxia did not influence the primary tumor growth as well as lung metastasis. Interestingly, the number of CD31-positive cells in hypoxic tumors was lower than in control tumors.
Importantly, several studies tried to correlate HIF-1 expression with the survival rate of cancer patients. Although the general idea indicates that HIF-1 promotes angiogenic-dependent tumor progression, there are also studies presenting opposite results.
Volm and Koomagi have analyzed 96 paraffin-embedded sections obtained from patients with non-small cell lung carcinomas (NSCLCs) [104] and reported no relationship between HIF-1α or HIF-1β and proliferation. On the other hand, significant correlation between HIF-1 expression and apoptotic markers was detected. Surprisingly, patients with HIF-positive carcinomas had significantly longer median survival times than patients with HIF-negative carcinomas. These interesting results prompted the authors to check the expression of oncogene and tumor suppressor products and proliferative, apoptotic, and angiogenic factors in 216 patients with long-term surviving NSCLCs patients. Again, from several factors differentially expressed between control and studied patients, high level of HIF-1β was observed in carcinomas of long-term survivors [105]. Finally, recent data suggest also that activation of HIFs due to the inhibition of PHD2, can paradoxically improve the effectiveness of antitumor therapy due to tumor vessel normalization, increasing the delivery of chemotherapeutics [106].
6.3 Hypoxia-Regulated Angiogenic Inhibitors
As mentioned before, the regulation of pro-angiogenic factors by hypoxia/HIFs is complicated and still not fully discovered process. Moreover, not only activators but also inhibitors of angiogenesis (Fig. 8.5) could be regulated in HIF-dependent manner. Generally, the results of different studies indicate the inhibitory effect of HIFs activation on the regulation of angiogenic inhibitors. Thrombospondin-1 (TSP-1), a glycoprotein with major roles in cellular adhesion and vascular smooth muscle proliferation and migration, was shown to be downregulated in hypoxic conditions in different in vitro and in vivo models [98, 107, 108]. In the human microvascular endothelial cells and pericytes, the expression of other potent anti-angiogenic factor, endostatin was shown to be inhibited by hypoxia [109]. Additionally, the expression of angiostatin was decreased in a swine model of neonatal hypoxia [110]. In contrast to the above studies, several work underline the importance of hypoxia-induced expression of anti-angiogenic and anti-apoptotic factors.
One of the newly recognized hypoxia-overexpressed angiogenic inhibitor is regulator of G protein signaling 5 (RGS5) [111]. The acceleration of RGS5 by hypoxia is HIF-1 dependent and occurs also after treatment with cobalt chloride (CoCl2), hypoxia-mimicking compound, stabilizing HIF as well as with prolyl hydroxylase inhibitors, dimethyloxalylglycine (DMOG), or ethyl-3,4-dihydroxybenzoate (DHB). Importantly, RGS5 attenuates growth and induces apoptosis of endothelial cells in vitro and hinders angiogenesis in vivo [111].
An and colleagues [112] reported hypoxic induction of response gene to complement-32 (RGC-32), important modulator of cell proliferation. Increase in RGC-32 expression is mediated by HIF-1, both at the transcriptional and posttranscriptional levels, and moreover, also VEGF stimulates RGC-32 expression. Notably, the overexpression of RGC-32 in endothelial cells inhibits cell proliferation and migration via downregulation of another major angiogenic protein, FGF-2. In vivo, anti-angiogenic effect of RGC-32 was tested in two different models: this factor suppressed SW480 melanoma tumor growth as well as attenuated the blood flow recovery after hindlimb ischemia. Additionally, the reduction of CD-31-positive endothelial cells was observed in both experimental models.
7 Conclusions
Although extensive studies on the identifying molecular mechanisms of hypoxia-regulated angiogenesis are performed in many laboratories worldwide, still much work remains to be done to discover fully the mechanisms of hypoxia/HIF-regulated expression of angiogenic factors and to transfer this knowledge to translational medicine. Based on this information, optimal HIF-acting therapeutic agents could be developed what will result in an improved clinical outcome.
References
Semenza GL, Nejfelt MK, Chi SM, Antonarakis SE (1991) Hypoxia-inducible nuclear factors bind to an enhancer element located 3′ to the human erythropoietin gene. Proc Natl Acad Sci USA 88(13):5680–5684
Pugh CW, Tan CC, Jones RW, Ratcliffe PJ (1991) Functional analysis of an oxygen-regulated transcriptional enhancer lying 3′ to the mouse erythropoietin gene. Proc Natl Acad Sci USA 88(23):10553–10557
Semenza GL, Jiang BH, Leung SW, Passantino R, Concordet JP, Maire P, Giallongo A (1996) Hypoxia response elements in the aldolase A, enolase 1, and lactate dehydrogenase A gene promoters contain essential binding sites for hypoxia-inducible factor 1. J Biol Chem 271(51):32529–32537
Kimura H, Weisz A, Ogura T, Hitomi Y, Kurashima Y, Hashimoto K, D’Acquisto F, Makuuchi M, Esumi H (2001) Identification of hypoxia-inducible factor 1 ancillary sequence and its function in vascular endothelial growth factor gene induction by hypoxia and nitric oxide. J Biol Chem 276(3):2292–2298
Wang GL, Jiang BH, Rue EA, Semenza GL (1995) Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA 92(12):5510–5514
Makino Y, Kanopka A, Wilson WJ, Tanaka H, Poellinger L (2002) Inhibitory PAS domain protein (IPAS) is a hypoxia-inducible splicing variant of the hypoxia-inducible factor-3alpha locus. J Biol Chem 277(36):32405–32408
Maynard MA, Qi H, Chung J, Lee EH, Kondo Y, Hara S, Conaway RC, Conaway JW, Ohh M (2003) Multiple splice variants of the human HIF-3 alpha locus are targets of the von Hippel-Lindau E3 ubiquitin ligase complex. J Biol Chem 278(13):11032–11040
Makino Y, Cao R, Svensson K, Bertilsson G, Asman M, Tanaka H, Cao Y, Berkenstam A, Poellinger L (2001) Inhibitory PAS domain protein is a negative regulator of hypoxia-inducible gene expression. Nature 414(6863):550–554
Jaakkola P, Mole DR, Tian YM, Wilson MI, Gielbert J, Gaskell SJ, von Kriegsheim A, Hebestreit HF, Mukherji M, Schofield CJ, Maxwell PH, Pugh CW, Ratcliffe PJ (2001) Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science 292(5516):468–472
Ivan M, Kondo K, Yang H, Kim W, Valiando J, Ohh M, Salic A, Asara JM, Lane WS, Kaelin WG Jr (2001) HIFalpha targeted for VHL-mediated destruction by proline hydroxylation: implications for O2 sensing. Science 292(5516):464–468
Epstein AC, Gleadle JM, McNeill LA, Hewitson KS, O’Rourke J, Mole DR, Mukherji M, Metzen E, Wilson MI, Dhanda A, Tian YM, Masson N, Hamilton DL, Jaakkola P, Barstead R, Hodgkin J, Maxwell PH, Pugh CW, Schofield CJ, Ratcliffe PJ (2001) C. elegans EGL-9 and mammalian homologs define a family of dioxygenases that regulate HIF by prolyl hydroxylation. Cell 107(1):43–54
Bruick RK, McKnight SL (2001) A conserved family of prolyl-4-hydroxylases that modify HIF. Science 294(5545):1337–1340
Berra E, Benizri E, Ginouves A, Volmat V, Roux D, Pouyssegur J (2003) HIF prolyl-hydroxylase 2 is the key oxygen sensor setting low steady-state levels of HIF-1alpha in normoxia. EMBO J 22(16):4082–4090
Appelhoff RJ, Tian YM, Raval RR, Turley H, Harris AL, Pugh CW, Ratcliffe PJ, Gleadle JM (2004) Differential function of the prolyl hydroxylases PHD1, PHD2, and PHD3 in the regulation of hypoxia-inducible factor. J Biol Chem 279(37):38458–38465
Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, Wykoff CC, Pugh CW, Maher ER, Ratcliffe PJ (1999) The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399(6733):271–275
Ohh M, Park CW, Ivan M, Hoffman MA, Kim TY, Huang LE, Pavletich N, Chau V, Kaelin WG (2000) Ubiquitination of hypoxia-inducible factor requires direct binding to the beta-domain of the von Hippel-Lindau protein. Nat Cell Biol 2(7):423–427
Cockman ME, Masson N, Mole DR, Jaakkola P, Chang GW, Clifford SC, Maher ER, Pugh CW, Ratcliffe PJ, Maxwell PH (2000) Hypoxia inducible factor-alpha binding and ubiquitylation by the von Hippel-Lindau tumor suppressor protein. J Biol Chem 275(33):25733–25741
Chilov D, Camenisch G, Kvietikova I, Ziegler U, Gassmann M, Wenger RH (1999) Induction and nuclear translocation of hypoxia-inducible factor-1 (HIF-1): heterodimerization with ARNT is not necessary for nuclear accumulation of HIF-1alpha. J Cell Sci 112(Pt 8):1203–1212
Lando D, Peet DJ, Gorman JJ, Whelan DA, Whitelaw ML, Bruick RK (2002) FIH-1 is an asparaginyl hydroxylase enzyme that regulates the transcriptional activity of hypoxia-inducible factor. Genes Dev 16(12):1466–1471
Brunelle JK, Bell EL, Quesada NM, Vercauteren K, Tiranti V, Zeviani M, Scarpulla RC, Chandel NS (2005) Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab 1(6):409–414
Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, Schumacker PT (2005) Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab 1(6):401–408
Mansfield KD, Guzy RD, Pan Y, Young RM, Cash TP, Schumacker PT, Simon MC (2005) Mitochondrial dysfunction resulting from loss of cytochrome c impairs cellular oxygen sensing and hypoxic HIF-alpha activation. Cell Metab 1(6):393–399
Masson N, Singleton RS, Sekirnik R, Trudgian DC, Ambrose LJ, Miranda MX, Tian YM, Kessler BM, Schofield CJ, Ratcliffe PJ (2012) The FIH hydroxylase is a cellular peroxide sensor that modulates HIF transcriptional activity. EMBO Rep 13(3):251–257
Finkel T, Deng CX, Mostoslavsky R (2009) Recent progress in the biology and physiology of sirtuins. Nature 460(7255):587–591
Finley LW, Carracedo A, Lee J, Souza A, Egia A, Zhang J, Teruya-Feldstein J, Moreira PI, Cardoso SM, Clish CB, Pandolfi PP, Haigis MC (2011) SIRT3 opposes reprogramming of cancer cell metabolism through HIF1alpha destabilization. Cancer Cell 19(3):416–428
Bell EL, Emerling BM, Ricoult SJ, Guarente L (2011) SirT3 suppresses hypoxia inducible factor 1alpha and tumor growth by inhibiting mitochondrial ROS production. Oncogene 30(26):2986–2996
Yu W, Dittenhafer-Reed KE, Denu JM (2012) SIRT3 protein deacetylates isocitrate dehydrogenase 2 (IDH2) and regulates mitochondrial redox status. J Biol Chem 287(17):14078–14086
Tao R, Coleman MC, Pennington JD, Ozden O, Park SH, Jiang H, Kim HS, Flynn CR, Hill S, Hayes McDonald W, Olivier AK, Spitz DR, Gius D (2010) Sirt3-mediated deacetylation of evolutionarily conserved lysine 122 regulates MnSOD activity in response to stress. Mol Cell 40(6):893–904
Lim JH, Lee YM, Chun YS, Chen J, Kim JE, Park JW (2010) Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol Cell 38(6):864–878
Dioum EM, Chen R, Alexander MS, Zhang Q, Hogg RT, Gerard RD, Garcia JA (2009) Regulation of hypoxia-inducible factor 2alpha signaling by the stress-responsive deacetylase sirtuin 1. Science 324(5932):1289–1293
Rane S, He M, Sayed D, Vashistha H, Malhotra A, Sadoshima J, Vatner DE, Vatner SF, Abdellatif M (2009) Downregulation of miR-199a derepresses hypoxia-inducible factor-1alpha and Sirtuin 1 and recapitulates hypoxia preconditioning in cardiac myocytes. Circ Res 104(7):879–886
Chen R, Dioum EM, Hogg RT, Gerard RD, Garcia JA (2011) Hypoxia increases sirtuin 1 expression in a hypoxia-inducible factor-dependent manner. J Biol Chem 286(16):13869–13878
Zhong L, D’Urso A, Toiber D, Sebastian C, Henry RE, Vadysirisack DD, Guimaraes A, Marinelli B, Wikstrom JD, Nir T, Clish CB, Vaitheesvaran B, Iliopoulos O, Kurland I, Dor Y, Weissleder R, Shirihai OS, Ellisen LW, Espinosa JM, Mostoslavsky R (2010) The histone deacetylase Sirt6 regulates glucose homeostasis via Hif1alpha. Cell 140(2):280–293
Potente M, Ghaeni L, Baldessari D, Mostoslavsky R, Rossig L, Dequiedt F, Haendeler J, Mione M, Dejana E, Alt FW, Zeiher AM, Dimmeler S (2007) SIRT1 controls endothelial angiogenic functions during vascular growth. Genes Dev 21(20):2644–2658
Geng H, Harvey CT, Pittsenbarger J, Liu Q, Beer TM, Xue C, Qian DZ (2011) HDAC4 protein regulates HIF1alpha protein lysine acetylation and cancer cell response to hypoxia. J Biol Chem 286(44):38095–38102
Chen S, Sang N (2011) Histone deacetylase inhibitors: the epigenetic therapeutics that repress hypoxia-inducible factors. J Biomed Biotechnol 2011:197946
Karhausen J, Haase VH, Colgan SP (2005) Inflammatory hypoxia: role of hypoxia-inducible factor. Cell Cycle 4(2):256–258
Frede S, Stockmann C, Freitag P, Fandrey J (2006) Bacterial lipopolysaccharide induces HIF-1 activation in human monocytes via p44/42 MAPK and NF-kappaB. Biochem J 396(3):517–527
Jung YJ, Isaacs JS, Lee S, Trepel J, Neckers L (2003) IL-1beta-mediated up-regulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J 17(14):2115–2117
Frede S, Freitag P, Otto T, Heilmaier C, Fandrey J (2005) The proinflammatory cytokine interleukin 1beta and hypoxia cooperatively induce the expression of adrenomedullin in ovarian carcinoma cells through hypoxia inducible factor 1 activation. Cancer Res 65(11):4690–4697
Jung Y, Isaacs JS, Lee S, Trepel J, Liu ZG, Neckers L (2003) Hypoxia-inducible factor induction by tumour necrosis factor in normoxic cells requires receptor-interacting protein-dependent nuclear factor kappa B activation. Biochem J 370(Pt 3):1011–1017
Balkwill F, Mantovani A (2001) Inflammation and cancer: back to Virchow? Lancet 357(9255):539–545
Hanahan D, Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144(5):646–674
Hollander AP, Corke KP, Freemont AJ, Lewis CE (2001) Expression of hypoxia-inducible factor 1alpha by macrophages in the rheumatoid synovium: implications for targeting of therapeutic genes to the inflamed joint. Arthritis Rheum 44(7):1540–1544
Huerta-Yepez S, Baay-Guzman GJ, Bebenek IG, Hernandez-Pando R, Vega MI, Chi L, Riedl M, Diaz-Sanchez D, Kleerup E, Tashkin DP, Gonzalez FJ, Bonavida B, Zeidler M, Hankinson O (2011) Hypoxia inducible factor promotes murine allergic airway inflammation and is increased in asthma and rhinitis. Allergy 66(7):909–918
Parathath S, Mick SL, Feig JE, Joaquin V, Grauer L, Habiel DM, Gassmann M, Gardner LB, Fisher EA (2011) Hypoxia is present in murine atherosclerotic plaques and has multiple adverse effects on macrophage lipid metabolism. Circ Res 109(10):1141–1152
Belaiba RS, Bonello S, Zahringer C, Schmidt S, Hess J, Kietzmann T, Gorlach A (2007) Hypoxia up-regulates hypoxia-inducible factor-1alpha transcription by involving phosphatidylinositol 3-kinase and nuclear factor kappaB in pulmonary artery smooth muscle cells. Mol Biol Cell 18(12):4691–4697
van Uden P, Kenneth NS, Rocha S (2008) Regulation of hypoxia-inducible factor-1alpha by NF-kappaB. Biochem J 412(3):477–484
van Uden P, Kenneth NS, Webster R, Muller HA, Mudie S, Rocha S (2011) Evolutionary conserved regulation of HIF-1beta by NF-kappaB. PLoS Genet 7(1):e1001285
Tafani M, Pucci B, Russo A, Schito L, Pellegrini L, Perrone GA, Villanova L, Salvatori L, Ravenna L, Petrangeli E, Russo MA (2013) Modulators of HIF1alpha and NFkB in cancer treatment: is it a rational approach for controlling malignant progression? Front Pharmacol 4:13
Sandau KB, Faus HG, Brune B (2000) Induction of hypoxia-inducible-factor 1 by nitric oxide is mediated via the PI 3K pathway. Biochem Biophys Res Commun 278(1):263–267
Kimura H, Weisz A, Kurashima Y, Hashimoto K, Ogura T, D’Acquisto F, Addeo R, Makuuchi M, Esumi H (2000) Hypoxia response element of the human vascular endothelial growth factor gene mediates transcriptional regulation by nitric oxide: control of hypoxia-inducible factor-1 activity by nitric oxide. Blood 95(1):189–197
Sogawa K, Numayama-Tsuruta K, Ema M, Abe M, Abe H, Fujii-Kuriyama Y (1998) Inhibition of hypoxia-inducible factor 1 activity by nitric oxide donors in hypoxia. Proc Natl Acad Sci USA 95(13):7368–7373
Yin JH, Yang DI, Ku G, Hsu CY (2000) iNOS expression inhibits hypoxia-inducible factor-1 activity. Biochem Biophys Res Commun 279(1):30–34
Mateo J, Garcia-Lecea M, Cadenas S, Hernandez C, Moncada S (2003) Regulation of hypoxia-inducible factor-1alpha by nitric oxide through mitochondria-dependent and -independent pathways. Biochem J 376(Pt 2):537–544
Kimura H, Ogura T, Kurashima Y, Weisz A, Esumi H (2002) Effects of nitric oxide donors on vascular endothelial growth factor gene induction. Biochem Biophys Res Commun 296(4):976–982
Wang F, Zhang R, Xia T, Hsu E, Cai Y, Gu Z, Hankinson O (2007) Inhibitory effects of nitric oxide on invasion of human cancer cells. Cancer Lett 257(2):274–282
Quintero M, Brennan PA, Thomas GJ, Moncada S (2006) Nitric oxide is a factor in the stabilization of hypoxia-inducible factor-1alpha in cancer: role of free radical formation. Cancer Res 66(2):770–774
Nanni S, Benvenuti V, Grasselli A, Priolo C, Aiello A, Mattiussi S, Colussi C, Lirangi V, Illi B, D’Eletto M, Cianciulli AM, Gallucci M, De Carli P, Sentinelli S, Mottolese M, Carlini P, Strigari L, Finn S, Mueller E, Arcangeli G, Gaetano C, Capogrossi MC, Donnorso RP, Bacchetti S, Sacchi A, Pontecorvi A, Loda M, Farsetti A (2009) Endothelial NOS, estrogen receptor beta, and HIFs cooperate in the activation of a prognostic transcriptional pattern in aggressive human prostate cancer. J Clin Invest 119(5):1093–1108
Dulak J, Jozkowicz A, Dembinska-Kiec A, Guevara I, Zdzienicka A, Zmudzinska-Grochot D, Florek I, Wojtowicz A, Szuba A, Cooke JP (2000) Nitric oxide induces the synthesis of vascular endothelial growth factor by rat vascular smooth muscle cells. Arterioscler Thromb Vasc Biol 20(3):659–666
Jozkowicz A, Cooke JP, Guevara I, Huk I, Funovics P, Pachinger O, Weidinger F, Dulak J (2001) Genetic augmentation of nitric oxide synthase increases the vascular generation of VEGF. Cardiovasc Res 51(4):773–783
Namba T, Koike H, Murakami K, Aoki M, Makino H, Hashiya N, Ogihara T, Kaneda Y, Kohno M, Morishita R (2003) Angiogenesis induced by endothelial nitric oxide synthase gene through vascular endothelial growth factor expression in a rat hindlimb ischemia model. Circulation 108(18):2250–2257
Ghiso N, Rohan RM, Amano S, Garland R, Adamis AP (1999) Suppression of hypoxia-associated vascular endothelial growth factor gene expression by nitric oxide via cGMP. Invest Ophthalmol Vis Sci 40(6):1033–1039
Dulak J, Jozkowicz A (2003) Regulation of vascular endothelial growth factor synthesis by nitric oxide: facts and controversies. Antioxid Redox Signal 5(1):123–132
Metzen E, Zhou J, Jelkmann W, Fandrey J, Brune B (2003) Nitric oxide impairs normoxic degradation of HIF-1alpha by inhibition of prolyl hydroxylases. Mol Biol Cell 14(8):3470–3481
Park YK, Ahn DR, Oh M, Lee T, Yang EG, Son M, Park H (2008) Nitric oxide donor, (+/−)-S-nitroso-N-acetylpenicillamine, stabilizes transactive hypoxia-inducible factor-1alpha by inhibiting von Hippel-Lindau recruitment and asparagine hydroxylation. Mol Pharmacol 74(1):236–245
Ball KA, Nelson AW, Foster DG, Poyton RO (2012) Nitric oxide produced by cytochrome c oxidase helps stabilize HIF-1alpha in hypoxic mammalian cells. Biochem Biophys Res Commun 420(4):727–732
Castello PR, David PS, McClure T, Crook Z, Poyton RO (2006) Mitochondrial cytochrome oxidase produces nitric oxide under hypoxic conditions: implications for oxygen sensing and hypoxic signaling in eukaryotes. Cell Metab 3(4):277–287
Lee RC, Feinbaum RL, Ambros V (1993) The C. elegans heterochronic gene lin-4 encodes small RNAs with antisense complementarity to lin-14. Cell 75(5):843–854
Chan SY, Loscalzo J (2010) MicroRNA-210: a unique and pleiotropic hypoxamir. Cell Cycle 9(6):1072–1083
Lei Z, Li B, Yang Z, Fang H, Zhang GM, Feng ZH, Huang B (2009) Regulation of HIF-1alpha and VEGF by miR-20b tunes tumor cells to adapt to the alteration of oxygen concentration. PLoS One 4(10):e7629
Taguchi A, Yanagisawa K, Tanaka M, Cao K, Matsuyama Y, Goto H, Takahashi T (2008) Identification of hypoxia-inducible factor-1 alpha as a novel target for miR-17-92 microRNA cluster. Cancer Res 68(14):5540–5545
Zhang Z, Sun H, Dai H, Walsh RM, Imakura M, Schelter J, Burchard J, Dai X, Chang AN, Diaz RL, Marszalek JR, Bartz SR, Carleton M, Cleary MA, Linsley PS, Grandori C (2009) MicroRNA miR-210 modulates cellular response to hypoxia through the MYC antagonist MNT. Cell Cycle 8(17):2756–2768
Liu F, Lou YL, Wu J, Ruan QF, Xie A, Guo F, Cui SP, Deng ZF, Wang Y (2012) Upregulation of microRNA-210 regulates renal angiogenesis mediated by activation of VEGF signaling pathway under ischemia/perfusion injury in vivo and in vitro. Kidney Blood Press Res 35(3):182–191
Fasanaro P, D’Alessandra Y, Di Stefano V, Melchionna R, Romani S, Pompilio G, Capogrossi MC, Martelli F (2008) MicroRNA-210 modulates endothelial cell response to hypoxia and inhibits the receptor tyrosine kinase ligand Ephrin-A3. J Biol Chem 283(23):15878–15883
Lou YL, Guo F, Liu F, Gao FL, Zhang PQ, Niu X, Guo SC, Yin JH, Wang Y, Deng ZF (2012) miR-210 activates notch signaling pathway in angiogenesis induced by cerebral ischemia. Mol Cell Biochem 370(1–2):45–51
Levy AP, Levy NS, Wegner S, Goldberg MA (1995) Transcriptional regulation of the rat vascular endothelial growth factor gene by hypoxia. J Biol Chem 270(22):13333–13340
Shima DT, Deutsch U, D’Amore PA (1995) Hypoxic induction of vascular endothelial growth factor (VEGF) in human epithelial cells is mediated by increases in mRNA stability. FEBS Lett 370(3):203–208
Levy AP, Levy NS, Goldberg MA (1996) Post-transcriptional regulation of vascular endothelial growth factor by hypoxia. J Biol Chem 271(5):2746–2753
Ray PS, Jia J, Yao P, Majumder M, Hatzoglou M, Fox PL (2009) A stress-responsive RNA switch regulates VEGFA expression. Nature 457(7231):915–919
Essafi-Benkhadir K, Onesto C, Stebe E, Moroni C, Pages G (2007) Tristetraprolin inhibits Ras-dependent tumor vascularization by inducing vascular endothelial growth factor mRNA degradation. Mol Biol Cell 18(11):4648–4658
Fellows A, Griffin ME, Petrella BL, Zhong L, Parvin-Nejad FP, Fava R, Morganelli P, Robey RB, Nichols RC (2012) AUF1/hnRNP D represses expression of VEGF in macrophages. Mol Biol Cell 23(8):1414–1422
Levy NS, Chung S, Furneaux H, Levy AP (1998) Hypoxic stabilization of vascular endothelial growth factor mRNA by the RNA-binding protein HuR. J Biol Chem 273(11):6417–6423
de Vries S, Naarmann de Vries IS, Urlaub H, Lue H, Bernhagen J, Ostareck DH, Ostareck-Lederer A (2013) Identification of DEAD-box RNA helicase 6 (DDX6) as a cellular modulator of vascular endothelial growth factor expression under hypoxia. J Biol Chem 288(8):5815–5827
Goldberg-Cohen I, Furneauxb H, Levy AP (2002) A 40-bp RNA element that mediates stabilization of vascular endothelial growth factor mRNA by HuR. J Biol Chem 277(16):13635–13640
Dang LT, Lawson ND, Fish JE (2013) MicroRNA control of vascular endothelial growth factor signaling output during vascular development. Arterioscler Thromb Vasc Biol 33(2):193–200
Chang SH, Lu YC, Li X, Hsieh WY, Xiong Y, Ghosh M, Evans T, Elemento O, Hla T (2013) Antagonistic function of the RNA-binding protein HuR and miR-200b in post-transcriptional regulation of VEGF-A expression and angiogenesis. J Biol Chem 288:4908–4921
Weijts BG, Bakker WJ, Cornelissen PW, Liang KH, Schaftenaar FH, Westendorp B, de Wolf CA, Paciejewska M, Scheele CL, Kent L, Leone G, Schulte-Merker S, de Bruin A (2012) E2F7 and E2F8 promote angiogenesis through transcriptional activation of VEGFA in cooperation with HIF1. EMBO J 31(19):3871–3884
Li J, Ran C, Li E, Gordon F, Comstock G, Siddiqui H, Cleghorn W, Chen HZ, Kornacker K, Liu CG, Pandit SK, Khanizadeh M, Weinstein M, Leone G, de Bruin A (2008) Synergistic function of E2F7 and E2F8 is essential for cell survival and embryonic development. Dev Cell 14(1):62–75
Qin G, Kishore R, Dolan CM, Silver M, Wecker A, Luedemann CN, Thorne T, Hanley A, Curry C, Heyd L, Dinesh D, Kearney M, Martelli F, Murayama T, Goukassian DA, Zhu Y, Losordo DW (2006) Cell cycle regulator E2F1 modulates angiogenesis via p53-dependent transcriptional control of VEGF. Proc Natl Acad Sci USA 103(29):11015–11020
Shie JL, Wu G, Wu J, Liu FF, Laham RJ, Oettgen P, Li J (2004) RTEF-1, a novel transcriptional stimulator of vascular endothelial growth factor in hypoxic endothelial cells. J Biol Chem 279(24):25010–25016
Yan SF, Fujita T, Lu J, Okada K, Shan Zou Y, Mackman N, Pinsky DJ, Stern DM (2000) Egr-1, a master switch coordinating upregulation of divergent gene families underlying ischemic stress. Nat Med 6(12):1355–1361
Jin Y, Wu J, Song X, Song Q, Cully BL, Messmer-Blust A, Xu M, Foo SY, Rosenzweig A, Li J (2011) RTEF-1, an upstream gene of hypoxia-inducible factor-1alpha, accelerates recovery from ischemia. J Biol Chem 286(25):22699–22705
Messmer-Blust AF, Zhang C, Shie JL, Song Q, He P, Lubenec I, Liu Y, Sellke F, Li J (2012) Related transcriptional enhancer factor 1 increases endothelial-dependent microvascular relaxation and proliferation. J Vasc Res 49(3):249–259
Xu M, Jin Y, Song Q, Wu J, Philbrick MJ, Cully BL, An X, Guo L, Gao F, Li J (2011) The endothelium-dependent effect of RTEF-1 in pressure overload cardiac hypertrophy: role of VEGF-B. Cardiovasc Res 90(2):325–334
Stein I, Itin A, Einat P, Skaliter R, Grossman Z, Keshet E (1998) Translation of vascular endothelial growth factor mRNA by internal ribosome entry: implications for translation under hypoxia. Mol Cell Biol 18(6):3112–3119
Brahimi-Horn MC, Pouyssegur J (2005) The hypoxia-inducible factor and tumor progression along the angiogenic pathway. Int Rev Cytol 242:157–213
Loboda A, Jazwa A, Jozkowicz A, Molema G, Dulak J (2006) Angiogenic transcriptome of human microvascular endothelial cells: Effect of hypoxia, modulation by atorvastatin. Vascul Pharmacol 44(4):206–214
Loboda A, Stachurska A, Florczyk U, Rudnicka D, Jazwa A, Wegrzyn J, Kozakowska M, Stalinska K, Poellinger L, Levonen AL, Yla-Herttuala S, Jozkowicz A, Dulak J (2009) HIF-1 induction attenuates Nrf2-dependent IL-8 expression in human endothelial cells. Antioxid Redox Signal 11(7):1501–1517
Florczyk U, Czauderna S, Stachurska A, Tertil M, Nowak W, Kozakowska M, Poellinger L, Jozkowicz A, Loboda A, Dulak J (2011) Opposite effects of HIF-1alpha and HIF-2alpha on the regulation of IL-8 expression in endothelial cells. Free Radic Biol Med 51(10):1882–1892
Mizukami Y, Jo WS, Duerr EM, Gala M, Li J, Zhang X, Zimmer MA, Iliopoulos O, Zukerberg LR, Kohgo Y, Lynch MP, Rueda BR, Chung DC (2005) Induction of interleukin-8 preserves the angiogenic response in HIF-1alpha-deficient colon cancer cells. Nat Med 11(9):992–997
Yu L, Hales CA (2011) Long-term exposure to hypoxia inhibits tumor progression of lung cancer in rats and mice. BMC Cancer 11:331
Kalliomaki TM, McCallum G, Lunt SJ, Wells PG, Hill RP (2008) Analysis of the effects of exposure to acute hypoxia on oxidative lesions and tumour progression in a transgenic mouse breast cancer model. BMC Cancer 8:151
Volm M, Koomagi R (2000) Hypoxia-inducible factor (HIF-1) and its relationship to apoptosis and proliferation in lung cancer. Anticancer Res 20(3A):1527–1533
Volm M, Koomagi R, Mattern J, Efferth T (2002) Expression profile of genes in non-small cell lung carcinomas from long-term surviving patients. Clin Cancer Res 8(6):1843–1848
Leite de Oliveira R, Deschoemaeker S, Henze AT, Debackere K, Finisguerra V, Takeda Y, Roncal C, Dettori D, Tack E, Jonsson Y, Veschini L, Peeters A, Anisimov A, Hofmann M, Alitalo K, Baes M, D’Hooge J, Carmeliet P, Mazzone M (2012) Gene-targeting of Phd2 improves tumor response to chemotherapy and prevents side-toxicity. Cancer Cell 22(2):263–277
Fleitas T, Martinez-Sales V, Vila V, Reganon E, Mesado D, Martin M, Gomez-Codina J, Montalar J, Reynes G (2013) VEGF and TSP1 levels correlate with prognosis in advanced non-small cell lung cancer. Clin Transl Oncol 15(11):897–902
Tenan M, Fulci G, Albertoni M, Diserens AC, Hamou MF, El Atifi-Borel M, Feige JJ, Pepper MS, Van Meir EG (2000) Thrombospondin-1 is downregulated by anoxia and suppresses tumorigenicity of human glioblastoma cells. J Exp Med 191(10):1789–1798
Wu P, Yonekura H, Li H, Nozaki I, Tomono Y, Naito I, Ninomiya Y, Yamamoto H (2001) Hypoxia down-regulates endostatin production by human microvascular endothelial cells and pericytes. Biochem Biophys Res Commun 288(5):1149–1154
Emara M, Obaid L, Johnson S, Bigam DL, Cheung PY (2007) The effect of hypoxia on plasma angiostatin and related factors in newborn pigs. Proc West Pharmacol Soc 50:47–52
Jin Y, An X, Ye Z, Cully B, Wu J, Li J (2009) RGS5, a hypoxia-inducible apoptotic stimulator in endothelial cells. J Biol Chem 284(35):23436–23443
An X, Jin Y, Guo H, Foo SY, Cully BL, Wu J, Zeng H, Rosenzweig A, Li J (2009) Response gene to complement 32, a novel hypoxia-regulated angiogenic inhibitor. Circulation 120(7):617–627
Acknowledgments
The authors apologize for the inability to cite all of the important studies that have been performed in the field due to space limitations.
AL is supported by the Foundation for Polish Science—PARENT-BRIDGE Programme co-financed by the European Union within European Regional Development Fund (POMOST/2010-2/8) and she is the recipient of L'Oreal Poland for Women in Science Scholarship. The Faculty of Biochemistry, Biophysics and Biotechnology of the Jagiellonian University is a beneficiary of the structural funds from the European Union and the Polish Ministry of Science and Higher Education (grants No: POIG.02.01.00-12 064/08, POIG 01.01.02-00-109/09, POIG.02.02.00-014/08 and 01.01.02-00-069/09).
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2013 Springer-Verlag Wien
About this chapter
Cite this chapter
Łoboda, A., Józkowicz, A., Dulak, J. (2013). Molecular Mechanisms of Hypoxia-Regulated Angiogenesis. In: Dulak, J., Józkowicz, A., Łoboda, A. (eds) Angiogenesis and Vascularisation. Springer, Vienna. https://doi.org/10.1007/978-3-7091-1428-5_8
Download citation
DOI: https://doi.org/10.1007/978-3-7091-1428-5_8
Published:
Publisher Name: Springer, Vienna
Print ISBN: 978-3-7091-1427-8
Online ISBN: 978-3-7091-1428-5
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)