
Abstract
TWIK-related potassium channel-1 (TREK1, KCNK2) is the most extensively studied member of the two-pore domain potassium (K2P) channel family. Recent studies have already demonstrated a key role in the pathophysiology of depression, pain and neurodegenerative damage pointing towards an important role in a broad spectrum of CNS disorders. The mammalian blood–brain barrier (BBB) is a highly specialized structure and an integral part of the neurovascular unit, which controls the transition of cells and molecules into the CNS. While BBB dysregulation is common in neurologic diseases, the molecular mechanisms involved in this process remain largely unknown. Recently, we were able to describe a role of TREK1 in this context. TREK1 was downregulated in murine and human BBB upon inflammation. Blocking of TREK1 increased lymphocyte migration, while activation had the opposite effect. In TREK1-deficient (Trek1 −/− ) mice, brain endothelial cells displayed an inflammatory phenotype and leukocyte trafficking was facilitated, as demonstrated in experimental autoimmune encephalomyelitis (EAE), an animal model for multiple sclerosis. Here we summarize these findings and discuss the implications in diseases related to BBB dysfunction.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The blood–brain barrier (BBB) constitutes a dynamic interface in the context of the so-called neurovascular unit (NVU) controlling the transition of fluids and cells between cerebral blood vessels and the central nervous system (CNS) (Neuwelt et al. 2011). The NVU comprises endothelial cells interconnected by complex tight junctions resting on a parenchymal basement membrane ensheathed by pericytes, smooth muscle cells and enveloping end foot processes of astrocytes. Circulating blood cells, such as leukocytes, complete the dynamic complex of the NVU. Dysfunction of the BBB, e.g. in autoimmune inflammatory neurodegeneration, results in increased vascular permeability or extravasation of blood cells, extracellular fluid and macromolecules into the CNS parenchyma, leading to edema, inflammation and demyelination (Fig. 1; (Engelhardt 2006; Bittner and Meuth 2013)).

The pathophysiology of multiple sclerosis. Autoreactive T cells are able to cross the activated blood–brain barrier. Grey: oligodendrocytes; yellow: neurons; green: T cells; blue: macrophages; orange: astrocytes; red: endothelial cells; purple: antigen-presenting cells, turquoise: B cells/plasma cells. Modified from (Bittner and Meuth 2013)
In general, BBB dysfunction is considered to be a common feature of several neurologic disorders associated with inflammatory responses, e.g. stroke or neurodegenerative diseases. Accordingly, many therapeutics aim to stabilize the BBB. However, in most cases, such agents (e.g. corticosteroids, interferon (IFN)-β) act non-specifically, have limited success (Kleinschnitz et al. 2011; Dhib-Jalbut and Marks 2010), or severe side effects (Diotti et al. 2013; Steinman 2005). Molecular mechanisms underlying BBB dysfunction are incompletely understood, and very few specific targets have been identified at the brain-vasculature interface (Engelhardt and Sorokin 2009).
Regulated and selective transport of ions mediated by channels across biological membranes is crucial for numerous fundamental physiological processes. The physiological importance of ion channels is underlined by their involvement in a wide range of pathologies spanning all major therapeutic areas (Clare 2010; Kaczorowski et al. 2008). Ion channels have gained attention as potential pharmaceutical targets in several neurologic diseases so far mainly due to their ability to electrically modulate neuronal activity or axonal conductance (Overington et al. 2006). Popular neurological drug targets involve voltage-gated calcium channels as targets for neuropathic pain (e.g. gabapentin), voltage-gated sodium channels for epilepsy or bipolar disorder (e.g. carbamazepine, lamotrigine or topiramate) or Kv7 channels for epilepsy (e.g. flupirtine). In contrast, a putative therapeutic influence of ion channels on primarily non-excitable cells within the CNS, e.g. astrocytes or endothelial cells, remains widely unclear. Ion channels are nowadays the second largest target class for approved drugs (Overington et al. 2006). Research efforts in exploring novel ion channel-modifying drugs for known and novel indications are still increasing.
In this perspective, we discuss recent findings about the two-pore domain (K2P) potassium channel TREK1 on brain endothelial cells modulating the function of the BBB and introduce this channel as a potentially new therapeutic target in several neurological disorders.
Two-Pore Domain Potassium Channels
Ion channels form pores across biological membranes that facilitate the passive diffusion of different ions alongside their electrochemical gradient, leading to changes in the plasma membrane potential. They exert many different functions in a diversity of cell types such as generation of action potentials, cell proliferation, cell differentiation, immune responses, pH regulation, insulin secretion or apoptosis (Hille et al. 1999). More than 140 genes encode for human ion channels, of which 92 are K+ channels.
While K+-selective leak currents were described around 60 years ago (Goldman 1943; Hodgkin and Huxley 1952; Hodgkin and Katz 1949), the first example of a two-pore domain potassium channel (K2P) was found in Saccharomyces cerevisae (TOK1) (Ketchum et al. 1995) in 1995. K2P channels are potassium selective ion channels that are constitutively open at rest and share the same 4TM/2P (transmembrane domain, TM; pore domain, P) structure as well as a long extracellular loop between TM1 and TM2. They can be arranged as either homo- or heterodimers forming the final configuration of a tetrameric channel.
K2P channels are highly expressed in the CNS as well as in other tissues (Honore 2007; Kim et al. 2010) nowadays, there is an increasing knowledge pointing towards an essential role, not only in setting the resting membrane potential, but also in regulating numerous physiological and pathophysiological processes (Patel and Honore 2001). 15 different K2P family (KCNK) members are subgrouped into six subfamilies based on sequence homology and similar functional properties that include chemical, thermal, and mechanical modalities. The subfamilies are called TWIK, TASK, TREK, THIK, TALK and TRESK. The grade of sequence homology between the subfamilies is very low, indicating their functional diversity apart from setting the resting membrane potential.
K2P channels have been shown to play fundamental roles in physiological and pathophysiological conditions, becoming important targets in a range of neuronal and cardiovascular diseases, pain and cancer (Alloui et al. 2006; Barel et al. 2008; Heurteaux et al. 2006b; Williams et al. 2013). Although functional expression of these channels has been described in various tissues, their role within the immune system has only recently begun to be understood (Bittner et al. 2010).
Involvement of TREK1 in Disease Pathologies
TWIK-related potassium channel-1 (TREK1, also known as KCNK2, K2P.2.1) is the most extensively studied K2P channel. It is predominantly expressed in the brain (GABA-containing neurons) and spinal cord, as well as in the prefrontral cortex, foetal brain, amygdala and thalamus (Hervieu et al. 2001), but also in peripheral tissues (Medhurst et al. 2001). Moreover, TREK1 was also found in prostate cancer (Voloshyna et al. 2008) and recently, a role in the progression of ovarian cancer (Innamaa et al. 2013) has been shown.
TREK1 activity is modulated by various stimuli including physical (membrane stretch, temperature) and chemical (arachidonic acid (AA), phospholipids or polyunsaturated fatty acids (PUFAs)) stimuli (Honore 2007). A proposed vasoactive role of TREK1 channels in skin microvessels, and cerebral basilar and mesenteric arteries remains controversial (Garry et al. 2007). In these vessels, deletion of the TREK-1 channel was shown to be associated with impairment of the nitrogen oxide (NO)-producing cascade, leading to endothelial dysfunction. TREK1 also participates in the beneficial effects of PUFAs on cerebral blood flow (Blondeau et al. 2007). Recent evidences suggest that neuronal TREK1 may have a role in depression (Heurteaux et al. 2006b), chronic pain (Alloui et al. 2006), or ischemic brain injury (Heurteaux et al. 2006a), rendering TREK1 as an attractive target for the development of new therapeutic agents.
Do You Want to Join Me?—Interaction Partners of TREK1
Like other K+ channels, TREK1 channels possess a specialized C-terminus, which is involved in the regulation by PUFAs, phospholipids, stretch and intracellular acidification. Moreover the C-Terminus is also critical in the interaction with other proteins such as A-kinase-anchoring protein (AKAP150) and microtubule-associated protein 2 (Mtap2), which promote the number of active TREK channels in the plasma membrane (Sandoz et al. 2006, 2008). Indeed, it has been demonstrated that the carboxy terminus of TREK1 is responsible for the voltage- and time- dependent gating (Maingret et al. 2002). Additionally, a Coat Protein Complex I (COPI) binding site was found at the N-terminus playing a role in the anterograde transport of TREK1 to the plasma membrane, evidencing the dual roles (anterograde and retrograde transport) of COPI in the trafficking of ion channels (Kim et al. 2010). Additionally, some residues have been shown to play an important role in TREK1 gating, e.g. residue E306 and S333, which is critical for the phosphorylation by protein kinase A (PKA) (Patel et al. 1998). It has also been shown, that these two sites participate in the interaction of TREK1 and the actin cytoskeleton. Mutations of these sites lead to an increased channel activity resulting in changes of cell shape (Lauritzen et al. 2005).
The identification of different proteins interacting with and regulating TREK1 channels opens up the possibility to find further, yet unidentified protein partners and to understand the involvement of TREK1 in pathological conditions.
A Novel Role of TREK1 Channels at the BBB
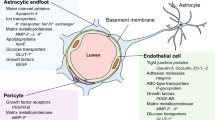
In a recent study, we characterized a novel and unexpected role for TREK1 in leukocyte trafficking across the BBB into the CNS (Bittner et al. 2013) (Fig. 2).

TREK1 on endothelial cells. Activated T lymphocytes (green) bind to endothelial cells via interaction of ICAM1 and LFA-1 or VCAM1 and VLA-4. Endothelial cells express TREK1 (dark blue) besides other ion channels (light blue). Cells are connected by tight junctions that are build up e.g. by occludin (purple) or cadherins (yellow) interconnected by cytoskeleton elements
Expression studies revealed that TREK1, but not other K2P channels are expressed on human and mouse brain endothelial cells. In addition, no significant upregulation of the closely related ion channels TREK2 or TRAAK were detected on endothelial cells isolated from TREK1−/− mice. On the other hand, TREK1 was not expressed in different human and murine immune cell subtypes. Electrophysiological measurements were performed using different blockers and activators of TREK1 currents. The recently described TREK1-blocking peptide spadin (Mazella et al. 2010; Moha Ou Maati et al. 2012) was used, revealing a spadin-sensitive current which disappeared in TREK1−/− mice underlining its specificity. As expected, TREK1 was not the only ion channel present on brain endothelial cells as seen in TREK1−/− mice. The current profile is for example compatible with TRP channels, while this aspect has formally not been investigated in the present study.
Different in vitro assays were performed to gain insight into the functional role of TREK1 channels on endothelial cells. TREK1 deletion or pharmacological blockade resulted in a reduced migration of leukocytes across endothelial cell layers. The genotype of the endothelial cells but not of the immune cells was responsible for the observed effects. No preferential effect of a specific immune cell subset was observed. An elevated migration might be due to an involvement of a number of different pathways. No changes of endothelial layer integrity were observed as expression levels of tight junction proteins, transendothelial resistance and the application of tracer molecules in vitro and in vivo yielded comparable results. Secretion of chemokines such as MCP-1 showed no differences as well. In contrast, an upregulation of adhesion molecules (ICAM1, VCAM1, PECAM1) was registered in TREK1−/− endothelial cells under inflammatory (i.e. treatment with IFNγ and TNFα) but not under basal conditions.
As a murine model of inflammation-induced BBB dysfunction, experimental autoimmune encephalomyelitis (EAE), an animal model for multiple sclerosis, was chosen. TREK1−/− mice showed a worsened clinical phenotype accompanied by an elevated infiltration of immune cells into the CNS while the peripheral immune response was comparable. Bone-marrow chimeras and adaptive transfer EAE experiments underlined a specific effect of TREK1 on endothelial cells. Further analysis showed again a stronger upregulation of ICAM1/VCAM1 in TREK1−/− on endothelial cells and experiments using blocking antibodies confirmed a functional involvement of this pathway in TREK1−/− mice. Pharmacological modulation of the EAE phenotype was performed both by TREK1 blockade using spadin and by TREK1 activation using alpha-linolenic acid or riluzole. Finally, brain sections from human MS patients corroborated our results underlining a potential role of TREK1 as a novel target structure for therapeutic interventions.
Role of Blood–Brain Barrier Dysfunction in Neurological Disorders
Multifocal perivascular infiltrates, predominantly of lymphocytes and macrophages, are found in brains of patients with MS. Dysregulation of the BBB and transendothelial migration of activated leukocytes are among the earliest abnormalities seen in MS brains and are therefore considered to be one of the hallmarks in its pathological cascade (Larochelle et al. 2011; Correale and Villa 2007). During the last years, several crucial components involved in leukocyte migration could be revealed. These observations were translated into the development of the humanized anti-α4 integrin antibody natalizumab, the first drug specifically targeting processes at the BBB (Steinman 2014). Natalizumab blocks the binding of lymphocytes to VCAM and osteopontin on inflamed brain endothelial cells resulting in a markedly reduced immune cell infiltration into the CNS. While natalizumab has a clear clinical benefit for MS patients and can be considered as highly effective, it is crucial to pay particular attention to the risk of developing progressive multifocal leukencephalopathy (PML) as a rare yet severe side effect (Sorensen et al. 2012; Fox 2011). These experiences encourage future research efforts directed at modulating the BBB.
It has long been known that dysfunction of the BBB is not a specific feature of neuroinflammatory disorders such as MS, but that it has been associated with the pathophysiology of numerous neurologic disorders, e.g. epilepsia, cerebral ischemia or even neurodegenerative diseases.
While clinical experience with clot-lysing drugs has confirmed expectations that early reperfusion improves clinical outcome in stroke patients, a large number of putative neuroprotective agents have failed in clinical trials (Cheng et al. 2004; Lo et al. 2003; Drummond et al. 2000). The reasons for these disappointments are a matter of ongoing debates and both the shortcomings of clinical trials themselves and possible reasons for the discrepancy between preclinical mouse studies and human trials are discussed intensively. Additionally, the research focus has been shifted to novel aspects of stroke pathophysiology, such as potential BBB-based treatments for cerebral ischemia. BBB changes during cerebral ischemia and reperfusion are complex involving e.g. interactions between platelets, neutrophils, the endothelium, matrix metalloproteinases and infiltration of immune cells that amplify brain tissue injury.
Alzheimer’s disease (AD) is a progressive, neurodegenerative disorder characterized by a decline in cognitive function. Despite thorough research, the etiology and pathogenesis are still not fully understood. A cascade of molecular events results in neurodegeneration initiated by deposition of amyloid-β proteins. Dysfunction of the BBB has been repeatedly proposed to contribute to AD through a number of different mechanisms, among others BBB disruption, impairment of transporters and pathological influences of inflammation and oxidative stress (Kelleher and Soiza 2013; Erickson and Banks 2013). Our current knowledge, however, is still insufficient to decide whether neuroinflammation and BBB disturbance is an underlying cause, a promoting factor or just an epiphenomenon (Enciu and Popescu 2013).
Progressive degeneration of nigrostriatal dopaminergic neurons is a hallmark of Parkinson’s disease (PD), another common neurodegenerative disease. Microglial activation and infiltration of immune cells over the BBB contributing to neuronal degeneration has been found in both murine and human studies (Chung et al. 2010). Increased BBB permeability was observed in independent mouse models for PD. Moreover, CD4 and CD8 T lymphocytes can be detected in brain sections from PD patients.
In summary, CNS inflammation determines the severity and disease course of numerous neurologic disorders and can both cause and result from BBB dysfunction. Loss of BBB integrity can allow cytokines and immune cells to enter the CNS, which results in activation of glial cells and alterations in the extracellular milieu. On the other hand, an inflammatory response in the brain might lead to endothelial cell damage and increased BBB permeability (Kim et al. 2012). Therapeutic interventions aiming at disturbances of BBB function might therefore have a promising effect in a broad spectrum of neurological disorders.
What is Known About Channels at the BBB?
Until now, a comprehensive overview about ion channel expression on brain endothelial cells is missing and the role of ion channels in BBB inflammation and dysfunction are poorly understood. During the last years, aquaporin-4 (AQP4) has certainly received a great interest from both basic researchers and clinicians due to its involvement in the pathophysiology of neuromyelitis optica (NMO; (Nagelhus and Ottersen 2013)). AQP4 tetramers form a pore that is highly selective for water molecules and is assumed to be constitutively open. While the closely related AQP1 molecule has also been implicated in ion transport under certain conditions, the same has not yet been investigated for AQP4 (Anthony et al. 2000; Boassa et al. 2006). It is strongly expressed in astrocytic membranes at the blood–brain and brain-liquor interfaces. In NMO, which is an inflammatory demyelinating disorder of the CNS, serum autoantibodies specifically target external epitopes of AQP4 (Papadopoulos and Verkman 2012). These autoantibodies are considered to be pathogenic as they cause astrocyte damage by complement activation followed by an inflammatory cascade involving granulocyte and macrophage infiltration resulting in oligodendrocyte and neuronal cell death. New emerging treatments for NMO target specific components of disease pathogenesis; for example aquaporumab is a non-pathogenic, high-affinity monoclonal antibody preventing the binding of pathogenic AQP4-antibodies (Tradtrantip et al. 2012). However, the small number of patients and heterogenic disease courses will probably remain major challenges for future clinical trials.
Apart from that, a wide variety of ion channels has been described on different endothelial cells in various target organs (Nilius and Droogmans 2001). A functional role has been described for cell growth, calcium signaling, secretion of vasoactive molecules, regulation of vessel permeability and cell migration. However, the ion channel repertoire of endothelial cells shows huge differences depending on the target organ. Although endothelial cell lines may serve as a valuable tool for investigating various functional properties in vitro, their differing ion channel expression pattern is also a critical issue. When comparing different findings throughout the literature, the comparability is therefore often limited and data on a functional role of ion channels on primary human and mouse endothelial cells is limited.
Changes in intracellular calcium levels play a central role in the regulation of endothelial cell functions (Tiruppathi et al. 2006). A rise in intracellular calcium can activate signaling pathways resulting for example in the rearrangement of the cytoskeleton or the regulation of tight junction molecules. An expression of a diverse spectrum of non-selective cation channels of the transient receptor potential (TRP) family has been frequently described (Brown et al. 2008; Yamazaki et al. 2006; Balbuena et al. 2012; Csanady and Adam-Vizi 2003). Specific members have been associated with BBB changes e.g. TRPV1 during focal ischemia (Hu et al. 2005), TRPP2 and TRPC1 upon mechanical stress (Berrout et al. 2012) or TRPC during hypoxic stress (Hicks et al. 2010). Calcium release-activated calcium (CRAC) channels consisting of Orai and STIM molecules have been suggested as an alternative calcium entry route in endothelial cells (Li et al. 2011; Abdullaev et al. 2008) while it is currently unclear whether this might also be true for endothelial cells of the BBB.
Potassium channels have also been described on brain endothelial cells. It has been shown that the potassium channels SK and Kir2.1 are expressed in the cell line t-BBEC117 influencing cell proliferation. Moreover, cell stress can upregulate Kir2.1 facilitating cell death (Kito et al. 2011; Yamazaki et al. 2006). Another study has described various voltage-gated and inward-rectifying potassium channels on rat brain endothelial cells (Millar et al. 2008).
It can certainly be seen that ion channels are functionally expressed on brain endothelial cells and are involved in the regulation of BBB integrity. However, an overview about the ion channel repertoire and its functional role on the BBB under physiological conditions as well as its implications in autoimmune inflammation and other pathological conditions will be a task for future research efforts.
Regulation of Endothelial Cell Function by TREK1: The Known and the Unknown
How do TREK1 channels exert their biological effects on a molecular level? The regulation of signaling pathways by TREK1 within endothelial cells is still largely unresolved while a number of ideas may be derived from existing studies (Nilius and Droogmans 2001). Most often, an increase in endothelial calcium levels, e.g. by thrombin or by cytokines, has been proposed to increase BBB permeability. Myosin light-chain phosphorylation is thought to be a main target mediating changes in the cytoskeleton and cellular contraction (Dejana et al. 1995). A calcium/calmodulin-dependent activation of an endothelial cell-specific myosin light chain kinase (MLCK) has been described to regulate vascular permeability and neutrophil leukocyte migration (Garcia et al. 1988; Verin et al. 1998). An inhibition of MLCK after traumatic brain injury was shown to reduce brain edema formation in different mouse models (Luh et al. 2010; Rossi et al. 2013). In another study, treatment of MOG peptide-induced EAE with ML-7, a specific inhibitor of MLCK, resulted in a reduction of CNS-invading leukocytes ad clinical disease symptoms (Huppert et al. 2010). This effect has been associated with a reduction of IL17-mediated BBB disruption (Huppert et al. 2010; Kebir et al. 2007), as Th17 cells have been associated with the downregulation of tight junction molecules. However, a calcium-independent pathway has been suggested in this context as binding of IL17A to its receptor on endothelial cells is followed by increased oxidative stress mediated by ROS (reactive oxygen species) production by NADPH oxidases. ROS production leads to MLCK inactivation independently of calcium/calmodulin. Alternatively, TRPC channels might also influence MLCK in hypoxic conditions (Hicks et al. 2010).
While these findings are of great interest as they describe a novel cytokine-induced pathway at the BBB, an involvement of TREK1 in these pathways seems unlikely. We were recently able to show that TREK1 modulation has no significant effect neither on expression levels of tight junction molecules nor on TEER and permeability for tracer molecules in vitro and in vivo nor on ROS production.
Interaction of leukocytes with endothelial cells induces the formation of microvilli-like membrane protrusions that are known as endothelial docking structures or transmigratory cups (Barreiro et al. 2002; Carman et al. 2003). These structures contain accumulated ICAM1, VCAM1, as well as cytoskeletal and adaptor molecules. Moreover, these protein complexes induce endothelial signaling important for leukocyte migration (Kanters et al. 2008). The cytoplasmic domain of ICAM1 itself participates actively in signaling pathways regulating transendothelial migration (Lyck et al. 2003) including protein tyrosine phosphorylation, rho protein activation and modulation of the cytoskeleton. It is commonly assumed, that all these processes work bidirectional and also signal towards ICAM1 clustering. The actin cross-linking molecule filamin B (Kanters et al. 2008) as well as cortactin (Kelley et al. 2011) are two exemplary proteins that regulate ICAM1 mobility and clustering, while other known binding partners include nonmuscular α-actinin, ezrin, radixin, moesin (ERM) proteins, β-tubulin, GAPDH or PIP2 (Celli et al. 2006). Interestingly, TREK1 has been shown to influence cytoskeleton remodeling independently of channel permeation (Lauritzen et al. 2005), possibly via direct interaction with ERM proteins.
Furthermore, an elevation of intracellular calcium levels associated with a calcium-dependent activation of protein kinase C and Src tyrosine kinases/cortactin has also been proposed as intracellular ICAM1-signaling pathway (Etienne-Manneville et al. 2000; Fernandez-Borja et al. 2010; van Buul et al. 2007). Both potential pathways open up a link between ICAM1-enriched docking structures and TREK1 channels either by influencing calcium signals or by direct protein interaction and await further experimental clarification.
TREK1 Channels as a Potential Drug Target
Our study identified for the first time a distinct ion channel that regulates the barrier function in the CNS. Furthermore, it suggests modulation of TREK1 as a novel strategy to treat diseases related to BBB dysfunction.
A major challenge for translational ion channel research is the development of suitable, clinically applicable, highly specific drugs. A complex and species- or even cell-specific physiology and a broad expression in different target organs with a risk of off-target effects are just two major challenges in the validation of new ion channel drug targets.
The modulation of ion channels is frequently poorly tolerated in cell lines, e.g. an over-expression can lead to increased cytotoxicity by effects on calcium homeostasis or apoptosis signaling pathways (Clare 2010). Recently, a human TREK-1/HEK cell line with stable overexpression and preserved sensitivity to known blockers and activators was developed (Moha ou Maati et al. 2011). This cell line might represent a valuable tool for large-scale screenings of novel compounds.
So far, the pharmacology of K2P channels has been largely unexplored due to several reasons: Firstly, this channel group is the youngest potassium channel family discovered nearly 20 years ago. The pharmacology of K2P channels differs from other potassium channel families, as they are insensitive towards “classical” potassium channel blocking substances such as TEA (Tetraetyhlammonium; (Goldstein et al. 2001)). Furthermore, the electrophysiological characteristics of K2P channels, which display voltage-independent currents, are difficult to assess by automated electrophysiological screening assays. Recently, an elegant approach was chosen in order to overcome these problems (Bagriantsev et al. 2013) using a yeast-based screen in combination with electrophysiological analysis. Therein, the survival of a potassium-uptake-deficient yeast strain expressing TREK1 was assessed by fluorescence signals of the vital dye resazurin which only viable cells convert to a fluorescent form. After screening a library of >100,000 substances, a novel selective activator (ML67-33) was identified while its biological activity in vivo has not yet been investigated. These recent developments might yield in further insights into regulation and (patho)physiological roles of TREK1 possibly resulting in novel therapeutic strategies for neurological diseases.
References
Abdullaev IF, Bisaillon JM, Potier M, Gonzalez JC, Motiani RK, Trebak M (2008) Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ Res 103(11):1289–1299. doi:10.1161/01.RES.0000338496.95579.56
Alloui A, Zimmermann K, Mamet J, Duprat F, Noel J, Chemin J, Guy N, Blondeau N, Voilley N, Rubat-Coudert C, Borsotto M, Romey G, Heurteaux C, Reeh P, Eschalier A, Lazdunski M (2006) TREK-1, a K+ channel involved in polymodal pain perception. EMBO J 25(11):2368–2376. doi:10.1038/sj.emboj.7601116
Anthony TL, Brooks HL, Boassa D, Leonov S, Yanochko GM, Regan JW, Yool AJ (2000) Cloned human aquaporin-1 is a cyclic GMP-gated ion channel. Mol Pharmacol 57(3):576–588
Bagriantsev SN, Ang KH, Gallardo-Godoy A, Clark KA, Arkin MR, Renslo AR, Minor DL Jr (2013) A high-throughput functional screen identifies small molecule regulators of temperature- and mechano-sensitive K2P channels. ACS Chem Biol 8(8):1841–1851. doi:10.1021/cb400289x
Balbuena P, Li W, Rzigalinski BA, Ehrich M (2012) Malathion/oxon and lead acetate increase gene expression and protein levels of transient receptor potential canonical channel subunits TRPC1 and TRPC4 in rat endothelial cells of the blood–brain barrier. Int J Toxicol 31(3):238–249. doi:10.1177/1091581812442688
Barel O, Shalev SA, Ofir R, Cohen A, Zlotogora J, Shorer Z, Mazor G, Finer G, Khateeb S, Zilberberg N, Birk OS (2008) Maternally inherited Birk Barel mental retardation dysmorphism syndrome caused by a mutation in the genomically imprinted potassium channel KCNK9. Am J Hum Genet 83(2):193–199. doi:10.1016/j.ajhg.2008.07.010
Barreiro O, Yanez-Mo M, Serrador JM, Montoya MC, Vicente-Manzanares M, Tejedor R, Furthmayr H, Sanchez-Madrid F (2002) Dynamic interaction of VCAM-1 and ICAM-1 with moesin and ezrin in a novel endothelial docking structure for adherent leukocytes. J Cell Biol 157(7):1233–1245. doi:10.1083/jcb.200112126
Berrout J, Jin M, O’Neil RG (2012) Critical role of TRPP2 and TRPC1 channels in stretch-induced injury of blood–brain barrier endothelial cells. Brain Res 1436:1–12. doi:10.1016/j.brainres.2011.11.044
Bittner S, Meuth SG (2013) Targeting ion channels for the treatment of autoimmune neuroinflammation. Ther Adv Neurol Disord 6(5):322–336. doi:10.1177/1756285613487782
Bittner S, Bobak N, Herrmann AM, Gobel K, Meuth P, Hohn KG, Stenner MP, Budde T, Wiendl H, Meuth SG (2010) Upregulation of K2P5.1 potassium channels in multiple sclerosis. Ann Neurol 68(1):58–69. doi:10.1002/ana.22010
Bittner S, Ruck T, Schuhmann MK, Herrmann AM, Moha ou Maati H, Bobak N, Gobel K, Langhauser F, Stegner D, Ehling P, Borsotto M, Pape HC, Nieswandt B, Kleinschnitz C, Heurteaux C, Galla HJ, Budde T, Wiendl H, Meuth SG (2013) Endothelial TWIK-related potassium channel-1 (TREK1) regulates immune-cell trafficking into the CNS. Nat Med 19(9):1161–1165. doi:10.1038/nm.3303
Blondeau N, Petrault O, Manta S, Giordanengo V, Gounon P, Bordet R, Lazdunski M, Heurteaux C (2007) Polyunsaturated fatty acids are cerebral vasodilators via the TREK-1 potassium channel. Circ Res 101(2):176–184. doi:10.1161/CIRCRESAHA.107.154443
Boassa D, Stamer WD, Yool AJ (2006) Ion channel function of aquaporin-1 natively expressed in choroid plexus. J Neurosci 26(30):7811–7819. doi:10.1523/JNEUROSCI.0525-06.2006
Brown RC, Wu L, Hicks K, O’Neil RG (2008) Regulation of blood–brain barrier permeability by transient receptor potential type C and type v calcium-permeable channels. Microcirculation 15(4):359–371. doi:10.1080/10739680701762656
Carman CV, Jun CD, Salas A, Springer TA (2003) Endothelial cells proactively form microvilli-like membrane projections upon intercellular adhesion molecule 1 engagement of leukocyte LFA-1. J Immunol 171(11):6135–6144
Celli L, Ryckewaert JJ, Delachanal E, Duperray A (2006) Evidence of a functional role for interaction between ICAM-1 and nonmuscle alpha-actinins in leukocyte diapedesis. J Immunol 177(6):4113–4121
Cheng YD, Al-Khoury L, Zivin JA (2004) Neuroprotection for ischemic stroke: two decades of success and failure. NeuroRx 1(1):36–45. doi:10.1602/neurorx.1.1.36
Chung YC, Ko HW, Bok E, Park ES, Huh SH, Nam JH, Jin BK (2010) The role of neuroinflammation on the pathogenesis of Parkinson’s disease. BMB Rep 43(4):225–232
Clare JJ (2010) Targeting ion channels for drug discovery. Discov Med 9(46):253–260
Correale J, Villa A (2007) The blood–brain-barrier in multiple sclerosis: functional roles and therapeutic targeting. Autoimmunity 40(2):148–160. doi:10.1080/08916930601183522
Csanady L, Adam-Vizi V (2003) Ca(2+)- and voltage-dependent gating of Ca(2+)- and ATP-sensitive cationic channels in brain capillary endothelium. Biophys J 85(1):313–327. doi:10.1016/S0006-3495(03)74476-2
Dejana E, Corada M, Lampugnani MG (1995) Endothelial cell-to-cell junctions. FASEB J 9(10):910–918
Dhib-Jalbut S, Marks S (2010) Interferon-beta mechanisms of action in multiple sclerosis. Neurology 74(Suppl 1):S17–S24. doi:10.1212/WNL.0b013e3181c97d99
Diotti RA, Nakanishi A, Clementi N, Mancini N, Criscuolo E, Solforosi L, Clementi M (2013) JC polyomavirus (JCV) and monoclonal antibodies: friends or potential foes? Clin Dev Immunol 2013:967581. doi:10.1155/2013/967581
Drummond JC, Piyash PM, Kimbro JR (2000) Neuroprotection failure in stroke. Lancet 356(9234):1032–1033. doi:10.1016/S0140-6736(05)72654-4
Enciu AM, Popescu BO (2013) Is there a causal link between inflammation and dementia? Biomed Res Int 2013:316495. doi:10.1155/2013/316495
Engelhardt B (2006) Molecular mechanisms involved in T cell migration across the blood–brain barrier. J Neural Transm 113(4):477–485. doi:10.1007/s00702-005-0409-y
Engelhardt B, Sorokin L (2009) The blood–brain and the blood-cerebrospinal fluid barriers: function and dysfunction. Semin Immunopathol 31(4):497–511. doi:10.1007/s00281-009-0177-0
Erickson MA, Banks WA (2013) Blood–brain barrier dysfunction as a cause and consequence of Alzheimer’s disease. J Cereb Blood Flow Metab 33(10):1500–1513. doi:10.1038/jcbfm.2013.135
Etienne-Manneville S, Manneville JB, Adamson P, Wilbourn B, Greenwood J, Couraud PO (2000) ICAM-1-coupled cytoskeletal rearrangements and transendothelial lymphocyte migration involve intracellular calcium signaling in brain endothelial cell lines. J Immunol 165(6):3375–3383
Fernandez-Borja M, van Buul JD, Hordijk PL (2010) The regulation of leucocyte transendothelial migration by endothelial signalling events. Cardiovasc Res 86(2):202–210. doi:10.1093/cvr/cvq003
Fox R (2011) Advances in the management of PML: focus on natalizumab. Cleve Clin J Med 78(Suppl 2):S33–S37. doi:10.3949/ccjm.78.s2.08
Garcia JG, Verin AD, Herenyiova M, English D (1988) Adherent neutrophils activate endothelial myosin light chain kinase: role in transendothelial migration. J Appl Physiol (1985) 84(5):1817–1821
Garry A, Fromy B, Blondeau N, Henrion D, Brau F, Gounon P, Guy N, Heurteaux C, Lazdunski M, Saumet JL (2007) Altered acetylcholine, bradykinin and cutaneous pressure-induced vasodilation in mice lacking the TREK1 potassium channel: the endothelial link. EMBO Rep 8(4):354–359. doi:10.1038/sj.embor.7400916
Goldman DE (1943) Potential, impedance, and rectification in membranes. J Gen Physiol 27(1):37–60
Goldstein SA, Bockenhauer D, O’Kelly I, Zilberberg N (2001) Potassium leak channels and the KCNK family of two-P-domain subunits. Nat Rev Neurosci 2(3):175–184. doi:10.1038/35058574
Hervieu GJ, Cluderay JE, Gray CW, Green PJ, Ranson JL, Randall AD, Meadows HJ (2001) Distribution and expression of TREK-1, a two-pore-domain potassium channel, in the adult rat CNS. Neuroscience 103(4):899–919
Heurteaux C, Laigle C, Blondeau N, Jarretou G, Lazdunski M (2006a) Alpha-linolenic acid and riluzole treatment confer cerebral protection and improve survival after focal brain ischemia. Neuroscience 137(1):241–251. doi:10.1016/j.neuroscience.2005.08.083
Heurteaux C, Lucas G, Guy N, El Yacoubi M, Thummler S, Peng XD, Noble F, Blondeau N, Widmann C, Borsotto M, Gobbi G, Vaugeois JM, Debonnel G, Lazdunski M (2006b) Deletion of the background potassium channel TREK-1 results in a depression-resistant phenotype. Nat Neurosci 9(9):1134–1141. doi:10.1038/nn1749
Hicks K, O’Neil RG, Dubinsky WS, Brown RC (2010) TRPC-mediated actin-myosin contraction is critical for BBB disruption following hypoxic stress. Am J Physiol Cell Physiol 298(6):C1583–C1593. doi:10.1152/ajpcell.00458.2009
Hille B, Armstrong CM, MacKinnon R (1999) Ion channels: from idea to reality. Nat Med 5(10):1105–1109. doi:10.1038/13415
Hodgkin AL, Huxley AF (1952) A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol 117(4):500–544
Hodgkin AL, Katz B (1949) The effect of sodium ions on the electrical activity of giant axon of the squid. J Physiol 108(1):37–77
Honore E (2007) The neuronal background K2P channels: focus on TREK1. Nat Rev Neurosci 8(4):251–261. doi:10.1038/nrn2117
Hu DE, Easton AS, Fraser PA (2005) TRPV1 activation results in disruption of the blood–brain barrier in the rat. Br J Pharmacol 146(4):576–584. doi:10.1038/sj.bjp.0706350
Huppert J, Closhen D, Croxford A, White R, Kulig P, Pietrowski E, Bechmann I, Becher B, Luhmann HJ, Waisman A, Kuhlmann CR (2010) Cellular mechanisms of IL-17-induced blood–brain barrier disruption. FASEB J 24(4):1023–1034. doi:10.1096/fj.09-141978
Innamaa A, Jackson L, Asher V, van Shalkwyk G, Warren A, Keightley A, Hay D, Bali A, Sowter H, Khan R (2013) Expression and effects of modulation of the K2P potassium channels TREK-1 (KCNK2) and TREK-2 (KCNK10) in the normal human ovary and epithelial ovarian cancer. Clin Transl Oncol 15(11):910–918. doi:10.1007/s12094-013-1022-4
Kaczorowski GJ, McManus OB, Priest BT, Garcia ML (2008) Ion channels as drug targets: the next GPCRs. J Gen Physiol 131(5):399–405. doi:10.1085/jgp.200709946
Kanters E, van Rijssel J, Hensbergen PJ, Hondius D, Mul FP, Deelder AM, Sonnenberg A, van Buul JD, Hordijk PL (2008) Filamin B mediates ICAM-1-driven leukocyte transendothelial migration. J Biol Chem 283(46):31830–31839. doi:10.1074/jbc.M804888200
Kebir H, Kreymborg K, Ifergan I, Dodelet-Devillers A, Cayrol R, Bernard M, Giuliani F, Arbour N, Becher B, Prat A (2007) Human TH17 lymphocytes promote blood–brain barrier disruption and central nervous system inflammation. Nat Med 13(10):1173–1175. doi:10.1038/nm1651
Kelleher RJ, Soiza RL (2013) Evidence of endothelial dysfunction in the development of Alzheimer’s disease: Is Alzheimer’s a vascular disorder? Am J Cardiovasc Dis 3(4):197–226
Kelley LC, Hayes KE, Ammer AG, Martin KH, Weed SA (2011) Revisiting the ERK/Src cortactin switch. Commun Integr Biol 4(2):205–207. doi:10.4161/cib.4.2.14420
Ketchum KA, Joiner WJ, Sellers AJ, Kaczmarek LK, Goldstein SA (1995) A new family of outwardly rectifying potassium channel proteins with two pore domains in tandem. Nature 376(6542):690–695. doi:10.1038/376690a0
Kim E, Hwang EM, Yarishkin O, Yoo JC, Kim D, Park N, Cho M, Lee YS, Sun CH, Yi GS, Yoo J, Kang D, Han J, Hong SG, Park JY (2010) Enhancement of TREK1 channel surface expression by protein-protein interaction with beta-COP. Biochem Biophys Res Commun 395(2):244–250. doi:10.1016/j.bbrc.2010.03.171
Kim SY, Buckwalter M, Soreq H, Vezzani A, Kaufer D (2012) Blood–brain barrier dysfunction-induced inflammatory signaling in brain pathology and epileptogenesis. Epilepsia 53(Suppl 6):37–44. doi:10.1111/j.1528-1167.2012.03701.x
Kito H, Yamazaki D, Ohya S, Yamamura H, Asai K, Imaizumi Y (2011) Up-regulation of K(ir)2.1 by ER stress facilitates cell death of brain capillary endothelial cells. Biochem Biophys Res Commun 411(2):293–298. doi:10.1016/j.bbrc.2011.06.128
Kleinschnitz C, Blecharz K, Kahles T, Schwarz T, Kraft P, Gobel K, Meuth SG, Burek M, Thum T, Stoll G, Forster C (2011) Glucocorticoid insensitivity at the hypoxic blood–brain barrier can be reversed by inhibition of the proteasome. Stroke 42(4):1081–1089. doi:10.1161/STROKEAHA.110.592238
Larochelle C, Alvarez JI, Prat A (2011) How do immune cells overcome the blood–brain barrier in multiple sclerosis? FEBS Lett 585(23):3770–3780. doi:10.1016/j.febslet.2011.04.066
Lauritzen I, Chemin J, Honore E, Jodar M, Guy N, Lazdunski M, Jane Patel A (2005) Cross-talk between the mechano-gated K2P channel TREK-1 and the actin cytoskeleton. EMBO Rep 6(7):642–648. doi:10.1038/sj.embor.7400449
Li J, Cubbon RM, Wilson LA, Amer MS, McKeown L, Hou B, Majeed Y, Tumova S, Seymour VA, Taylor H, Stacey M, O’Regan D, Foster R, Porter KE, Kearney MT, Beech DJ (2011) Orai1 and CRAC channel dependence of VEGF-activated Ca2+ entry and endothelial tube formation. Circ Res 108(10):1190–1198. doi:10.1161/CIRCRESAHA.111.243352
Lo EH, Dalkara T, Moskowitz MA (2003) Mechanisms, challenges and opportunities in stroke. Nat Rev Neurosci 4(5):399–415. doi:10.1038/nrn1106
Luh C, Kuhlmann CR, Ackermann B, Timaru-Kast R, Luhmann HJ, Behl C, Werner C, Engelhard K, Thal SC (2010) Inhibition of myosin light chain kinase reduces brain edema formation after traumatic brain injury. J Neurochem 112(4):1015–1025. doi:10.1111/j.1471-4159.2009.06514.x
Lyck R, Reiss Y, Gerwin N, Greenwood J, Adamson P, Engelhardt B (2003) T-cell interaction with ICAM-1/ICAM-2 double-deficient brain endothelium in vitro: the cytoplasmic tail of endothelial ICAM-1 is necessary for transendothelial migration of T cells. Blood 102(10):3675–3683. doi:10.1182/blood-2003-02-0358
Maingret F, Honore E, Lazdunski M, Patel AJ (2002) Molecular basis of the voltage-dependent gating of TREK-1, a mechano-sensitive K(+) channel. Biochem Biophys Res Commun 292(2):339–346. doi:10.1006/bbrc.2002.6674
Mazella J, Petrault O, Lucas G, Deval E, Beraud-Dufour S, Gandin C, El-Yacoubi M, Widmann C, Guyon A, Chevet E, Taouji S, Conductier G, Corinus A, Coppola T, Gobbi G, Nahon JL, Heurteaux C, Borsotto M (2010) Spadin, a sortilin-derived peptide, targeting rodent TREK-1 channels: a new concept in the antidepressant drug design. PLoS Biol 8(4):e1000355. doi:10.1371/journal.pbio.1000355
Medhurst AD, Rennie G, Chapman CG, Meadows H, Duckworth MD, Kelsell RE, Gloger II, Pangalos MN (2001) Distribution analysis of human two pore domain potassium channels in tissues of the central nervous system and periphery. Brain Res Mol Brain Res 86(1–2):101–114
Millar ID, Wang S, Brown PD, Barrand MA, Hladky SB (2008) Kv1 and Kir2 potassium channels are expressed in rat brain endothelial cells. Pflugers Arch 456(2):379–391. doi:10.1007/s00424-007-0377-1
Moha ou Maati H, Peyronnet R, Devader C, Veyssiere J, Labbal F, Gandin C, Mazella J, Heurteaux C, Borsotto M (2011) A human TREK-1/HEK cell line: a highly efficient screening tool for drug development in neurological diseases. PLoS One 6(10):e25602. doi:10.1371/journal.pone.0025602
Moha Ou Maati H, Veyssiere J, Labbal F, Coppola T, Gandin C, Widmann C, Mazella J, Heurteaux C, Borsotto M (2012) Spadin as a new antidepressant: absence of TREK-1-related side effects. Neuropharmacology 62(1):278–288. doi:10.1016/j.neuropharm.2011.07.019
Nagelhus EA, Ottersen OP (2013) Physiological roles of aquaporin-4 in brain. Physiol Rev 93(4):1543–1562. doi:10.1152/physrev.00011.2013
Neuwelt EA, Bauer B, Fahlke C, Fricker G, Iadecola C, Janigro D, Leybaert L, Molnar Z, O’Donnell ME, Povlishock JT, Saunders NR, Sharp F, Stanimirovic D, Watts RJ, Drewes LR (2011) Engaging neuroscience to advance translational research in brain barrier biology. Nat Rev Neurosci 12(3):169–182. doi:10.1038/nrn2995
Nilius B, Droogmans G (2001) Ion channels and their functional role in vascular endothelium. Physiol Rev 81(4):1415–1459
Overington JP, Al-Lazikani B, Hopkins AL (2006) How many drug targets are there? Nat Rev Drug Discov 5(12):993–996. doi:10.1038/nrd2199
Papadopoulos MC, Verkman AS (2012) Aquaporin 4 and neuromyelitis optica. Lancet Neurol 11(6):535–544. doi:10.1016/S1474-4422(12)70133-3
Patel AJ, Honore E (2001) Properties and modulation of mammalian 2P domain K+ channels. Trends Neurosci 24(6):339–346
Patel AJ, Honore E, Maingret F, Lesage F, Fink M, Duprat F, Lazdunski M (1998) A mammalian two pore domain mechano-gated S-like K+ channel. EMBO J 17(15):4283–4290. doi:10.1093/emboj/17.15.4283
Rossi JL, Todd T, Bazan NG, Belayev L (2013) Inhibition of Myosin light-chain kinase attenuates cerebral edema after traumatic brain injury in postnatal mice. J Neurotrauma 30(19):1672–1679. doi:10.1089/neu.2013.2898
Sandoz G, Thummler S, Duprat F, Feliciangeli S, Vinh J, Escoubas P, Guy N, Lazdunski M, Lesage F (2006) AKAP150, a switch to convert mechano-, pH- and arachidonic acid-sensitive TREK K(+) channels into open leak channels. EMBO J 25(24):5864–5872. doi:10.1038/sj.emboj.7601437
Sandoz G, Tardy MP, Thummler S, Feliciangeli S, Lazdunski M, Lesage F (2008) Mtap2 is a constituent of the protein network that regulates twik-related K(+) channel expression and trafficking. J Neurosci 28(34):8545–8552. doi:10.1523/Jneurosci.1962-08.2008
Sorensen PS, Bertolotto A, Edan G, Giovannoni G, Gold R, Havrdova E, Kappos L, Kieseier BC, Montalban X, Olsson T (2012) Risk stratification for progressive multifocal leukoencephalopathy in patients treated with natalizumab. Mult Scler 18(2):143–152. doi:10.1177/1352458511435105
Steinman L (2005) Blocking adhesion molecules as therapy for multiple sclerosis: natalizumab. Nat Rev Drug Discov 4(6):510–518. doi:10.1038/nrd1752
Steinman L (2014) Immunology of Relapse and Remission in Multiple Sclerosis. Annu Rev Immunol. doi:10.1146/annurev-immunol-032713-120227
Tiruppathi C, Ahmmed GU, Vogel SM, Malik AB (2006) Ca2+ signaling, TRP channels, and endothelial permeability. Microcirculation 13(8):693–708. doi:10.1080/10739680600930347
Tradtrantip L, Zhang H, Saadoun S, Phuan PW, Lam C, Papadopoulos MC, Bennett JL, Verkman AS (2012) Anti-aquaporin-4 monoclonal antibody blocker therapy for neuromyelitis optica. Ann Neurol 71(3):314–322. doi:10.1002/ana.22657
van Buul JD, Kanters E, Hordijk PL (2007) Endothelial signaling by Ig-like cell adhesion molecules. Arterioscler Thromb Vasc Biol 27(9):1870–1876. doi:10.1161/ATVBAHA.107.145821
Verin AD, Lazar V, Torry RJ, Labarrere CA, Patterson CE, Garcia JG (1998) Expression of a novel high molecular-weight myosin light chain kinase in endothelium. Am J Respir Cell Mol Biol 19(5):758–766. doi:10.1165/ajrcmb.19.5.3125
Voloshyna I, Besana A, Castillo M, Matos T, Weinstein IB, Mansukhani M, Robinson RB, Cordon-Cardo C, Feinmark SJ (2008) TREK-1 is a novel molecular target in prostate cancer. Cancer Res 68(4):1197–1203. doi:10.1158/0008-5472.Can-07-5163
Williams S, Bateman A, O’Kelly I (2013) Altered expression of two-pore domain potassium (K2P) channels in cancer. PLoS One 8(10):e74589. doi:10.1371/journal.pone.0074589
Yamazaki D, Aoyama M, Ohya S, Muraki K, Asai K, Imaizumi Y (2006) Novel functions of small conductance Ca2+−activated K+ channel in enhanced cell proliferation by ATP in brain endothelial cells. J Biol Chem 281(50):38430–38439. doi:10.1074/jbc.M603917200
Acknowledgments
This work was supported by the Deutsche Forschungsgemeinschaft (SFB TR128, TP B6 to S.G.M., FOR1086, TP2 to S.G.M. and ME3283/2-1 to S.G.M.), the Else-Kröner-Fresenius Stiftung (S.B., S.G.M.), the Interdisciplinary Center for Clinical Research (IZKF) Münster (SEED 03/12 to S.B.) and the excellence cluster ‘Cells in motion’ (CIM, to S.G.M., H.W., S.B.).
Disclosure
The authors have no conflicts of interest or financial disclosures to make.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Bittner, S., Ruck, T., Fernández-Orth, J. et al. TREK-King the Blood–Brain-Barrier. J Neuroimmune Pharmacol 9, 293–301 (2014). https://doi.org/10.1007/s11481-014-9530-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11481-014-9530-8