
Abstract
HIV infection of the central nervous system (CNS) can result in neurologic dysfunction with devastating consequences in a significant number of individuals with AIDS. Two main CNS complications in individuals with HIV are encephalitis and dementia, which are characterized by leukocyte infiltration into the CNS, microglia activation, aberrant chemokine expression, blood–brain barrier (BBB) disruption, and eventual damage and/or loss of neurons. One of the major mediators of NeuroAIDS is the transmigration of HIV-infected leukocytes across the BBB into the CNS. This review summarizes new key findings that support a critical role of the BBB in regulating leukocyte transmigration. In addition, we discuss studies on communication among cells of the immune system, BBB, and the CNS parenchyma, and suggest how these interactions contribute to the pathogenesis of NeuroAIDS. We also describe some of the animal models that have been used to study and characterize important mechanisms that have been proposed to be involved in HIV-induced CNS dysfunction. Finally, we review the pharmacologic interventions that address neuroinflammation, and the effect of substance abuse on HIV-1 related neuroimmunity.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
An early consequence of human immunodeficiency virus type 1 (HIV-1, which we will refer to as “HIV”) infection is entry of the virus into the central nervous system (CNS), resulting in several neurological sequelae. Neuropathological conditions that result from HIV entry into the CNS include HIV-associated encephalitis (HIVE), which is characterized by the formation of multinucleated giant cells, inflammation, microglial activation, astrogliosis, myelin pallor, loss of neurons and astrocytes, and the development of a syndrome of cognitive and motor dysfunction often leading to HIV-associated dementia (HAD) (Gendelman et al. 2005). Prior to the availability of highly active antiretroviral treatment (HAART), HAD was detected in approximately 20% of individuals with AIDS (McArthur et al. 1993). Initially, there was a drop in the incidence of dementia and other neurological manifestations as a result of HAART. However, the prevalence of neurologic disease in people with HIV has actually increased because individuals are living much longer, and therapeutic drugs are relatively ineffective in crossing the blood–brain barrier (BBB) (McArthur et al. 2003; Anthony et al. 2005). Thus infection and inflammation persist in the CNS despite HAART, suggesting that CNS infection and inflammation are independent of peripheral HIV status. The mechanism of HIV-induced CNS infection and inflammation is complex. In this review, we summarize several reports that provide an understanding of leukocyte transmigration across the BBB and viral entry into the CNS, and their role in the pathogenesis of cognitive impairment. Lastly, we describe some animal models that have contributed to our understanding of HIV entry into the CNS, therapeutics that could potentially improve or limit some of the neurological complications induced by HIV infection, and the contributions of substances of abuse to the pathogenesis of NeuroAIDS.
Mechanisms of viral entry across the BBB
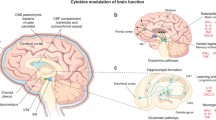
The molecular mechanisms by which HIV enters the CNS and contributes to both acute and chronic inflammatory processes that may culminate in neurologic dysfunction are still unclear. The most accepted hypothesis on viral entry into the CNS is the “Trojan Horse” model, in which infected monocytes and lymphocytes transport the virus from the blood into the CNS. After entry into the CNS, these infected cells release toxic molecules, including viral proteins, cytokines, and chemokines that recruit resident CNS cells, especially microglia and astrocytes, to areas of viral infiltration (Fig. 1). This recruitment allows for subsequent infection of these resident cells and the spread of HIV within the CNS. HIV infects perivascular macrophages and microglia productively, while astrocyte infection is restricted, resulting in the formation of viral reservoirs within CNS cells in which replication competent viral genomes persist in a stable state (Lavi et al. 1998). There is no evidence of direct HIV infection of neurons; therefore, the neuronal cell damage and death that occurs in HAD must be mediated by production and release of neurotoxic factors by other infected or uninfected cells within the CNS. In fact, neuronal dysfunction correlates more closely with inflammation and activated monocytes/microglia than with viral load (Sevigny et al. 2004).

Sequence of inflammatory events in NeuroAIDS. Gap junctions, integrins, adhesion molecules, tight junction proteins (TJPs), adherens junction proteins (AJPs), matrix metalloproteases (MMPs), and viral proteins such as Tat, gp120, Nef, and Vpr, participate in HIV-infected leukocyte adhesion to and transmigration across EC of the BBB. CCL2, and possibly other chemoattractants, significantly enhances this transmigration. Once within the CNS, the leukocytes produce soluble viral proteins, cytokines, chemokines, and other factors, thus recruiting, activating, and facilitating the infection of microglia and astrocytes. HIV-associated cognitive impairment begins with the initial transmigration of infected leukocytes into the CNS, which leads to BBB disruption, causing acute inflammation. During the subsequent chronic phase of inflammation and infection, leukocyte transmigration is an ongoing process, resulting in NeuroAIDS. Substances of abuse and alcohol contribute to the progression of this neuropathogenesis.
A second proposed mechanism of HIV entry into the CNS is the direct infection of BBB cells by cell-free HIV, providing a source of infectious particles in close proximity to the CNS parenchyma, thus promoting viral infection of resident CNS cells as well as the further release free virus. However, this theory does not explain how the extracellular virus in the circulation survives long enough to infect BBB cells. Recent studies demonstrated that proteoglycans on the surface of endothelial cells (EC) bound and protected circulating HIV from degradation for a prolonged period of time, providing an opportunity for EC to become infected (Argyris et al. 2003; Bobardt et al. 2004) through a CD4 and galactosylceramide-independent mechanism (Moses et al. 1993; Mankowski et al. 1994). This mechanism, however, still does not address how HIV crosses the structural components of the BBB, which includes astrocyte foot processes and pericytes, after EC infection, nor does it distinguish between viral uptake and productive infection. Furthermore, the issue on whether EC are productively infected by HIV remains controversial.
A third model of viral entry into the CNS is the internalization of the HIV virion by EC or astrocytic foot processes by macropinocytosis or endocytosis, with subsequent transfer of the virus to parenchymal CNS cells (Banks et al. 1998, 2001; Marechal et al. 2001; Liu et al. 2002a). Macropinocytosis in BBB EC is dependent on intact lipid rafts and MAPK signaling (Liu et al. 2002a). The possibility of macropinocytotic entry of HIV into CNS EC is supported by the presence of increased surface microvilli on EC, abundance of cytoplasmic vesicles, and inhibition of HIV entry by dimethylamiloride, an inhibitor of the Na+/H+ pump in membranes (Liu et al. 2002a). However, the presence of virions within EC or BBB cells may also be due to direct infection of these cell types, transcytosis of virions through the BBB, or the presence of HIV-infected leukocytes trapped in the BBB during the process of transmigration.
The last proposed mechanism for viral entry into the CNS is the nonspecific passage of HIV through the BBB and into the brain. This mechanism is a combination of the other mechanisms (previously described) that is dependent on the loss of BBB integrity due to exposure to cytokines, chemokines, viral proteins, or neurotransmitters, and/or the induction of apoptosis in EC and/or astrocytes. However, this theory is the least likely explanation, because it requires extensive BBB disruption early in the course of infection without repair. This seems unlikely given that HIV-infected individuals now live longer, in a chronic phase of infection, as a result of HAART.
In all of these proposed models, the integrity and function of the BBB is essential in regulating the passage of the virus into the CNS parenchyma. Alterations in the structural integrity of the BBB result in increased EC permeability. The mechanisms involved in this increase in permeability are unknown, but there is a close correlation between elevated blood and cerebral spinal fluid (CSF) cytokine levels induced by injury and the breakdown of the BBB, again suggesting that inflammation plays an important role (Martiney et al. 1992; Hurwitz et al. 1994; Fiala et al. 1997, 1998; Dallasta et al. 1999; Rubin and Staddon 1999; Gloor et al. 2001; Strelow et al. 2001; Eugenin et al. 2006a).
Blood–brain barrier function
BBB is a physical and metabolic barrier that separates the CNS from the periphery (see Persidsky et al. in this issue for details on BBB biology). It is not rigid and is composed of dynamic vessels that are capable of responding to rapid changes in the brain or blood (Huber et al. 2001). BBB is mainly composed of EC in close contact with astrocytic end foot processes (Fig. 1). BBB cells express very specific systems of transport of metabolites, as well as tight junction proteins (TJP), which seal the intercellular gaps between EC–EC and EC–astrocytes, resulting in impermeability to most macromolecules and blood cells (Pardridge and Choi 1986; Goldstein 1988; Pardridge et al. 1990; Risau and Wolburg 1990; Risau 1991; Rubin and Staddon 1999).
The major functions of BBB are to transmit biochemical signals from the blood to the brain, to exclude leukocytes and soluble factors, and to select specific nutrients to transport into or out of the brain (Pardridge and Choi 1986; Pardridge et al. 1990; Risau and Wolburg 1990). The concept of the CNS as an “immune privileged” site requires that the cells of the BBB and CNS parenchyma be shielded from physical and chemical interactions with circulating cells of the immune system. However, cells of the BBB, CNS parenchyma, and circulating leukocytes all express many common cell surface receptors that respond to soluble factors released by these cell types. Many studies demonstrated crosstalk among BBB cells, cells within the CNS expressing immune cell receptors, and immune cells expressing receptors common to CNS cells, warranting the reevaluation of the extent to which the CNS is “immune privileged” (Fig. 2).

Neuroimmune interactions in NeuroAIDS. A simplified representation of extensive interactions among BBB cells, and immune and central nervous systems. These interactions allow each system to detect changes in others. Most of the communication among these systems is between gap junctions, ions (sodium, potassium, and calcium), receptor–ligand interactions, and soluble factors, including cytokines, chemokines, neurotransmitters, neurotrophic factors, and adhesion molecules, both intact on the cell surface and/or in soluble forms. HIV infection in both the CNS and the periphery can alter the ability of these systems to detect change.
The role of BBB as an active participant in CNS function is still not completely understood. However, the recent findings that leukocytes and CNS cells share proteins originally believed to be expressed uniquely on one or the other, may enhance our understanding of viral entry into the CNS, as well as explain how the neurological complications seen in NeuroAIDS are continuing during the chronic phase of infection and become highly prominent during the late stages of HIV disease pathogenesis. Some of the newly described mechanisms by which BBB cells may have a proactive role in the modulation of CNS function are: (1) calcium waves in BBB cells, supported by ATP receptors and gap junctions; (2) neurotransmitters originating from the CNS or blood acting on BBB cells; (3) intercellular communication between BBB cells and leukocytes through gap junctions; (4) BBB activation of accessory cells, such as pericytes and perivascular macrophages.
Calcium waves
This new communication system was initially identified in liver. It is faster than chemical synapses and slower than electrical synapses (Gaspers and Thomas 2005). Calcium waves are inositol triphosphate (IP3)-dependent and can involve large areas of tissue (mm) (Nathanson et al. 1995). One study demonstrated that BBB cells establish an active and coordinated communication system mediated by calcium waves that can be visualized by using confocal microscopy, multiphoton, and calcium imaging. This study also showed that the purinergic receptors P2Y(2) and P2Y(4) and gap junctions, mainly Connexin 43 (Cx43), are present in astrocytic end feet of the BBB and, once activated, trigger changes in intercellular calcium signaling, altering some of the biological characteristics of BBB EC (Simard et al. 2003). This suggests that ATP and gap junctions may contribute to the regulation of BBB functions, such as blood flow, metabolic trafficking, and water homeostasis. ATP in the CNS is normally released by neurons and astrocytes; therefore, this finding also suggests that alterations in the CNS parenchyma can directly affect the functional properties of the BBB. It was recently suggested that signaling through P2X7 receptor (a member of the P2X family of purinergic receptors), which forms a pore in response to ligand stimulation and regulates cell permeability and cytokine release, may modulate the astrocytic response to inflammation in the CNS (Narcisse et al. 2005). Interestingly, ATP and gap junctions are the two pathways that propagate calcium waves in the CNS. These calcium waves are bidirectional (EC ↔ astrocyte) and involve IP3 generation (Braet et al. 2001; Paemeleire 2002a,b).
Neurotransmitters originating from the CNS or blood acting on BBB cells
This communication system of neurons and leukocytes has been extensively studied; however, only a few studies have examined the role of neurotransmitters in the regulation of BBB function. Despite conflicting reports, the possibility of active communication by the BBB in response to neurotransmitters under normal and pathological conditions merits consideration (Fig. 2). In this work, we review some representative studies.
Glutamate
Glutamate is the major excitatory neurotransmitter in the CNS. In excess, glutamate has also been found to be highly toxic to neuronal and glial cells. Most of the glutamate present in the brain is synthesized de novo by astrocytes (Hertz et al. 1998). Some reports indicate that cerebral EC express NMDA receptors (NMDAR), and that exposure of these cells to excitotoxic levels of glutamate leads to a breakdown of the BBB through activation of NMDAR (Krizbai et al. 1998; Sharp et al. 2003, 2005a). Glutamate–glutamate receptor interactions can up-regulate P-glycoprotein (P-gp) expression in rat microvessel EC (Zhu and Liu 2004), suggesting that glutamate treatment results in the enhanced capability of the BBB to transport P-gp substrates. The most likely mechanism of glutamate modulation of BBB function is that high neuronal activity releases glutamate into the extracellular space in the CNS that acts on the BBB. However, it has also been demonstrated that neutrophil-derived glutamate regulates BBB integrity, suggesting that glutamate from the bloodstream may also be important (Collard et al. 2002). Increased glutamate was detected in the CSF and plasma of individuals with HAD. In addition, HIV-infected macrophages release quinolinic acid, a glutamate receptor agonist that can enhance glutamate toxicity in neurons (Heyes et al. 2001). These and other data suggest a pathogenic role for glutamate in HIV dementia, thereby supporting the use of glutamate receptor antagonists as an alternative therapeutic treatment to maintain BBB integrity.
Dopamine
Dopamine is a neurotransmitter that is synthesized both peripherally and in the brain. T cells express dopamine receptors (Ilani et al. 2004). A subset of T cells, highly activated T cells (termed blast cells), expresses dopamine receptors and has been shown to cross the BBB (Owens et al. 1998; Ilani et al. 2004). Thus, these cells can encounter and bind dopamine in the brain. Studies investigating substances of abuse and HIV infection suggest that drugs accelerate neuronal loss in HIV infection, and that dopamine, in addition to its typical neurotransmission role, can alter BBB integrity and promote leukocyte activation (see Substance abuse, HIV, and the BBB for a more comprehensive discussion of dopamine) (Reyes et al. 1991; Bennett et al. 1995; Herz 1995; Czub et al. 2004; Wang et al. 2004).
Substance P
The neuropeptide substance P (SP) is a critical link between the nervous and immune systems (Ho and Douglas 2004). Several lines of evidence demonstrate that SP may play an important role in the pathophysiology of neuropsychiatric disorders, including stress and depression, in HIV-infected individuals. SP can activate and damage brain EC in the presence of proinflammatory cytokines that have been found to be elevated in the CSF of HIV-infected individuals (Annunziata et al. 2002). Monocyte-derived macrophages and lymphocytes from both placental cord blood and adult peripheral blood were demonstrated to express SP, which was significantly increased by HIV infection, suggesting that HIV enhances SP expression in immune cells (Ho et al. 2002). In addition, SP was shown to enhance HIV replication in a latently infected promonocytic cell line and in a T lymphocyte cell line (Li et al. 2001). In conjunction with gp120, an HIV protein, SP can compromise the BBB (Annunziata et al. 1998). SP binds to neurokinin-1 receptor, a G-protein coupled receptor in neurons and immune cells (Ho and Douglas 2004). Understanding the mechanisms of SP activity and its interaction with its receptor may provide a novel approach to the treatment of CNS disorders induced by HIV infection.
Neuropeptide Y
Neuropeptide Y (NPY) is one of the most abundant peptides in the central and peripheral nervous systems, where it plays a role in the regulation of cardiovascular, metabolic, endocrine, immunological, and cognitive functions (Malessa et al. 1996). NPY is a trophic/angiogenic factor at concentrations below those required for vasocontraction (Zukowska-Grojec et al. 1998). In addition, NPY alters the release of neurotransmitters, such as norepinephrine (Bitran et al. 1999). These data suggest that NPY may play a role in angiogenesis during development and pathological conditions. Increased NPY-like immunoreactivity in the CSF and plasma of HIV positive individuals has been detected and positively correlated with the degree of HIV encephalopathy, suggesting its role in HIV neuropathogenesis (Malessa et al. 1996).
Neurotrophic factors
Neurotrophic factors such as nerve growth factor (NGF), brain-derived neurotrophic factor (BDNF), and neurotrophin-3 (NT-3) belong to the neurotrophin family of growth factors. They are produced by neurons and glial cells to promote neuronal survival and growth (Encinas et al. 2000). Fibroblast growth factor-2 (FGF2) and vascular endothelial growth factor (VEGF) are angiogenic factors produced by astrocytes and EC, promoting survival, proliferation, and differentiation of brain microvascular cells (Sobue et al. 1999). In HIV infection, BDNF is neuroprotective. It activates NF-κB, thereby inducing expression of the antiapoptotic Bcl-2 gene, which protects neurons from the proapoptotic effects of HIV Tat (Ramirez et al. 2001). In addition, BDNF reduces gp120 neurotoxicity (Nosheny et al. 2005). Another study showed that FGF levels are increased in tissue sections obtained from individuals with HIVE who exhibited little neuronal cell death, and that FGF protects primary cultured neurons from the neurotoxic effects of gp120 (Everall et al. 2001). These factors therefore provide a mechanism whereby bidirectional communication between the blood and CNS cells may be facilitated by the BBB. The direct or indirect mechanisms by which these factors regulate BBB integrity require further study.
Intercellular communication between BBB cells and leukocytes through gap junctions
This new avenue of investigation does not involve soluble factors or indirect mechanisms of cellular communication. We previously reported that leukocytes express functional gap junction channels during the process of transmigration across BBB cells (Eugenin et al. 2003a). Gap junctions provide an additional mechanism for neighboring cells to communicate. The gap junction protein Cx43 is expressed homotypically and heterotypically between EC and astrocyte foot processes in the BBB (Virgintino et al. 2001; Simard et al. 2003). Interactions through gap junctions may facilitate the bidirectional communication between peripheral blood leukocytes and BBB cells, as well as between BBB and CNS cells, thus participating in the BBB function of selective transport of metabolites (Braet et al. 2001). We demonstrated that tumor necrosis factor alpha (TNF-α) plus interferon gamma (IFN-γ) induced gap junctional communication between human monocytes/macrophages mediated by Cx43. During transmigration, Cx43 was also detected at cellular interfaces between monocytes and EC in our in vitro model of the BBB, and blocking these channels with chemical inhibitors resulted in reduced monocyte transmigration across the BBB. Therefore, we proposed that transient expression of Cx43 by monocytes/macrophages allows for the formation of gap junction channels that mediate intercellular communication required for different cellular responses and functions during inflammation (Eugenin et al. 2003a).
BBB activation of accessory cells, such as pericytes and perivascular macrophages
In addition to the direct effects of cell contact and soluble factors, the presence of other cell types, not often considered a part of the BBB, is also essential in regulating BBB permeability. The activation of pericytes and perivascular macrophages triggers BBB disruption by their elaboration of factors that compromise BBB integrity. Pericytes wrap around the EC in the BBB. These cells provide structural support and regulate the microvasculature. Pericytes express contractile proteins that help regulate capillary flow (Bandopadhyay et al. 2001). In hypoxia and traumatic brain injury, pericytes migrate away from the BBB, resulting in increased BBB permeability (Dore-Duffy et al. 2000; Gonul et al. 2002). Pericytes have roles in aneurysm formation in PDGF-B-deficient mice (Lindahl et al. 1997), retinal microaneurysm formation in diabetes mellitus (Kern and Engerman 1996), hereditary cerebral hemorrhage with amyloidosis, and Alzheimer's disease (Verbeek et al. 1997). In addition, pericyte-derived angiopoetin can induce endothelial expression of occludin, a major constituent of BBB tight junctions (Hori et al. 2004). Taken together, these data suggest that pericytes can induce/maintain BBB properties. Pericytes and perivascular macrophages express a number of immune and CNS receptors and mediators, including catecholamines, angiotensin, VIP, ET-1, MHC class I and II, CD4, Fc receptor, CR3 complement receptor, and vasopressin (van Zwieten et al. 1988; Elfont et al. 1989; Healy and Wilk 1993; Benagiano et al. 1996; Dehouck et al. 1997; Thomas 1999). Thus, these cells can sense CNS and immune alterations. The activation of these cells is similar to that of cells of the macrophage lineage, causing the release of cytokines/chemokines and other factors that can alter BBB integrity.
Transmigration of HIV-infected leukocytes across the BBB
Leukocyte transmigration across the BBB during normal immune surveillance is an active process not only for the leukocyte but also for the BBB cells (Fig. 1). It has been characterized as a dynamic multistep process involving the initial “rolling” of cells on vessel endothelium in response to locally produced proinflammatory mediators, and subsequent firm adhesion to, and diapedesis across the vasculature. The rolling of leukocytes along the endothelial surface is mediated by weak interactions of selectin molecules and their corresponding glycoprotein ligands, expressed by activated EC and leukocytes. Rolling leukocytes are then stimulated by chemokines and other chemotactic molecules, resulting in activation of the β1 integrin, VLA-4, and the β2 integrin, LFA-1. Ultimately, leukocyte arrest occurs, mediated by strong interactions between LFA-1 and its EC counter receptor, intercellular adhesion molecule-1 (ICAM-1), and between VLA-4 and its EC counter receptor, vascular cell adhesion molecule-1 (VCAM-1) (Muller 2003; Liu et al. 2004; van Buul and Hordijk 2004). Leukocyte diapedesis across the blood vessel endothelium is a process mediated, in part, by homophilic and heterophilic binding molecules including junctional adhesion molecules (JAM), platelet endothelial cell adhesion molecule 1 (PECAM-1), and CD99 (Martin-Padura et al. 1998; Del Maschio et al. 1999; Ostermann et al. 2002; Schenkel et al. 2002; Ebnet et al. 2004). Although other adhesion proteins and integrins are involved in leukocyte transmigration, data suggest that those mentioned above may be associated with the development of HIV CNS pathology (Seilhean et al. 1997; Eugenin et al. 2006b).
The BBB excludes most circulating leukocytes, although there is baseline trafficking of lymphocytes and monocytes. Therefore regulatory mechanisms must exist to minimize the deleterious effects of inflammatory events in the CNS. There are a few studies that address how HIV infection alters the dynamic interaction between leukocytes and EC. An immunohistochemical study of HIVE tissue showed increased expression of adhesion proteins on astrocytes (Seilhean et al. 1997). HIV infection of human monocytes increased their expression of LFA-1 (Stent and Crowe 1997), and another study showed that the adhesion of HIV-infected monocytes to EC induced the expression of E selectin and VCAM-1 (Nottet et al. 1996). Recently, we demonstrated that in brain tissue from individuals with HIVE there is an accumulation of cleaved, soluble forms of the extracellular region of PECAM-1 (sPECAM-1) (Eugenin et al. 2006b). In addition, we assayed sera from individuals with HIV for sPECAM, and found elevated levels. These findings suggest that this sPECAM-1 production by HIV-infected cells can alter PECAM interactions between EC–EC and EC–leukocytes and thus contribute to enhanced transmigration of HIV-infected leukocytes into the CNS and changes in BBB permeability during the pathogenesis of NeuroAIDS.
There is evidence in end-stage HIV disease of abnormal BBB structure and altered expression of the TJP ZO-1 and occludin on sites where leukocyte infiltration and HIV infection were detected (Dallasta et al. 1999; Boven et al. 2000). Using an in vitro model of the BBB and blood-derived primary monocytes infected with the R5 HIV strain, HIV-1ADA, it was shown that the state of immune activation of cells that constituted the BBB dictated the ability of monocytes to traverse the BBB (Persidsky et al. 1997). Using a similar in vitro model of the BBB, we demonstrated increased BBB permeability and a loss of TJP when HIV-infected monocytes transmigrated across the BBB in response to the chemokine CCL2. The mechanism of BBB disruption and enhanced transmigration of HIV-infected cells was specifically CCL2-dependent, because other chemokines did not elicit the same effect (Eugenin et al. 2006a). We also found that BBB disruption was associated with enhanced expression of matrix metalloprotease-2 (MMP-2) and matrix metalloprotease-9 (MMP-9) (Eugenin et al. 2006a). We propose that during acute infection, HIV-infected cells disrupt the BBB as they gain access to the CNS in vivo. This transient disruption facilitates ongoing inflammation within the CNS. The advent of HAART has created a chronic phase of CNS infection/inflammation in which this ongoing inflammation is subtle, but continues to cause CNS damage over an extended time period. In late-stage disease, extensive inflammation occurs, and may result in overt dementia. Future studies will investigate the role of CCL2 in HIV entry into the CNS by characterizing the transmigration of HIV-infected monocytes in the absence and presence of CCL2, and by examining the contribution of BBB cells and HIV-infected peripheral blood mononuclear cells to CCL2 enhanced transmigration.
Cytokines/chemokines that contribute to neuropathogenicity of HIV
Blood–brain barrier dysfunction during HIV infection may be attributable to many host factors. Cytokines/chemokines, excitatory amino acids, neuropeptides, and other factors have been shown to cause BBB disruption, increased leukocyte transmigration, and EC and neuron apoptosis. Cytokines are a large and diverse group of small polypeptides that have a broad range of effects on different cell types. Chemokines are a subclass of cytokines that chemoattract and activate immune and nonimmune cells both in vivo and in vitro. Neurons, astrocytes, microglia, and oligodendrocytes produce many of these molecules, and all cell types in the brain express an array of corresponding receptors that provide responsiveness to a variety of different cytokines/chemokines (Cartier et al. 2005). Cytokines including TNF-α, IFN-γ, interleukin (IL)-1β and IL-6, and the chemokine CCL2 have been shown to be increased in sera, brain tissue, and CSF of individuals with HIVE and to modulate the BBB.
Although the signals that mediate the early transmigration of HIV-infected leukocytes into the brain are unknown, it has been proposed by our group and others that chemokines, especially CCL2, play a critical role during this process. Chemokines and their receptors are more highly expressed in HIV and simian immunodeficiency virus (SIV) encephalitic brain tissue as compared to normal brain, as well as in cultured microglia, astrocytes, and neurons in the presence of HIV (Sasseville et al. 1996; Conant et al. 1998; Gabuzda et al. 1998; Kelder et al. 1998; Lavi et al. 1998; Sanders et al. 1998; Albright et al. 1999; McManus et al. 2000). Experiments in transgenic mice and humans indicated that CCL2 is an essential chemokine for monocyte transmigration into the brain parenchyma (Fuentes et al. 1995; Gonzalez et al. 2002). Moreover, it has been demonstrated that HIV-infected individuals homozygous for the MCP-1-2578G allele have elevated levels of MCP-1/CCL2 in the CSF, and a 4.5-fold increased risk of developing HAD (Gonzalez et al. 2002; Letendre et al. 2004). In addition, elevated levels of CCL2 have been detected in the CSF of individuals with HAD and HIVE (Conant et al. 1998; Kelder et al. 1998). In SIV, an animal model of HIV (see later sections for more details), CSF levels of CCL2 are biphasic, rising early after infection, and again later during the course of CNS disease (Zink et al. 2001; Mankowski et al. 2004). When minocycline, an antibiotic with potent anti-inflammatory and neuroprotective properties, was given beginning 21 days postinoculation, there was reduced severity of encephalitis, suppressed viral load in the brain, and decreased expression of CNS inflammatory markers (Zink et al. 2005). These SIV studies have shown that the second phase of increased CCL2 CSF levels may correlate with the severity of injury. As noted above, we demonstrated the enhanced transmigration of HIV-infected leukocytes across the BBB, and BBB disruption, in response to CCL2. The expression by EC of CCR2, the CCL2 receptor, may also play a role in this process (Dzenko et al. 2005).
Interestingly, CCL2 may also have a novel role as a protective factor in human neurons and astrocytes from NMDA and HIV-Tat induced apoptosis (Eugenin et al. 2003b). Thus, CCL2 has a complex pivotal role in the pathogenesis of NeuroAIDS. For a detailed discussion on cytokine and chemokine expression induced or modulated by HIV in the CNS, see Speth et al. (2005).
Viral factors and the BBB
Many laboratories examined the direct effects of HIV-specific proteins on the human BBB and CNS cells. Viral proteins in the brain are released or secreted from free viral particles and HIV-infected cells. These proteins induce functional changes in their host cells that further contribute to neuronal injury and BBB disruption (Fig. 1).
gp120
The viral protein gp120 is a part of the envelope protein of HIV that binds the coreceptors, CCR5 and/or CXCR4, facilitating entry of the virus into cells. Chemokine receptors on the surface of neurons interact with gp120, causing an increase in intracellular Ca2+ concentration, which induces neuronal apoptosis (see review Martin-Garcia et al. 2002). Macrophages/microglia are activated by gp120, causing them to secrete TNFα and chemokines that cause neuronal injury (see Yi et al. 2004; D'Aversa et al. 2005). Treatment of EC with gp120 induces cell death, as well as increases the permeability of endothelial monolayers by disrupting and down-regulating TJP important to the BBB, such as ZO-1, ZO-2, and occludin (Kanmogne et al. 2002, 2005). Exposure of astrocytes to gp120 decreases their capacity to uptake glutamate (Wang et al. 2003b), and alters gene expression patterns (Galey et al. 2003).
Tat
Many studies examined the effects of Tat on different cell types of the CNS. Tat is released by HIV-infected cells and can be taken up by uninfected cells, including neurons (Liu et al. 2000). In human neurons, Tat can induce an increase in the levels of intracellular calcium and the generation of reactive oxygen species that eventually causes cell death through a mechanism that is attributable, in part, to the activation of NMDA and non-NMDA receptors (reviewed by Peruzzi et al. 2005). We propose that Tat induces apoptosis in neurons through the formation of a plasma membrane complex that includes Tat, low-density lipoprotein receptor-related protein (LRP), PSD-95, and NMDAR channels (Eugenin and Berman, unpublished data; King et al. 2006). Tat has also been demonstrated to affect chemokine production and function in the CNS. We demonstrated that Tat facilitates microglia migration by inducing autocrine CCL2 release (Eugenin et al. 2005), and induces CCL2 expression in astrocytes (Weiss et al. 1999). We also demonstrated that Tat induces chemokine expression by microglia, which may be involved in inflammatory cell influx into the CNS (D'Aversa et al. 2004). Exposure of brain microvascular EC (BMVEC) to Tat resulted in the disruption of TJP (Andras et al. 2003; Buckner and Berman, unpublished data). Tat treatment of astrocytes induced inducible nitric oxide synthase (iNOS) and increased production of NO (Liu et al. 2002b). Tat facilitated the infiltration of monocytes into the brain through its up-regulation of VCAM-1 and ICAM-1 production by astrocytes and EC (Woodman et al. 1999; Pu et al. 2003). Thus, Tat may play a central role in viral entry into the CNS and disruption of the BBB. (For a detailed review of Tat and its neurotoxic effects in the CNS, see King et al. 2006).
Nef and Vpr
Nef and Vpr are viral proteins that induce the release of proinflammatory cytokines and cause cell death in the CNS. Nef downmodulates host immune cell receptors, thereby enabling the virus to escape the host defense and increase viral infectivity. It also enhances viral replication and infection through a combination of different effector functions (Joseph et al. 2005). Nef has been detected in supernatants of HIV-infected cell cultures and in the sera of individuals with AIDS (Fujii et al. 1996). Extracellular Nef may interfere directly with neuronal survival or functions, as recombinant Nef treatment of cultured neuronal cells was toxic (Trillo-Pazos et al. 2000). Exposure of human monocytes/macrophages to Nef induced the release of proinflammatory factors (Olivetta et al. 2003), and supernatants from Nef-expressing macrophages induced both the chemotaxis and activation of resting T lymphocytes, facilitating productive HIV infection. This suggests a role for Nef in lymphocyte recruitment and activation at sites of viral replication (Swingler et al. 1999). Vpr has been implicated in the regulation of cellular functions including apoptosis, cell cycle arrest, differentiation, and immune suppression (Muthumani et al. 2005). In the CNS, it has been shown to induce apoptosis in neuronal cells (Patel et al. 2000), as well as to be involved in the induction of CCL5 in microglial cells (Si et al. 2002). CCL5 is a chemokine that has been shown to limit HIV infection by binding to CCR5, a coreceptor for HIV, thereby blocking viral entry into the cell (Cocchi et al. 1995).
Stress and HIV
There have been several studies examining the effects of depression and stress on AIDS progression. Although there are conflicting data due to differences in research design, duration of follow-up, number and quality of control variables, and measurement of depression or study outcome, evidence suggests that depression and stress may be predictive of, or contribute to, HIV disease progression (Burack et al. 1993; Lyketsos et al. 1993; Leserman et al. 1997, 1999, 2000; Ickovics et al. 2001; Leserman et al. 2002). Many studies demonstrated the negative effects of stress and depression on cells of the immune system that are also affected by HIV (Evans et al. 1989; Weisse 1992). Comorbid clinical depression and some symptoms of AIDS are similar, such as poor appetite, weight loss, and cognitive impairment. It has been shown that inflammatory cytokines can contribute to these symptoms (Dantzer and Kelley 1989; Clerici et al. 1997).
Inflammation within the CNS results in the elaboration of cytokines that may act on neurons and other cells within the CNS to cause changes in neuronal function, thereby contributing to cognitive impairment or dementia. HAART may not be effective in reducing neuroinflammation (Anthony et al. 2005), so psychiatric disorders may increase. In addition, the hypothalamic–pituitary–adrenal (HPA) axis and sympathetic nervous system (SNS) have been implicated in mediating the relationship among stress, depression, and HIV. It is proposed that hormones of the HPA axis and SNS are affected by stress and depression, and that dysregulation of these systems may, in turn, have negative immunologic consequences in HIV infection and contribute to disease progression (Leserman 2003). Further research is needed to address these hypotheses as well as to determine if treatment of depression, in addition to enhancing the mental health of individuals with HIV, may also limit disease progression and mortality.
Animal models of NeuroAIDS
Macaque, mouse, and feline animal models have advanced the study of the progression and pathology of NeuroAIDS. In this section, we discuss recent findings using these models, placing emphasis on their contributions to the understanding of neuroimmunity and the BBB in NeuroAIDS, with the recognition that none of these models fully replicate the disease condition exhibited in humans.
The macaque model
The macaque is a major animal model of HIV infection and NeuroAIDS, allowing studies of the CNS and CSF that cannot be performed in living humans. SIV, like HIV, causes fatal immunodeficiency syndrome in rhesus macaques. SIV crossed species and diverged to establish both HIV-1 and HIV-2 (see review by Sharp et al. 2005b). SIV is a well-accepted alternative virus for the study of HIV because SIV and HIV share morphologic and antigenic similarities, phylogenetic proximity, and parallel neuropathogenic disease progression (Desrosiers et al. 1989; Murray et al. 1992; Zink et al. 1998; Apetrei et al. 2004; Weed and Steward 2005). HIV and SIV affect several of the same cells of the CNS and elicit similar immunological responses (Clements et al. 1994; Lackner et al. 1994; Mankowski et al. 1994; Fox et al. 1997; Zink et al. 1998). Both SIV and HIV predominantly utilize CD4 for entry (Clapham et al. 1989, 1991). Unlike HIV, which can switch tropism from R5 to X4, SIV is an R5 virus. Some strains of SIV use other coreceptors, such as GPR15 (Bob) and STRL33 (Bonzo) (Marx and Chen 1998; Gabuzda and Wang 1999; Unutmaz et al. 2000). Here, we review applications of various macaque models, each of which has made its own unique contribution to the understanding of the role of neuroinflammation in NeuroAIDS.
One of the most common strains of study is SIVmac251; its injection causes SIV encephalitis in rhesus macaques. Studies using this viral strain determined that perivascular macrophages are one of the primary infected cells in SIV neuropathogenesis (Williams et al. 2001), and that SIV infection causes disruption of the BBB (Maclean et al. 2005). RT-PCR was used to quantify IL-1β, TNF-α, and IFN-γ during acute and chronic infection in this model. IL-1β was found in both infected and uninfected macaques. TNF-α and IFN-γ were detected in several areas of the brain in infected macaques within 1–2 weeks of SIV injection as well as at terminal infection (Orandle et al. 2002), but were not present in uninfected animals.
Two SIV strains are used simultaneously to model NeuroAIDS in pigtailed macaques. Infection with both the neurovirulent SIV/17E-Fr strain and the immunosuppressive SIV/deltaB670 strain recapitulates the acute phase of NeuroAIDS (Zink and Clements 2000, 2002). The neurovirulent strain quickly causes SIV encephalitis (Edinger et al. 1997), and 90% of infected macaques show CNS symptoms and pathology consistent with NeuroAIDS. The severity of encephalitis in the infected macaques correlates with high CNS viral load, not plasma viral load (Zink et al. 1999). The role of chemokines and inflammation in the CNS associated with NeuroAIDS has been examined in this model. Because monocyte and lymphocyte activity in the model is similar to that in NeuroAIDS, the CNS/plasma ratio of CCL2, a potent monocyte chemotactic factor that also recruits activated T cells, was investigated. Encephalitic macaques had a higher CNS/plasma ratio of CCL2 than those that had mild to no encephalitis, or were uninfected controls (Zink et al. 2001). Thus, in this model, CCL2 is a predictor of SIV encephalitis. CCL3, CCL4, and CCL5 cause T cell and monocyte chemotaxis as well. When their role in SIV encephalitis was examined, no significant differences between infected and uninfected macaques were found (Edinger et al. 1998).
This two-virus model of NeuroAIDS also facilitates the study of HAART. HAART has not been effective in reducing the prevalence of HIV-related neurodegenerative disease (see review by Perry et al. 2005), suggesting the existence of a viral reservoir in the CNS. This was found in the two-virus model of NeuroAIDS; infected macaques treated with a compound incapable of crossing the BBB were found to have higher CNS viral loads than those that received a compound that could (Clements et al. 2005). The finding that the CNS is a reservoir for SIV in this model underscores its ability to measure the effectiveness of current and future clinical interventions, such as minocycline, which will be described later.
Another macaque model achieves infection by using an anti-CD8 antibody at the time of infection (with SIVmac251), depleting the animal of CD8-mediated immunity and allowing rapid infection (Schmitz et al. 1999). This method can mimic acute infection, showing effects on IL-6, IFNα, and CCL2 production (Madden et al. 2004). This model has been used to identify novel amino acids of interest in neurovirulent strains of SIV required for CNS infectivity (Gaskill et al. 2005), and was also used for microarray analysis of frontal lobes of macaques exhibiting signs of SIV encephalitis (Roberts et al. 2003). Increased cyclin D3, tissue transglutaminase, α1-anti-chymotrypsin, and STAT1 were found—all of which are involved in pathways promoting migration of macrophages to the brain (Roberts et al. 2003). A similar study showed that IL-6 and other inflammatory factors were up-regulated in both the acute and long-term phases of infection (Roberts et al. 2004). Further investigation using this model may clarify whether the activities of IL-6 are neuroprotective or damaging in chronic infection.
A chimera of SIV and HIV termed SHIV is also used for the study of HIV-1 and HIV-2 infection in macaque models. SHIV is generated by the substitution of HIV genes with the similar genes from SIV. The HIV-1 env gene was the first gene replaced to study its protein product in nonhuman primates (Shibata et al. 1991; Shibata and Adachi 1992). Many strains of SHIV have been developed; the highly neuropathic SHIV-KU2 strain was produced by passaging an earlier neurovirulent strain (Raghavan et al. 1997; Liu et al. 1999). Another study examined the role of PDGF, which induces CCL2, in SHIV-KU2-infected cell lines and encephalitic macaques (Potula et al. 2004). This study showed that up-regulation of PDGF was associated with encephalitis.
Microarray analysis of total RNA extracted from basal ganglia of SHIV89.6P-infected encephalitic macaques showed significant up-regulation of a number of inflammatory mediators, including PDGF-B, CCL2, and IL-4 (Sui et al. 2003; see review Buch et al. 2004). Another study, using the pathogenic SHIV50OLNV strain, showed that BBB disruption in the SHIV model was transient. Zona Occludens-1, a TJP, was absent in brain vascular EC during the early acute phase of infection, but was restored at the end stage of infection (Stephens et al. 2003). The results of these studies support the hypothesis that dysregulation of cytokines and chemokines during SHIV infection may be responsible for disruption of the BBB as well as for infiltration of monocytes.
The rodent model
Rodent models utilizing transgenic mice, specialized viral chimeras, and severe combined immunodeficient (SCID) mice have also contributed to the understanding of neurological disease associated with HIV infection (for a comprehensive review, see Persidsky et al. 2005). One mouse model was developed by using SCID mice that are injected with infected human primary macrophages to mimic the acute phase of NeuroAIDS (Tyor et al. 1993; Persidsky et al. 1996) as well as chronic infection, causing both behavioral and cognitive deficits (Avgeropoulos et al. 1998; Griffin et al. 2004). This model was used in conjunction with an in vitro cell culture BBB model, and postmortem histopathology of human tissue from individuals with HIVE, to investigate the role of chemokines in monocyte migration across the BBB in HAD. This approach demonstrated that production of the chemokines CCL2, CCL3, CCL4, and CCL5 by microglia, astrocytes, and macrophages was responsible for migration of monocytes across the BBB (Persidsky et al. 1999). Additionally, the neuropathology in this model was attributed to viral infection of the macrophages and their subsequent production of inflammatory mediators including TNFα and IL-6 (Nukuna et al. 2004).
Another mouse model of HIV infection and AIDS was developed by constructing a JR-CSF transgenic mouse line and crossing it with a SCID mouse line. The JR-CSF line itself was produced by microinjection of a full molecular clone of HIV-1JR-CSF, under its natural LTR promoter, into FVB × C57/B6 fertilized embryos (Browning Paul et al. 2000; Wang et al. 2002). In this model, HIV was detected in all organs tested, including the spleen, thymus, lymph node, bone marrow, and brain (Browning Paul et al. 2000). This model has been used to study the pathways by which inflammatory responses, such as the production of GM-CSF, can affect latently infected monocytes, causing them to produce virus (Osiecki et al. 2005). This model was also used to examine infected microglia (Wang et al. 2003a), and demonstrated that when stimulated by LPS, JR-CSF transgenic microglia not only expressed a greater level of CCL2, but also produced significantly more viruses. Recently, these mice were crossed with hu-cycT1 mice, to allow expression of the human cyclin T1 gene under a CD4+ promoter (Sun et al. 2006). The T lymphocytes, monocytes, and macrophages of JR-CSF/hu-cycT1 mice expressed cyclin T1, facilitating Tat-mediated transactivation. These mice exhibited even greater inflammatory responses, and more closely paralleled aspects of NeuroAIDS when challenged with LPS. Thus, this model provides a transgenic mouse line through which the interplay of inflammatory cells in the periphery and neurological inflammation has a role in productive HIV-infection and its associated neurodegeneration and pathologies.
Another informative mouse model of HIV-related disease progression and pathology uses a specialized chimera of HIV-1/NL4 and ecotropic murine leukemia virus (termed EcoHIV). This model allows for infection of adult immunocompetent mice, mimicking HIV infection of humans (Potash et al. 2005). EcoHIV contains an element of gp80 from MLV that substitutes for a portion of gp120, bypassing the normal receptor requirements of HIV. This pseudotyped virus produced inflammatory and immunological responses observed in archetypal human HIV infection; the virus was detectable in the spleens, macrophages, and brains of infected mice (Potash et al. 2005). The authors also showed that a similarly produced chimeric virus (EcoNDK) caused the expression of IL-1β, CCL2, and STAT-1 in the brains of infected mice. These pseudotyped chimeric viruses produced markers of neuroinflammation, therefore providing a useful mouse model for NeuroAIDS.
The feline model
FIV reproduces a number of significant aspects of HIV infection as well as its associated neurological disease (see Fox and Phillips 2002). Its potential contribution is underscored by the findings that FIV is capable of infecting microglia, macrophages, and astrocytes in its natural host (Poli et al. 1999; Hein et al. 2001). Cats have been used to study a number of immunological aspects of retroviral infection and neurodegeneration, including the role of PBMCs/monocytes, N-acetylaspartyl glutamate, and CD40 ligand (see Fox and Phillips 2002).
Pharmacology of HIV infection and the BBB
The pharmacological challenge: crossing the blood–brain barrier
Highly active antiretroviral therapy, although successful in maintaining low viral load in the periphery, may not effectively eradicate hidden viral reservoirs. This dichotomy is most striking in the increased prevalence of HAD since the advent of HAART (McArthur et al. 2003). While infected individuals are living longer, the prevalence of HAD suggests that drugs used in combination therapies are ineffective in the CNS. Zidovudine (AZT) has been found to be effective in treating symptoms of NeuroAIDS; however, this effect may be short-lived (Sidtis et al. 1993; Baldeweg et al. 1998). Various approaches have been developed to address these challenges. Examination of the compartmentalization of drug resistance mutations in HIV to specific regions of the brain suggested that subpopulations of drug resistant virus may reseed, and spread through the circulation, thus disseminating drug resistant virus (Smit et al. 2004). One clinical study indicated that HAART was able to improve neurocognitive function in spite of its poor ability to penetrate the BBB (Robertson et al. 2004).
Other studies emphasized the importance of characterizing drug transport into, and efflux from, the CNS itself as a primary mode of addressing poor penetration by therapeutic agents and the prevalence of neurocognitive disorders during the course of HIV infection. P-glycoprotein was identified as one of the transporters capable of preventing antiretroviral therapies from accessing the brain (Strazielle et al. 2004; Loscher and Potschka 2005). In one study, postmortem tissue samples from encephalitic brains of HIV-infected individuals were immunostained for P-gp. Results showed that individuals with HIVE had more EC P-gp staining than those without HIVE (Langford et al. 2004). Those with HIVE also showed significantly enhanced staining of astrocytes and microglia, thus identifying these cells as primary participants in the exclusion of antiretroviral treatments in individuals with HIVE (Langford et al. 2004). These data may lead to the development of methods that improve the transport of NeuroAIDS therapies across the BBB.
Adjunctive therapy
Treatment of HIV-related neurodegenerative diseases requires combating the virus and its direct effects, as well as controlling the inflammatory response that mediates much of the CNS pathology. Therefore, the pharmaceutical focus should expand to include adjunctive therapies that may prevent HIV infection of immune cells that infiltrate the BBB, decrease neurocognitive impairment through neuronal protection, and target and control neurotoxic mediators of CNS inflammation (Lane et al. 1996; Nuovo and Alfieri 1996; Gartner 2000; Diesing et al. 2002; Perry et al. 2005).
Receptor antagonists
Chemokine antagonists
The identification of CXCR4 and CCR5 as coreceptors for HIV infection led to the study of the ligands of these receptors for their ability to prevent viral binding to, and infection of, CXCR4 and CCR5 positive cells. However, the finding that these ligands could also activate infected cells to produce virus precluded the use of these proteins as an antiviral therapeutic strategy (for comprehensive review, see Princen and Schols 2005). Thus, the focus of future studies centered on the identification of chemokine receptor antagonists. However, their ability to cross the BBB is not well characterized. AMD3465, a monomacrocyclic compound related to the X4 antagonist AMD3100, can strongly inhibit HIV entry through the CXCR4 receptor (Hatse et al. 2005). The structure of AMD3465 may translate into oral delivery and potential clinical use (Hatse et al. 2005). Another receptor antagonist, AMD3451, was shown to inhibit both X4 and R5 strains; however, more studies are needed to determine its therapeutic potential (Princen et al. 2004). A number of CCR5 antagonists are in clinical trials (Julg and Goebel 2005; Princen and Schols 2005). By preventing entry of the virus into cells that have the potential to cross the BBB, it may be possible to decrease incidence of NeuroAIDS.
NMDAR antagonists: memantine
NMDAR antagonists are a proposed therapy for HAD (Lipton 1994, 1998). Memantine, an NMDAR antagonist, has been successfully used for the treatment of Alzheimer's disease, a neurodegenerative disorder with several clinical manifestations similar to HAD (Reisberg et al. 2003). It is also prescribed for long-term treatment of Parkinson's disease (Lipton 1998). This drug was studied in the SCID mouse model of HIVE described above (Tyor et al. 1993; Persidsky et al. 1996; Anderson et al. 2004). Using monocyte-derived macrophages infected with HIV-1ADA, a well-characterized R5 strain, this model replicates the neurotoxicity and neuroinflammation associated with HIVE. The testing of long-term potentiation and other cognitive indicators, as well as histopathology, showed that the memantine-treated HIVE mice had a lesser degree of synaptic impairment than the HIVE mice that were not treated with memantine (Anderson et al. 2004). Thus, the authors conclude that this drug may be a candidate adjunctive therapy, capable of decreasing synaptic impairment in infected individuals by maintaining neuronal integrity in the face of the inflammatory response that is characteristic of NeuroAIDS.
Neuroprotective lithium
The neuroprotective potential of lithium and its pro- and antiapoptotic effects have been studied in a number of neurological diseases, including Alzheimer's disease. Lithium affects many neurological pathways through protective mechanisms, and is linked to each of the following apoptotic, signaling, and transduction molecules and pathways: phosphatidylinositol 3 (PI3)-kinase (PI3-K), Akt, MAPK, glycogen synthase kinase-3β (GSK-3β), Tau, β-catenin, heat shock factor 1, activator protein 1, cAMP, Bcl-2, BDNF, NMDAR activation, and calcium (see Dou et al. 2005). Immunohistochemical and long-term potentiation studies were used to investigate the neuroprotective potential of lithium in the previously described monocyte derived macrophage rodent model of HIVE (Dou et al. 2005). Histopathology showed that lithium was protective in the neurodegeneration associated with HIV infection, yet it had no specific antiretroviral effect. Thus, the authors theorize that lithium may protect neurons against neurotoxins associated with HIV. Further understanding of the effects of lithium on neuroinflammation are required.
Minocycline
Minocycline, a derivative of tetracycline, has been shown to have both antiretroviral and neuroprotective effects in macaques, dually infected with SIV (Zink et al. 2005). Minocycline-treated animals showed no or mild SIV encephalitis, whereas untreated macaques showed moderate to severe SIV encephalitis. The untreated macaques showed significantly decreased expression in the brain of the following markers of inflammation: major histocompatibility class II antigens, macrophage marker CD68, T-cell intracytoplasmic antigen I, and β-amyloid precursor protein. Activated p38 mitogen-activated protein kinase (MAPK) had lower expression in HIV-infected minocycline-treated T lymphocytes as well. After 28 days (during the second phase of increased CCL2 in the CSF, discussed earlier), minocycline-treated, SIV-infected macaques showed little CCL2 in CSF and brain samples, as compared to untreated, infected animals. These results show a correlation between minocycline treatment and a diminished inflammatory response (Zink et al. 2005).
In addition to its potent anti-inflammatory effects, minocycline inhibits the release of both R5 and X4R5 viruses in cultured human primary microglia (Si et al. 2004). Minocycline was found to affect LTR promoter activity in U38 cells, as well as NF-κB nuclear binding activity in cultured microglia (Si et al. 2004). Minocycline is a candidate for treatment of NeuroAIDS because it can cross the BBB, decrease viral release, and decrease CNS inflammation (Tikka et al. 2001; Colovic and Caccia 2003). Limiting neuroinflammation may positively affect the integrity of the BBB during the chronic HIV infection. A treatment strategy combining the ability of minocycline to reduce neuroinflammation with both antiretroviral and neuroprotective therapies may be most effective against the effects of NeuroAIDS.
Substance abuse, HIV, and the BBB
There is significant comorbidity in HIV-infected individuals who use substances of abuse. Thus, the effects of HIV infection and drug abuse on the BBB and viral entry into the CNS are a target of study (Fig. 1). (For a more comprehensive review, see Nath et al. 2002.)
Cocaine
The decline of neurocognitive function associated with cocaine abuse has similar properties to that associated with HAD (see Goodkin et al. 1998 for review of neuropathology). Cocaine was shown to induce inflammatory responses in the CNS through the up-regulation of cytokines (Fiala et al. 1996), and to alter the migration of monocytes across the BBB (Fiala et al. 1998). Thus, the role of cocaine in inflammation and monocyte trafficking into the CNS was significant and its effects on HIV infection were investigated. One study showed that cocaine increased the permeability of the BBB to the HIV-1JR-FL R5 strain (Zhang et al. 1998). Additionally, when BMVEC were treated with cocaine, there was increased production of IL-8/CXCL8, CCL2, CCL3, TNF-α, and interferon-inducible protein-10 (IP-10/CXCL10) (Zhang et al. 1998). Another report demonstrated that HIV binds BMVEC in a manner not yet identified, causing the formation of gaps in cell junctions (Fiala et al. 2005). Cocaine-treated BMVEC showed disruption of the actin cytoskeleton. The data further suggested that this disruption facilitated the aggregation of HIV into “cytoplasmic lakes,” altering cytoplasmic trafficking of the virus (Fiala et al. 2005). Changes in BBB permeability due to the toxic effects of cocaine, combined with the response of HIV-infected monocytes to inflammatory mediators, may explain some of the accelerated HAD and neuronal pathology described clinically in HIV-infected individuals who use substances of abuse, as well as in animal studies (Reyes et al. 1991; Nath et al. 2001, 2002).
Methamphetamine
Like cocaine, methamphetamine (METH) damages neurological function. A number of studies examined the effects of METH abuse in HIV-infected individuals; one study showed changes in expression of genes that are induced by interferon (Everall et al. 2005). Other studies showed that the presence of Tat and METH induced changes in expression of TNF-α, IL-1β, and ICAM-1 in the brain (Flora et al. 2003) as well as changes in dopaminergic activity (Maragos et al. 2002) and neuronal death (Turchan et al. 2001). Individuals who abuse METH have a higher plasma viral load, suggesting that the effectiveness of HAART is decreased with the use of this substance (Ellis et al. 2003). Thus, METH has a significant effect upon HIV-induced inflammation and progression of NeuroAIDS.
Morphine
The μ-opioid class of receptors (MOR) is associated with the effects of heroin, and its major metabolite, morphine (Matthes et al. 1996). Morphine increased the expression of the CXCR4 and CCR5 HIV coreceptors in T lymphocytes and monocytes, and the CCR5 receptor in primary human astrocytes (Mahajan et al. 2002; Steele et al. 2003). These findings suggest that morphine may increase both HIV binding to and infection of PBMCs and may be astrocytes (Mahajan et al. 2002; Steele et al. 2003). Studies of SIV-infected macaques that received morphine demonstrated increased CCR5 expression on PBMCs, as well as increased viral replication in the CNS (Suzuki et al. 2002; Kumar et al. 2004).
Morphine also affects chemokine production in astrocytes. When treated with Tat and morphine, human and mouse astrocytes increased their production of CCL2, which caused chemotaxis of microglia (El-Hage et al. 2005, 2006). PBMCs treated with DAMGO ([D-ala2, N-Me-Phe4, Gly-ol5] enkephalin), a ligand of MOR, produce CCL2, CCL5, and CXCL10 proinflammatory chemokines (Wetzel et al. 2000). When treated with 2-5AN6B, an inhibitor of morphine-potentiated HIV replication, PBMCs showed elevated expression of CCL2 and CCL5 (Homan et al. 2002). (For a more comprehensive review on opioid receptors and inflammation, see Rogers and Peterson 2003). These results support a mechanism by which inflammatory mediators of NeuroAIDS are significantly affected by morphine.
Alcohol
Alcohol abuse has been shown to cause neurological dysfunction and disruption of the BBB (for review, see Tyor and Middaugh 1999; Mann et al. 2001; Meyerhoff 2001). Its metabolism generated reactive oxygen species that caused phosphorylation of myosin light chain and TJP. This affected immune function, decreased the integrity of the BBB, and increased monocyte migration across a BBB model (Haorah et al. 2004, 2005a,b). HIV infection compounded these effects. Ethanol and Tat treatment caused a synergistic increase in CCL2 production in rat hippocampus and corpus striatum (Flora et al. 2005). Alcohol was also shown to increase CXCR4 expression in peripheral blood lymphocytes, and CCR5 in monocyte derived macrophages, suggesting increased susceptibility of these cells to infection and chemotactic signals (Wang et al. 2002; Liu et al. 2003).
Conclusion
Alterations in signaling between components of the BBB with either HIV proteins or factors produced in response to HIV infection, or with HIV-infected cells, disrupt BBB integrity and result in compromise, thereby promoting continued transmigration of activated monocytes and/or HIV-infected cells into the brain. A more complete understanding of the interactions among the BBB, nervous system, and immune system is critical to identification of the mechanisms that mediate NeuroAIDS. The use of animal models will facilitate this understanding as well as assist in the development of therapeutic strategies that will limit CNS dysfunction. Clarification of the contribution of substances of abuse to BBB disruption will enhance our understanding of the effects of HIV and drugs on neuroinflammation and comorbidity.
References
Albright AV, Shieh JT, Itoh T, Lee B, Pleasure D, O'Connor MJ, Doms RW, Gonzalez-Scarano F (1999) Microglia express CCR5, CXCR4, and CCR3, but of these, CCR5 is the principal coreceptor for human immunodeficiency virus type 1 dementia isolates. J Virol 73:205–213
Anderson ER, Gendelman HE, Xiong H (2004) Memantine protects hippocampal neuronal function in murine human immunodeficiency virus type 1 encephalitis. J Neurosci 24:7194–7198
Andras IE, Pu H, Deli MA, Nath A, Hennig B, Toboreck M (2003) HIV-1 Tat protein alters tight junction protein expresion and distribution in cultured brain endothelial cells. J Neurosci Res 72:255–265
Annunziata P, Cioni C, Toneatto S, Paccagnini E (1998) HIV-1 gp120 increases the permeability of rat brain endothelium cultures by a mechanism involving substance P. Aids 12:2377–2385
Annunziata P, Cioni C, Santonini R, Paccagnini E (2002) Substance P antagonist blocks leakage and reduces activation of cytokine-stimulated rat brain endothelium. J Neuroimmunol 131:41–49
Anthony IC, Ramage SN, Carnie FW, Simmonds P, Bell JE (2005) Influence of HAART on HIV-related CNS disease and neuroinflammation. J Neuropathol Exp Neurol 64:529–536
Apetrei C, Robertson DL, Marx PA (2004) The history of SIVS and AIDS: epidemiology, phylogeny and biology of isolates from naturally SIV infected non-human primates (NHP) in Africa. Front Biosci 9:225–254
Argyris EG, Acheampong E, Nunnari G, Mukhtar M, Williams KJ, Pomerantz RJ (2003) Human immunodeficiency virus type 1 enters primary human brain microvascular endothelial cells by a mechanism involving cell surface proteoglycans independent of lipid rafts. J Virol 77:12140–12151
Avgeropoulos N, Kelley B, Middaugh L, Arrigo S, Persidsky Y, Gendelman HE, Tyor WR (1998) SCID mice with HIV encephalitis develop behavioral abnormalities. J Acquir Immune Defic Syndr Hum Retrovirol 18:13–20
Baldeweg T, Catalan J, Gazzard BG (1998) Risk of HIV dementia and opportunistic brain disease in AIDS and zidovudine therapy. J Neurol Neurosurg Psychiatry 65:34–41
Bandopadhyay R, Orte C, Lawrenson JG, Reid AR, De Silva S, Allt G (2001) Contractile proteins in pericytes at the blood–brain and blood–retinal barriers. J Neurocytol 30:35–44
Banks WA, Akerstrom V, Kastin AJ (1998) Adsorptive endocytosis mediates the passage of HIV-1 across the blood–brain barrier: evidence for a post-internalization coreceptor. J Cell Sci 111(Pt 4):533–540
Banks WA, Freed EO, Wolf KM, Robinson SM, Franko M, Kumar VB (2001) Transport of human immunodeficiency virus type 1 pseudoviruses across the blood–brain barrier: role of envelope proteins and adsorptive endocytosis. J Virol 75:4681–4691
Benagiano V, Virgintino D, Maiorano E, Rizzi A, Palombo S, Roncali L, Ambrosi G (1996) VIP-like immunoreactivity within neurons and perivascular neuronal processes of the human cerebral cortex. Eur J Histochem 40:53–56
Bennett BA, Rusyniak DE, Hollingsworth CK (1995) HIV-1 gp120-induced neurotoxicity to midbrain dopamine cultures. Brain Res 705:168–176
Bitran M, Tapia W, Eugenin E, Orio P, Boric MP (1999) Neuropeptide Y induced inhibition of noradrenaline release in rat hypothalamus: role of receptor subtype and nitric oxide. Brain Res 851:87–93
Bobardt MD, Salmon P, Wang L, Esko JD, Gabuzda D, Fiala M, Trono D, Van der Schueren B, David G, Gallay PA (2004) Contribution of proteoglycans to human immunodeficiency virus type 1 brain invasion. J Virol 78:6567–6584
Boven LA, Middel J, Verhoef J, De Groot CJ, Nottet HS (2000) Monocyte infiltration is highly associated with loss of the tight junction protein zonula occludens in HIV-1-associated dementia. Neuropathol Appl Neurobiol 26:356–360
Braet K, Paemeleire K, D'Herde K, Sanderson MJ, Leybaert L (2001) Astrocyte–endothelial cell calcium signals conveyed by two signalling pathways. Eur J Neurosci 13:79–91
Browning Paul J, Wang EJ, Pettoello-Mantovani M, Raker C, Yurasov S, Goldstein MM, Horner JW, Chan J, Goldstein H (2000) Mice transgenic for monocyte-tropic HIV type 1 produce infectious virus and display plasma viremia: a new in vivo system for studying the postintegration phase of HIV replication. AIDS Res Hum Retroviruses 16:481–492
Buch S, Sui Y, Dhillon N, Potula R, Zien C, Pinson D, Li S, Dhillon S, Nicolay B, Sidelnik A, Li C, Villinger T, Bisarriya K, Narayan O (2004) Investigations on four host response factors whose expression is enhanced in X4 SHIV encephalitis. J Neuroimmunol 157:71–80
Burack JH, Barrett DC, Stall RD, Chesney MA, Ekstrand ML, Coates TJ (1993) Depressive symptoms and CD4 lymphocyte decline among HIV-infected men. JAMA 270:2568–2573
Cartier L, Hartley O, Dubois-Dauphin M, Krause KH (2005) Chemokine receptors in the central nervous system: role in brain inflammation and neurodegenerative diseases. Brain Res Brain Res Rev 48:16–42
Clapham PR, Weber JN, Whitby D, McIntosh K, Dalgleish AG, Maddon PJ, Deen KC, Sweet RW, Weiss RA (1989) Soluble CD4 blocks the infectivity of diverse strains of HIV and SIV for T cells and monocytes but not for brain and muscle cells. Nature 337:368–370
Clapham PR, Blanc D, Weiss RA (1991) Specific cell surface requirements for the infection of CD4-positive cells by human immunodeficiency virus types 1 and 2 and by simian immunodeficiency virus. Virology 181:703–715
Clements JE, Anderson MG, Zink MC, Joag SV, Narayan O (1994) The SIV model of AIDS encephalopathy. Role of neurotropic viruses in diseases. Res Publ Assoc Res Nerv Ment Dis 72:147–157
Clements JE, Li M, Gama L, Bullock B, Carruth LM, Mankowski JL, Zink MC (2005) The central nervous system is a viral reservoir in simian immunodeficiency virus-infected macaques on combined antiretroviral therapy: a model for human immunodeficiency virus patients on highly active antiretroviral therapy. J Neurovirol 11:180–189
Clerici M, Sarin A, Henkart PA, Shearer GM (1997) Apoptotic cell death and cytokine dysregulation in human immunodeficiency virus infection: pivotal factors in disease progression. Cell Death Differ 4:699–706
Cocchi F, DeVico AL, Garzino-Demo A, Arya SK, Gallo RC, Lusso P (1995) Identification of RANTES, MIP-1 alpha, and MIP-1 beta as the major HIV-suppressive factors produced by CD8+ T cells. Science 270:1811–1815
Collard CD, Park KA, Montalto MC, Alapati S, Buras JA, Stahl GL, Colgan SP (2002) Neutrophil-derived glutamate regulates vascular endothelial barrier function. J Biol Chem 277:14801–14811
Colovic M, Caccia S (2003) Liquid chromatographic determination of minocycline in brain-to-plasma distribution studies in the rat. J Chromatogr B Anal Technol Biomed Life Sci 791:337–343
Conant K, Garzino-Demo A, Nath A, McArthur JC, Halliday W, Power C, Gallo RC, Major EO (1998) Induction of monocyte chemoattractant protein-1 in HIV-1 Tat-stimulated astrocytes and elevation in AIDS dementia. Proc Natl Acad Sci USA 95:3117–3121
Czub S, Czub M, Koutsilieri E, Sopper S, Villinger F, Muller JG, Stahl-Hennig C, Riederer P, Ter Meulen V, Gosztonyi G (2004) Modulation of simian immunodeficiency virus neuropathology by dopaminergic drugs. Acta Neuropathol (Berl) 107:216–226
D'Aversa TG, Yu KO, Berman JW (2004) Expression of chemokines by human fetal microglia after treatment with the human immunodeficiency virus type 1 protein Tat. J Neurovirol 10:86–97
D'Aversa TG, Eugenin EA, Berman JW (2005) NeuroAIDS: contributions of the human immunodeficiency virus-1 proteins Tat and gp120 as well as CD40 to microglial activation. J Neurosci Res 81:436–446
Dallasta LM, Pisarov LA, Esplen JE, Werley JV, Moses AV, Nelson JA, Achim CL (1999) Blood–brain barrier tight junction disruption in human immunodeficiency virus-1 encephalitis. Am J Pathol 155:1915–1927
Dantzer R, Kelley KW (1989) Stress and immunity: an integrated view of relationships between the brain and the immune system. Life Sci 44:1995–2008
Dehouck MP, Vigne P, Torpier G, Breittmayer JP, Cecchelli R, Frelin C (1997) Endothelin-1 as a mediator of endothelial cell–pericyte interactions in bovine brain capillaries. J Cereb Blood Flow Metab 17:464–469
Del Maschio A, De Luigi A, Martin-Padura I, Brockhaus M, Bartfai T, Fruscella P, Adorini L, Martino G, Furlan R, De Simoni MG, Dejana E (1999) Leukocyte recruitment in the cerebrospinal fluid of mice with experimental meningitis is inhibited by an antibody to junctional adhesion molecule (JAM). J Exp Med 190:1351–1356
Desrosiers RC, Daniel MD, Li Y (1989) HIV-related lentiviruses of nonhuman primates. AIDS Res Hum Retroviruses 5:465–473
Diesing TS, Swindells S, Gelbard H, Gendelman HE (2002) HIV-1-associated dementia: a basic science and clinical perspective. AIDS Read 12:358–368
Dore-Duffy P, Owen C, Balabanov R, Murphy S, Beaumont T, Rafols JA (2000) Pericyte migration from the vascular wall in response to traumatic brain injury. Microvasc Res 60:55–69
Dou H, Ellison B, Bradley J, Kasiyanov A, Poluektova LY, Xiong H, Maggirwar S, Dewhurst S, Gelbard HA, Gendelman HE (2005) Neuroprotective mechanisms of lithium in murine human immunodeficiency virus-1 encephalitis. J Neurosci 25:8375–8385
Dzenko KA, Song L, Ge S, Kuziel WA, Pachter JS (2005) CCR2 expression by brain microvascular endothelial cells is critical for macrophage transendothelial migration in response to CCL2. Microvasc Res 70:53–64
Ebnet K, Suzuki A, Ohno S, Vestweber D (2004) Junctional adhesion molecules (JAMs): more molecules with dual functions? J Cell Sci 117:19–29
Edinger AL, Amedee A, Miller K, Doranz BJ, Endres M, Sharron M, Samson M, Lu ZH, Clements JE, Murphey-Corb M, Peiper SC, Parmentier M, Broder CC, Doms RW (1997) Differential utilization of CCR5 by macrophage and T cell tropic simian immunodeficiency virus strains. Proc Natl Acad Sci USA 94:4005–4010
Edinger AL, Hoffman TL, Sharron M, Lee B, Yi Y, Choe W, Kolson DL, Mitrovic B, Zhou Y, Faulds D, Collman RG, Hesselgesser J, Horuk R, Doms RW (1998) An orphan seven-transmembrane domain receptor expressed widely in the brain functions as a coreceptor for human immunodeficiency virus type 1 and simian immunodeficiency virus. J Virol 72:7934–7940
El-Hage N, Gurwell JA, Singh IN, Knapp PE, Nath A, Hauser KF (2005) Synergistic increases in intracellular Ca2+, and the release of MCP-1, RANTES, and IL-6 by astrocytes treated with opiates and HIV-1 Tat. Glia 50:91–106
El-Hage N, Wu G, Wang J, Ambati J, Knapp PE, Reed JL, Bruce-Keller AJ, Hauser KF (2006) HIV-1 Tat and opiate-induced changes in astrocytes promote chemotaxis of microglia through the expression of MCP-1 and alternative chemokines. Glia 53:132–146
Elfont RM, Sundaresan PR, Sladek CD (1989) Adrenergic receptors on cerebral microvessels: pericyte contribution. Am J Physiol 256:R224–230
Ellis RJ, Childers ME, Cherner M, Lazzaretto D, Letendre S, Grant I (2003) Increased human immunodeficiency virus loads in active methamphetamine users are explained by reduced effectiveness of antiretroviral therapy. J Infect Dis 188:1820–1826
Encinas M, Iglesias M, Liu Y, Wang H, Muhaisen A, Cena V, Gallego C, Comella JX (2000) Sequential treatment of SH-SY5Y cells with retinoic acid and brain-derived neurotrophic factor gives rise to fully differentiated, neurotrophic factor-dependent, human neuron-like cells. J Neurochem 75:991–1003
Eugenin EA, Branes MC, Berman JW, Saez JC (2003a) TNF-alpha plus IFN-gamma induce connexin43 expression and formation of gap junctions between human monocytes/macrophages that enhance physiological responses. J Immunol 170:1320–1328
Eugenin EA, D'Aversa TG, Lopez L, Calderon TM, Berman JW (2003b) MCP-1 (CCL2) protects human neurons and astrocytes from NMDA or HIV-tat-induced apoptosis. J Neurochem 85:1299–1311
Eugenin EA, Dyer G, Calderon TM, Berman JW (2005) HIV-1 tat protein induces a migratory phenotype in human fetal microglia by a CCL2 (MCP-1)-dependent mechanism: possible role in NeuroAIDS. Glia 49:501–510
Eugenin EA, Osiecki K, Lopez L, Goldstein H, Calderon TM, Berman JW (2006a) CCL2/monocyte chemoattractant protein-1 mediates enhanced transmigration of human immunodeficiency virus (HIV)-infected leukocytes across the blood–brain barrier: a potential mechanism of HIV-CNS invasion and NeuroAIDS. J Neurosci 26:1098–1106
Eugenin EA, Gamss R, Buckner C, Buono D, Klein RS, Schoenbaum EE, Calderon TM, Berman JW (2006b) Shedding of PECAM-1 during HIV infection: a potential role for soluble PECAM-1 in the pathogenesis of NeuroAIDS. J Leukoc Biol 79:444–452
Evans DL, Leserman J, Pedersen CA, Golden RN, Lewis MH, Folds JA, Ozer H (1989) Immune correlates of stress and depression. Psychopharmacol Bull 25:319–324
Everall IP, Trillo-Pazos G, Bell C, Mallory M, Sanders V, Masliah E (2001) Amelioration of neurotoxic effects of HIV envelope protein gp120 by fibroblast growth factor: a strategy for neuroprotection. J Neuropathol Exp Neurol 60:293–301
Everall I, Salaria S, Roberts E, Corbeil J, Sasik R, Fox H, Grant I, Masliah E (2005) Methamphetamine stimulates interferon inducible genes in HIV infected brain. J Neuroimmunol 170:158–171
Fiala AM, Gan XH, Newton T, Chiappelli F, Shapshak P, Kermani V, Kung MA, Diagne A, Martinez O, Way D, Weinand M, Witte M, Graves M (1996) Divergent effects of cocaine on cytokine production by lymphocytes and monocyte/macrophages: HIV-1 enhancement by cocaine within the blood–brain barrier. Adv Exp Med Biol 402:145–156
Fiala M, Looney DJ, Stins M, Way DD, Zhang L, Gan X, Chiappelli F, Schweitzer ES, Shapshak P, Weinand M, Graves MC, Witte M, Kim KS (1997) TNF-alpha opens a paracellular route for HIV-1 invasion across the blood–brain barrier. Mol Med 3:553–564
Fiala M, Gan XH, Zhang L, House SD, Newton T, Graves MC, Shapshak P, Stins M, Kim KS, Witte M, Chang SL (1998) Cocaine enhances monocyte migration across the blood–brain barrier. Cocaine's connection to AIDS dementia and vasculitis? Adv Exp Med Biol 437:199–205
Fiala M, Eshleman AJ, Cashman J, Lin J, Lossinsky AS, Suarez V, Yang W, Zhang J, Popik W, Singer E, Chiappelli F, Carro E, Weinand M, Witte M, Arthos J (2005) Cocaine increases human immunodeficiency virus type 1 neuroinvasion through remodeling brain microvascular endothelial cells. J Neurovirol 11:281–291
Flora G, Lee YW, Nath A, Hennig B, Maragos W, Toborek M (2003) Methamphetamine potentiates HIV-1 Tat protein-mediated activation of redox-sensitive pathways in discrete regions of the brain. Exp Neurol 179:60–70
Flora G, Pu H, Lee YW, Ravikumar R, Nath A, Hennig B, Toborek M (2005) Proinflammatory synergism of ethanol and HIV-1 Tat protein in brain tissue. Exp Neurol 191:2–12
Fox HS, Phillips TR (2002) FIV and neuroAIDS. J Neurovirol 8:155–157
Fox HS, Gold LH, Henriksen SJ, Bloom FE (1997) Simian immunodeficiency virus: a model for neuroAIDS. Neurobiol Dis 4:265–274
Fuentes ME, Durham SK, Swerdel MR, Lewin AC, Barton DS, Megill JR, Bravo R, Lira SA (1995) Controlled recruitment of monocytes and macrophages to specific organs through transgenic expression of monocyte chemoattractant protein-1. J Immunol 155:5769–5776
Fujii Y, Otake K, Tashiro M, Adachi A (1996) Human immunodeficiency virus type 1 Nef protein on the cell surface is cytocidal for human CD4+ T cells. FEBS Lett 393:105–108
Gabuzda D, Wang J (1999) Chemokine receptors and virus entry in the central nervous system. J Neurovirol 5:643–658
Gabuzda D, He J, Ohagen A, Vallat AV (1998) Chemokine receptors in HIV-1 infection of the central nervous system. Semin Immunol 10:203–213
Galey D, Becker K, Haughey N, Kalehua A, Taub D, Woodward J, Mattson MP, Nath A (2003) Differential transcriptional regulation by human immunodeficiency virus type 1 and gp120 in human astrocytes. J Neurovirol 9:358–371
Gartner S (2000) HIV infection and dementia. Science 287:602–604
Gaskill PJ, Watry DD, Burdo TH, Fox HS (2005) Development and characterization of positively selected brain-adapted SIV. Virol J 2:44
Gaspers LD, Thomas AP (2005) Calcium signaling in liver. Cell Calcium 38:329–342
Gendelman HE, Grant I, Lipton SA, Everall I, Swindells S (2005) The Neurology of AIDS. Oxford University Press, London
Gloor SM, Wachtel M, Bolliger MF, Ishihara H, Landmann R, Frei K (2001) Molecular and cellular permeability control at the blood–brain barrier. Brain Res Brain Res Rev 36:258–264
Goldstein GW (1988) Endothelial cell–astrocyte interactions. A cellular model of the blood–brain barrier. Ann N Y Acad Sci 529:31–39
Gonul E, Duz B, Kahraman S, Kayali H, Kubar A, Timurkaynak E (2002) Early pericyte response to brain hypoxia in cats: an ultrastructural study. Microvasc Res 64:116–119
Gonzalez E, Rovin BH, Sen L, Cooke G, Dhanda R, Mummidi S, Kulkarni H, Bamshad MJ, Telles V, Anderson SA, Walter EA, Stephan KT, Deucher M, Mangano A, Bologna R, Ahuja SS, Dolan MJ, Ahuja SK (2002) HIV-1 infection and AIDS dementia are influenced by a mutant MCP-1 allele linked to increased monocyte infiltration of tissues and MCP-1 levels. Proc Natl Acad Sci USA 99:13795–13800
Goodkin K, Shapshak P, Metsch LR, McCoy CB, Crandall KA, Kumar M, Fujimura RK, McCoy V, Zhang BT, Reyblat S, Xin KQ, Kumar AM (1998) Cocaine abuse and HIV-1 infection: epidemiology and neuropathogenesis. J Neuroimmunol 83:88–101
Griffin WC 3rd, Middaugh LD, Cook JE, Tyor WR (2004) The severe combined immunodeficient (SCID) mouse model of human immunodeficiency virus encephalitis: deficits in cognitive function. J Neurovirol 10:109–115
Haorah J, Heilman D, Diekmann C, Osna N, Donohue TM Jr, Ghorpade A, Persidsky Y (2004) Alcohol and HIV decrease proteasome and immunoproteasome function in macrophages: implications for impaired immune function during disease. Cell Immunol 229:139–148
Haorah J, Knipe B, Leibhart J, Ghorpade A, Persidsky Y (2005a) Alcohol-induced oxidative stress in brain endothelial cells causes blood–brain barrier dysfunction. J Leukoc Biol 78:1223–1232
Haorah J, Heilman D, Knipe B, Chrastil J, Leibhart J, Ghorpade A, Miller DW, Persidsky Y (2005b) Ethanol-induced activation of myosin light chain kinase leads to dysfunction of tight junctions and blood-brain barrier compromise. Alcohol Clin Exp Res 29:999–1009
Hatse S, Princen K, De Clercq E, Rosenkilde MM, Schwartz TW, Hernandez-Abad PE, Skerlj RT, Bridger GJ, Schols D (2005) AMD3465, a monomacrocyclic CXCR4 antagonist and potent HIV entry inhibitor. Biochem Pharmacol 70:752–761
Healy DP, Wilk S (1993) Localization of immunoreactive glutamyl aminopeptidase in rat brain. II. Distribution and correlation with angiotensin II. Brain Res 606:295–303
Hein A, Martin JP, Dorries R (2001) In vitro activation of feline immunodeficiency virus in ramified microglial cells from asymptomatically infected cats. J Virol 75:8090–8095
Hertz L, Peng L, Lai JC (1998) Functional studies in cultured astrocytes. Methods 16:293–310
Herz A (1995) Neurobiological principles of drug dependence. Exemplified by opioids and psychostimulants. Nervenarzt 66:3–14
Heyes MP, Ellis RJ, Ryan L, Childers ME, Grant I, Wolfson T, Archibald S, Jernigan TL (2001) Elevated cerebrospinal fluid quinolinic acid levels are associated with region-specific cerebral volume loss in HIV infection. Brain 124:1033–1042
Ho WZ, Douglas SD (2004) Substance P and neurokinin-1 receptor modulation of HIV. J Neuroimmunol 157:48–55
Ho WZ, Lai JP, Li Y, Douglas SD (2002) HIV enhances substance P expression in human immune cells. FASEB J 16:616–618
Homan JW, Steele AD, Martinand-Mari C, Rogers TJ, Henderson EE, Charubala R, Pfleiderer W, Reichenbach NL, Suhadolnik RJ (2002) Inhibition of morphine-potentiated HIV-1 replication in peripheral blood mononuclear cells with the nuclease-resistant 2-5A agonist analog, 2-5A(N6B). J Acquir Immune Defic Syndr 30:9–20
Hori S, Ohtsuki S, Hosoya K, Nakashima E, Terasaki T (2004) A pericyte-derived angiopoietin-1 multimeric complex induces occludin gene expression in brain capillary endothelial cells through Tie-2 activation in vitro. J Neurochem 89:503–513
Huber JD, Egleton RD, Davis TP (2001) Molecular physiology and pathophysiology of tight junctions in the blood–brain barrier. Trends Neurosci 24:719–725
Hurwitz AA, Berman JW, Lyman WD (1994) The role of the blood–brain barrier in HIV infection of the central nervous system. Adv Neuroimmunol 4:249–256
Ickovics JR, Hamburger ME, Vlahov D, Schoenbaum EE, Schuman P, Boland RJ, Moore J (2001) Mortality, CD4 cell count decline, and depressive symptoms among HIV-seropositive women: longitudinal analysis from the HIV Epidemiology Research Study. JAMA 285:1466–1474
Ilani T, Strous RD, Fuchs S (2004) Dopaminergic regulation of immune cells via D3 dopamine receptor: a pathway mediated by activated T cells. FASEB J 18:1600–1602
Joseph AM, Kumar M, Mitra D (2005) Nef: “necessary and enforcing factor” in HIV infection. Curr HIV Res 3:87–94
Julg B, Goebel FD (2005) What's New in HIV/AIDS? Chemokine receptor antagonists: a new era of HIV therapy? Infection 33:408–410
Kanmogne GD, Kennedy RC, Grammas P (2002) HIV-1 gp120 proteins and gp160 peptides are toxic to brain endothelial cells and neurons: possible pathway for HIV entry into the brain and HIV-associated dementia. J Neuropathol Exp Neurol 61:992–1000
Kanmogne GD, Primeaux C, Grammas P (2005) HIV-1 gp120 proteins alter tight junction protein expression and brain endothelial cell permeability: implications for the pathogenesis of HIV-associated dementia. J Neuropathol Exp Neurol 64:498–505
Kelder W, McArthur JC, Nance-Sproson T, McClernon D, Griffin DE (1998) beta-Chemokines MCP-1 and RANTES are selectively increased in cerebrospinal fluid of patients with human immunodeficiency virus-associated dementia. Ann Neurol 44:831–835
Kern TS, Engerman RL (1996) Capillary lesions develop in retina rather than cerebral cortex in diabetes and experimental galactosemia. Arch Ophthalmol 114:306–310
King J, Eugenin EA, Buckner C, Berman JW (2006) HIV-tat and neurotoxicity. Microbes Infect, in press
Krizbai IA, Deli MA, Pestenacz A, Siklos L, Szabo CA, Andras I, Joo F (1998) Expression of glutamate receptors on cultured cerebral endothelial cells. J Neurosci Res 54:814–819
Kumar R, Torres C, Yamamura Y, Rodriguez I, Martinez M, Staprans S, Donahoe RM, Kraiselburd E, Stephens EB, Kumar A (2004) Modulation by morphine of viral set point in rhesus macaques infected with simian immunodeficiency virus and simian–human immunodeficiency virus. J Virol 78:11425–11428
Lackner AA, Vogel P, Ramos RA, Kluge JD, Marthas M (1994) Early events in tissues during infection with pathogenic (SIVmac239) and nonpathogenic (SIVmac1A11) molecular clones of simian immunodeficiency virus. Am J Pathol 145:428–439
Lane TE, Buchmeier MJ, Watry DD, Fox HS (1996) Expression of inflammatory cytokines and inducible nitric oxide synthase in brains of SIV-infected rhesus monkeys: applications to HIV-induced central nervous system disease. Mol Med 2:27–37
Langford D, Grigorian A, Hurford R, Adame A, Ellis RJ, Hansen L, Masliah E (2004) Altered P-glycoprotein expression in AIDS patients with HIV encephalitis. J Neuropathol Exp Neurol 63:1038–1047
Lavi E, Kolson DL, Ulrich AM, Fu L, Gonzalez-Scarano F (1998) Chemokine receptors in the human brain and their relationship to HIV infection. J Neurovirol 4:301–311
Leserman J (2003) HIV disease progression: depression, stress, and possible mechanisms. Biol Psychiatry 54:295–306
Leserman J, Petitto JM, Perkins DO, Folds JD, Golden RN, Evans DL (1997) Severe stress, depressive symptoms, and changes in lymphocyte subsets in human immunodeficiency virus-infected men. A 2-year follow-up study. Arch Gen Psychiatry 54:279–285
Leserman J, Jackson ED, Petitto JM, Golden RN, Silva SG, Perkins DO, Cai J, Folds JD, Evans DL (1999) Progression to AIDS: the effects of stress, depressive symptoms, and social support. Psychosom Med 61:397–406
Leserman J, Petitto JM, Golden RN, Gaynes BN, Gu H, Perkins DO, Silva SG, Folds JD, Evans DL (2000) Impact of stressful life events, depression, social support, coping, and cortisol on progression to AIDS. Am J Psychiatry 157:1221–1228
Leserman J, Petitto JM, Gu H, Gaynes BN, Barroso J, Golden RN, Perkins DO, Folds JD, Evans DL (2002) Progression to AIDS, a clinical AIDS condition and mortality: psychosocial and physiological predictors. Psychol Med 32:1059–1073
Letendre S, Marquie-Beck J, Singh KK, de Almeida S, Zimmerman J, Spector SA, Grant I, Ellis R (2004) The monocyte chemotactic protein-1-2578G allele is associated with elevated MCP-1 concentrations in cerebrospinal fluid. J Neuroimmunol 157:193–196
Li Y, Douglas SD, Song L, Sun S, Ho WZ (2001) Substance P enhances HIV-1 replication in latently infected human immune cells. J Neuroimmunol 121:67–75
Lindahl P, Johansson BR, Leveen P, Betsholtz C (1997) Pericyte loss and microaneurysm formation in PDGF-B-deficient mice. Science 277:242–245
Lipton SA (1994) HIV-related neuronal injury. Potential therapeutic intervention with calcium channel antagonists and NMDA antagonists. Mol Neurobiol 8:181–196
Lipton SA (1998) Neuronal injury associated with HIV-1: approaches to treatment. Annu Rev Pharmacol Toxicol 38:159–177
Liu ZQ, Muhkerjee S, Sahni M, McCormick-Davis C, Leung K, Li Z, Gattone VH, 2nd, Tian C, Doms RW, Hoffman TL, Raghavan R, Narayan O, Stephens EB (1999) Derivation and biological characterization of a molecular clone of SHIV(KU-2) that causes AIDS, neurological disease, and renal disease in rhesus macaques. Virology 260:295–307
Liu Y, Jones M, Hingtgen CM, Bu G, Laribee N, Tanzi RE, Moir RD, Nath A, He JJ (2000) Uptake of HIV-1 tat protein mediated by low-density lipoprotein receptor-related protein disrupts the neuronal metabolic balance of the receptor ligands. Nat Med 6:1380–1387
Liu NQ, Lossinsky AS, Popik W, Li X, Gujuluva C, Kriederman B, Roberts J, Pushkarsky T, Bukrinsky M, Witte M, Weinand M, Fiala M (2002a) Human immunodeficiency virus type 1 enters brain microvascular endothelia by macropinocytosis dependent on lipid rafts and the mitogen-activated protein kinase signaling pathway. J Virol 76:6689–6700
Liu X, Jana M, Dasgupta S, Koka S, He J, Wood C, Pahan K (2002b) Human immunodeficiency virus type 1 (HIV-1) tat induces nitric-oxide synthase in human astroglia. J Biol Chem 277:39312–39319
Liu X, Zha J, Nishitani J, Chen H, Zack JA (2003) HIV-1 infection in peripheral blood lymphocytes (PBLs) exposed to alcohol. Virology 307:37–44
Liu Y, Shaw SK, Ma S, Yang L, Luscinskas FW, Parkos CA (2004) Regulation of leukocyte transmigration: cell surface interactions and signaling events. J Immunol 172:7–13
Loscher W, Potschka H (2005) Drug resistance in brain diseases and the role of drug efflux transporters. Nat Rev Neurosci 6:591–602
Lyketsos CG, Hoover DR, Guccione M, Senterfitt W, Dew MA, Wesch J, VanRaden MJ, Treisman GJ, Morgenstern H (1993) Depressive symptoms as predictors of medical outcomes in HIV infection. Multicenter AIDS Cohort Study. JAMA 270:2563–2567
Maclean AG, Belenchia GE, Bieniemy DN, Moroney-Rasmussen TA, Lackner AA (2005) Simian immunodeficiency virus disrupts extended lengths of the blood–brain barrier. J Med Primatol 34:237–242
Madden LJ, Zandonatti MA, Flynn CT, Taffe MA, Marcondes MC, Schmitz JE, Reimann KA, Henriksen SJ, Fox HS (2004) CD8+ cell depletion amplifies the acute retroviral syndrome. J Neurovirol 10(Suppl 1):58–66
Mahajan SD, Schwartz SA, Shanahan TC, Chawda RP, Nair MP (2002) Morphine regulates gene expression of alpha- and beta-chemokines and their receptors on astroglial cells via the opioid mu receptor. J Immunol 169:3589–3599
Malessa R, Heimbach M, Brockmeyer NH, Hengge U, Rascher W, Michel MC (1996) Increased neuropeptide Y-like immunoreactivity in cerebrospinal fluid and plasma of human immunodeficiency virus-infected patients: relationship to HIV encephalopathy. J Neurol Sci 136:154–158
Mankowski JL, Spelman JP, Ressetar HG, Strandberg JD, Laterra J, Carter DL, Clements JE, Zink MC (1994) Neurovirulent simian immunodeficiency virus replicates productively in endothelial cells of the central nervous system in vivo and in vitro. J Virol 68:8202–8208
Mankowski JL, Queen SE, Clements JE, Zink MC (2004) Cerebrospinal fluid markers that predict SIV CNS disease. J Neuroimmunol 157:66–70
Mann K, Agartz I, Harper C, Shoaf S, Rawlings RR, Momenan R, Hommer DW, Pfefferbaum A, Sullivan EV, Anton RF, Drobes DJ, George MS, Bares R, Machulla HJ, Mundle G, Reimold M, Heinz A (2001) Neuroimaging in alcoholism: ethanol and brain damage. Alcohol Clin Exp Res 25:104S–109S
Maragos WF, Young KL, Turchan JT, Guseva M, Pauly JR, Nath A, Cass WA (2002) Human immunodeficiency virus-1 Tat protein and methamphetamine interact synergistically to impair striatal dopaminergic function. J Neurochem 83:955–963
Marechal V, Prevost MC, Petit C, Perret E, Heard JM, Schwartz O (2001) Human immunodeficiency virus type 1 entry into macrophages mediated by macropinocytosis. J Virol 75:11166–11177
Martin-Garcia J, Kolson DL, Gonzalez-Scarano F (2002) Chemokine receptors in the brain: their role in HIV infection and pathogenesis. Aids 16:1709–1730
Martin-Padura I, Lostaglio S, Schneemann M, Williams L, Romano M, Fruscella P, Panzeri C, Stoppacciaro A, Ruco L, Villa A, Simmons D, Dejana E (1998) Junctional adhesion molecule, a novel member of the immunoglobulin superfamily that distributes at intercellular junctions and modulates monocyte transmigration. J Cell Biol 142:117–127
Martiney JA, Berman JW, Brosnan CF (1992) Chronic inflammatory effects of interleukin-1 on the blood–retina barrier. J Neuroimmunol 41:167–176
Marx PA, Chen Z (1998) The function of simian chemokine receptors in the replication of SIV. Semin Immunol 10:215–223
Matthes HW, Maldonado R, Simonin F, Valverde O, Slowe S, Kitchen I, Befort K, Dierich A, Le Meur M, Dolle P, Tzavara E, Hanoune J, Roques BP, Kieffer BL (1996) Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature 383:819–823
McArthur JC, Hoover DR, Bacellar H, Miller EN, Cohen BA, Becker JT, Graham NM, McArthur JH, Selnes OA, Jacobson LP et al (1993) Dementia in AIDS patients: incidence and risk factors. Multicenter AIDS Cohort Study. Neurology 43:2245–2252
McArthur JC, Haughey N, Gartner S, Conant K, Pardo C, Nath A, Sacktor N (2003) Human immunodeficiency virus-associated dementia: an evolving disease. J Neurovirol 9:205–221
McManus CM, Weidenheim K, Woodman SE, Nunez J, Hesselgesser J, Nath A, Berman JW (2000) Chemokine and chemokine-receptor expression in human glial elements: induction by the HIV protein, Tat, and chemokine autoregulation. Am J Pathol 156:1441–1453
Meyerhoff DJ (2001) Effects of alcohol and HIV infection on the central nervous system. Alcohol Res Health 25:288–298
Moses AV, Bloom FE, Pauza CD, Nelson JA (1993) Human immunodeficiency virus infection of human brain capillary endothelial cells occurs via a CD4/galactosylceramide-independent mechanism. Proc Natl Acad Sci USA 90:10474–10478
Muller WA (2003) Leukocyte–endothelial-cell interactions in leukocyte transmigration and the inflammatory response. Trends Immunol 24:327–334
Murray EA, Rausch DM, Lendvay J, Sharer LR, Eiden LE (1992) Cognitive and motor impairments associated with SIV infection in rhesus monkeys. Science 255:1246–1249
Muthumani K, Choo AY, Premkumar A, Hwang DS, Thieu KP, Desai BM, Weiner DB (2005) Human immunodeficiency virus type 1 (HIV-1) Vpr-regulated cell death: insights into mechanism. Cell Death Differ 12(Suppl 1):962–970
Narcisse L, Scemes E, Zhao Y, Lee SC, Brosnan CF (2005) The cytokine IL-1beta transiently enhances P2X7 receptor expression and function in human astrocytes. Glia 49:245–258
Nath A, Maragos WF, Avison MJ, Schmitt FA, Berger JR (2001) Acceleration of HIV dementia with methamphetamine and cocaine. J Neurovirol 7:66–71
Nath A, Hauser KF, Wojna V, Booze RM, Maragos W, Prendergast M, Cass W, Turchan JT (2002) Molecular basis for interactions of HIV and drugs of abuse. J Acquir Immune Defic Syndr 31(Suppl 2):S62–S69
Nathanson MH, Burgstahler AD, Mennone A, Fallon MB, Gonzalez CB, Saez JC (1995) Ca2+ waves are organized among hepatocytes in the intact organ. Am J Physiol 269:G167–G171
Nosheny RL, Mocchetti I, Bachis A (2005) Brain-derived neurotrophic factor as a prototype neuroprotective factor against HIV-1-associated neuronal degeneration. Neurotox Res 8:187–198
Nottet HS, Persidsky Y, Sasseville VG, Nukuna AN, Bock P, Zhai QH, Sharer LR, McComb RD, Swindells S, Soderland C, Gendelman HE (1996) Mechanisms for the transendothelial migration of HIV-1-infected monocytes into brain. J Immunol 156:1284–1295
Nukuna A, Gendelman HE, Limoges J, Rasmussen J, Poluektova L, Ghorpade A, Persidsky Y (2004) Levels of human immunodeficiency virus type 1 (HIV-1) replication in macrophages determines the severity of murine HIV-1 encephalitis. J Neurovirol 10(Suppl 1):82–90
Nuovo GJ, Alfieri ML (1996) AIDS dementia is associated with massive, activated HIV-1 infection and concomitant expression of several cytokines. Mol Med 2:358–366
Olivetta E, Percario Z, Fiorucci G, Mattia G, Schiavoni I, Dennis C, Jager J, Harris M, Romeo G, Affabris E, Federico M (2003) HIV-1 Nef induces the release of inflammatory factors from human monocyte/macrophages: involvement of Nef endocytotic signals and NF-kappa B activation. J Immunol 170:1716–1727
Orandle MS, MacLean AG, Sasseville VG, Alvarez X, Lackner AA (2002) Enhanced expression of proinflammatory cytokines in the central nervous system is associated with neuroinvasion by simian immunodeficiency virus and the development of encephalitis. J Virol 76:5797–5802
Osiecki K, Xie L, Zheng JH, Squires R, Pettoello-Mantovani M, Goldstein H (2005) Identification of granulocyte–macrophage colony-stimulating factor and lipopolysaccharide-induced signal transduction pathways that synergize to stimulate HIV type 1 production by monocytes from HIV type 1 transgenic mice. AIDS Res Hum Retroviruses 21:125–139
Ostermann G, Weber KS, Zernecke A, Schroder A, Weber C (2002) JAM-1 is a ligand of the beta(2) integrin LFA-1 involved in transendothelial migration of leukocytes. Nat Immunol 3:151–158
Owens T, Tran E, Hassan-Zahraee M, Krakowski M (1998) Immune cell entry to the CNS—a focus for immunoregulation of EAE. Res Immunol 149:781–789; discussion 844–786, 855–760
Paemeleire K (2002a) The cellular basis of neurovascular metabolic coupling. Acta Neurol Belg 102:153–157
Paemeleire K. (2002b) Calcium signaling in and between brain astrocytes and endothelial cells. Acta Neurol Belg 102:137–140
Pardridge WM, Choi TB (1986) Neutral amino acid transport at the human blood–brain barrier. Fed Proc 45:2073–2078
Pardridge WM, Triguero D, Yang J, Cancilla PA (1990) Comparison of in vitro and in vivo models of drug transcytosis through the blood–brain barrier. J Pharmacol Exp Ther 253:884–891
Patel CA, Mukhtar M, Pomerantz RJ (2000) Human immunodeficiency virus type 1 Vpr induces apoptosis in human neuronal cells. J Virol 74:9717–9726
Perry SW, Norman JP, Gelbard HA (2005) Adjunctive therapies for HIV-1 associated neurologic disease. Neurotox Res 8:161–166
Persidsky Y, Limoges J, McComb R, Bock P, Baldwin T, Tyor W, Patil A, Nottet HS, Epstein L, Gelbard H, Flanagan E, Reinhard J, Pirruccello SJ, Gendelman HE (1996) Human immunodeficiency virus encephalitis in SCID mice. Am J Pathol 149:1027–1053
Persidsky Y, Stins M, Way D, Witte MH, Weinand M, Kim KS, Bock P, Gendelman HE, Fiala M (1997) A model for monocyte migration through the blood–brain barrier during HIV-1 encephalitis. J Immunol 158:3499–3510
Persidsky Y, Ghorpade A, Rasmussen J, Limoges J, Liu XJ, Stins M, Fiala M, Way D, Kim KS, Witte MH, Weinand M, Carhart L, Gendelman HE (1999) Microglial and astrocyte chemokines regulate monocyte migration through the blood–brain barrier in human immunodeficiency virus-1 encephalitis. Am J Pathol 155:1599–1611
Persidsky Y, Potula R, Haorah J (2005) Rodent model systems for studies of HIV-1 associated dementia. Neurotox Res 8:91–106
Peruzzi F, Bergonzini V, Aprea S, Reiss K, Sawaya BE, Rappaport J, Amini S, Khalili K (2005) Cross talk between growth factors and viral and cellular factors alters neuronal signaling pathways: implication for HIV-associated dementia. Brain Res Brain Res Rev 50:114–125
Poli A, Pistello M, Carli MA, Abramo F, Mancuso G, Nicoletti E, Bendinelli M (1999) Tumor necrosis factor-alpha and virus expression in the central nervous system of cats infected with feline immunodeficiency virus. J Neurovirol 5:465–473
Potash MJ, Chao W, Bentsman G, Paris N, Saini M, Nitkiewicz J, Belem P, Sharer L, Brooks AI, Volsky DJ (2005) A mouse model for study of systemic HIV-1 infection, antiviral immune responses, and neuroinvasiveness. Proc Natl Acad Sci USA 102:3760–3765
Potula R, Dhillion N, Sui Y, Zien CA, Funa K, Pinson D, Mayo MS, Singh DK, Narayan O, Buch S (2004) Association of platelet-derived growth factor-B chain with simian human immunodeficiency virus encephalitis. Am J Pathol 165:815–824
Princen K, Schols D (2005) HIV chemokine receptor inhibitors as novel anti-HIV drugs. Cytokine Growth Factor Rev 16:659–677
Princen K, Hatse S, Vermeire K, Aquaro S, De Clercq E, Gerlach LO, Rosenkilde M, Schwartz TW, Skerlj R, Bridger G, Schols D (2004) Inhibition of human immunodeficiency virus replication by a dual CCR5/CXCR4 antagonist. J Virol 78:12996–13006
Pu H, Tian J, Flora G, Lee YW, Nath A, Hennig B, Toborek M (2003) HIV-1 Tat protein upregulates inflammatory mediators and induces monocyte invasion into the brain. Mol Cell Neurosci 24:224–237
Raghavan R, Stephens EB, Joag SV, Adany I, Pinson DM, Li Z, Jia F, Sahni M, Wang C, Leung K, Foresman L, Narayan O (1997) Neuropathogenesis of chimeric simian/human immunodeficiency virus infection in pig-tailed and rhesus macaques. Brain Pathol 7:851–861
Ramirez SH, Sanchez JF, Dimitri CA, Gelbard HA, Dewhurst S, Maggirwar SB (2001) Neurotrophins prevent HIV Tat-induced neuronal apoptosis via a nuclear factor-kappaB (NF-kappaB)-dependent mechanism. J Neurochem 78:874–889
Reisberg B, Doody R, Stoffler A, Schmitt F, Ferris S, Mobius HJ (2003) Memantine in moderate-to-severe Alzheimer's disease. N Engl J Med 348:1333–1341
Reyes MG, Faraldi F, Senseng CS, Flowers C, Fariello R (1991) Nigral degeneration in acquired immune deficiency syndrome (AIDS). Acta Neuropathol (Berl) 82:39–44
Risau W (1991) Induction of blood–brain barrier endothelial cell differentiation. Ann N Y Acad Sci 633:405–419
Risau W, Wolburg H (1990) Development of the blood–brain barrier. Trends Neurosci 13:174–178
Roberts ES, Zandonatti MA, Watry DD, Madden LJ, Henriksen SJ, Taffe MA, Fox HS (2003) Induction of pathogenic sets of genes in macrophages and neurons in NeuroAIDS. Am J Pathol 162:2041–2057
Roberts ES, Burudi EM, Flynn C, Madden LJ, Roinick KL, Watry DD, Zandonatti MA, Taffe MA, Fox HS (2004) Acute SIV infection of the brain leads to upregulation of IL6 and interferon-regulated genes: expression patterns throughout disease progression and impact on neuroAIDS. J Neuroimmunol 157:81–92
Robertson KR, Robertson WT, Ford S, Watson D, Fiscus S, Harp AG, Hall CD (2004) Highly active antiretroviral therapy improves neurocognitive functioning. J Acquir Immune Defic Syndr 36:562–566
Rogers TJ, Peterson PK (2003) Opioid G protein-coupled receptors: signals at the crossroads of inflammation. Trends Immunol 24:116–121
Rubin LL, Staddon JM (1999) The cell biology of the blood–brain barrier. Annu Rev Neurosci 22:11–28
Sanders VJ, Pittman CA, White MG, Wang G, Wiley CA, Achim CL (1998) Chemokines and receptors in HIV encephalitis. Aids 12:1021–1026
Sasseville VG, Smith MM, Mackay CR, Pauley DR, Mansfield KG, Ringler DJ, Lackner AA (1996) Chemokine expression in simian immunodeficiency virus-induced AIDS encephalitis. Am J Pathol 149:1459–1467
Schenkel AR, Mamdouh Z, Chen X, Liebman RM, Muller WA (2002) CD99 plays a major role in the migration of monocytes through endothelial junctions. Nat Immunol 3:143–150
Schmitz JE, Kuroda MJ, Santra S, Sasseville VG, Simon MA, Lifton MA, Racz P, Tenner-Racz K, Dalesandro M, Scallon BJ, Ghrayeb J, Forman MA, Montefiori DC, Rieber EP, Letvin NL, Reimann KA (1999) Control of viremia in simian immunodeficiency virus infection by CD8+ lymphocytes. Science 283:857–860
Seilhean D, Dzia-Lepfoundzou A, Sazdovitch V, Cannella B, Raine CS, Katlama C, Bricaire F, Duyckaerts C, Hauw JJ (1997) Astrocytic adhesion molecules are increased in HIV-1-associated cognitive/motor complex. Neuropathol Appl Neurobiol 23:83–92
Sevigny JJ, Albert SM, McDermott MP, McArthur JC, Sacktor N, Conant K, Schifitto G, Selnes OA, Stern Y, McClernon DR, Palumbo D, Kieburtz K, Riggs G, Cohen B, Epstein LG, Marder K (2004) Evaluation of HIV RNA and markers of immune activation as predictors of HIV-associated dementia. Neurology 63:2084–2090
Sharp CD, Hines I, Houghton J, Warren A, Jackson THt, Jawahar A, Nanda A, Elrod JW, Long A, Chi A, Minagar A, Alexander JS (2003) Glutamate causes a loss in human cerebral endothelial barrier integrity through activation of NMDA receptor. Am J Physiol Heart Circ Physiol 285:H2592–H2598
Sharp CD, Houghton J, Elrod JW, Warren A, Jackson THt, Jawahar A, Nanda A, Minagar A, Alexander JS (2005a) N-Methyl-d-aspartate receptor activation in human cerebral endothelium promotes intracellular oxidant stress. Am J Physiol Heart Circ Physiol 288:H1893–H1899
Sharp PM, Shaw GM, Hahn BH (2005b) Simian immunodeficiency virus infection of chimpanzees. J Virol 79:3891–3902
Shibata R, Adachi A (1992) SIV/HIV recombinants and their use in studying biological properties. AIDS Res Hum Retroviruses 8:403–409
Shibata R, Kawamura M, Sakai H, Hayami M, Ishimoto A, Adachi A (1991) Generation of a chimeric human and simian immunodeficiency virus infectious to monkey peripheral blood mononuclear cells. J Virol 65:3514–3520
Si Q, Kim MO, Zhao ML, Landau NR, Goldstein H, Lee S (2002) Vpr- and Nef-dependent induction of RANTES/CCL5 in microglial cells. Virology 301:342–353
Si Q, Cosenza M, Kim MO, Zhao ML, Brownlee M, Goldstein H, Lee S (2004) A novel action of minocycline: inhibition of human immunodeficiency virus type 1 infection in microglia. J Neurovirol 10:284–292
Sidtis JJ, Gatsonis C, Price RW, Singer EJ, Collier AC, Richman DD, Hirsch MS, Schaerf FW, Fischl MA, Kieburtz K et al (1993) Zidovudine treatment of the AIDS dementia complex: results of a placebo-controlled trial. AIDS Clinical Trials Group. Ann Neurol 33:343–349
Simard M, Arcuino G, Takano T, Liu QS, Nedergaard M (2003) Signaling at the gliovascular interface. J Neurosci 23:9254–9262
Smit TK, Brew BJ, Tourtellotte W, Morgello S, Gelman BB, Saksena NK (2004) Independent evolution of human immunodeficiency virus (HIV) drug resistance mutations in diverse areas of the brain in HIV-infected patients, with and without dementia, on antiretroviral treatment. J Virol 78:10133–10148
Sobue K, Yamamoto N, Yoneda K, Hodgson ME, Yamashiro K, Tsuruoka N, Tsuda T, Katsuya H, Miura Y, Asai K, Kato T (1999) Induction of blood–brain barrier properties in immortalized bovine brain endothelial cells by astrocytic factors. Neurosci Res 35:155–164
Speth C, Dierich MP, Sopper S (2005) HIV-infection of the central nervous system: the tightrope walk of innate immunity. Mol Immunol 42:213–228
Steele AD, Henderson EE, Rogers TJ (2003) Mu-opioid modulation of HIV-1 coreceptor expression and HIV-1 replication. Virology 309:99–107
Stent G, Crowe SM (1997) Effects of HIV-1 on the surface expression of LFA-1 on cultured monocytes. J Acquir Immune Defic Syndr Hum Retrovirol 15:95–103
Stephens EB, Singh DK, Kohler ME, Jackson M, Pacyniak E, Berman NE (2003) The primary phase of infection by pathogenic simian–human immunodeficiency virus results in disruption of the blood–brain barrier. AIDS Res Hum Retroviruses 19:837–846
Strazielle N, Khuth ST, Ghersi-Egea JF (2004) Detoxification systems, passive and specific transport for drugs at the blood–CSF barrier in normal and pathological situations. Adv Drug Deliv Rev 56:1717–1740
Strelow LI, Janigro D, Nelson JA (2001) The blood–brain barrier and AIDS. Adv Virus Res 56:355–388
Sui Y, Potula R, Pinson D, Adany I, Li Z, Day J, Buch E, Segebrecht J, Villinger F, Liu Z, Huang M, Narayan O, Buch S (2003) Microarray analysis of cytokine and chemokine genes in the brains of macaques with SHIV-encephalitis. J Med Primatol 32:229–239
Sun J, Osiecki K, Zheng J, Falkin L, Santambrogio L, Littman DR, Goldstein H (2006) CD4-specific transgenic expression of human cyclin T1 markedly increases HIV-1 production by CD4+ lymphocytes and myeloid committed cells in mice trasgenic for a provirus encoding a monocyte-tropic HIV-1 isolate. J Virol 80:1850–1862
Suzuki S, Chuang AJ, Chuang LF, Doi RH, Chuang RY (2002) Morphine promotes simian acquired immunodeficiency syndrome virus replication in monkey peripheral mononuclear cells: induction of CC chemokine receptor 5 expression for virus entry. J Infect Dis 185:1826–1829
Swingler S, Mann A, Jacque J, Brichacek B, Sasseville VG, Williams K, Lackner AA, Janoff EN, Wang R, Fisher D, Stevenson M (1999) HIV-1 Nef mediates lymphocyte chemotaxis and activation by infected macrophages. Nat Med 5:997–1103
Thomas WE (1999) Brain macrophages: on the role of pericytes and perivascular cells. Brain Res Brain Res Rev 31:42–57
Tikka T, Fiebich BL, Goldsteins G, Keinanen R, Koistinaho J (2001) Minocycline, a tetracycline derivative, is neuroprotective against excitotoxicity by inhibiting activation and proliferation of microglia. J Neurosci 21:2580–2588
Trillo-Pazos G, McFarlane-Abdulla E, Campbell IC, Pilkington GJ, Everall IP (2000) Recombinant nef HIV-IIIB protein is toxic to human neurons in culture. Brain Res 864:315–326
Turchan J, Anderson C, Hauser KF, Sun Q, Zhang J, Liu Y, Wise PM, Kruman I, Maragos W, Mattson MP, Booze R, Nath A (2001) Estrogen protects against the synergistic toxicity by HIV proteins, methamphetamine and cocaine. BMC Neurosci 2:3
Tyor WR, Middaugh LD (1999) Do alcohol and cocaine abuse alter the course of HIV-associated dementia complex? J Leukoc Biol 65:475–481
Tyor WR, Power C, Gendelman HE, Markham RB (1993) A model of human immunodeficiency virus encephalitis in scid mice. Proc Natl Acad Sci USA 90:8658–8662
Unutmaz D, Xiang W, Sunshine MJ, Campbell J, Butcher E, Littman DR (2000) The primate lentiviral receptor Bonzo/STRL33 is coordinately regulated with CCR5 and its expression pattern is conserved between human and mouse. J Immunol 165:3284–3292
van Buul JD, Hordijk PL (2004) Signaling in leukocyte transendothelial migration. Arterioscler Thromb Vasc Biol 24:824–833
van Zwieten EJ, Ravid R, Swaab DF, Van de Woude T (1988) Immunocytochemically stained vasopressin binding sites on blood vessels in the rat brain. Brain Res 474:369–373
Verbeek MM, de Waal RM, Schipper JJ, Van Nostrand WE (1997) Rapid degeneration of cultured human brain pericytes by amyloid beta protein. J Neurochem 68:1135–1141
Virgintino D, Robertson D, Errede M, Benagiano V, Bertossi M, Ambrosi G, Roncali L (2001) Expression of the gap junction protein connexin43 in human telencephalon microvessels. Microvasc Res 62:435–439
Wang X, Douglas SD, Metzger DS, Guo CJ, Li Y, O'Brien CP, Song L, Davis-Vogal A, Ho WZ (2002) Alcohol potentiates HIV-1 infection of human blood mononuclear phagocytes. Alcohol Clin Exp Res 26:1880–1886
Wang EJ, Sun J, Pettoello-Mantovani M, Anderson CM, Osiecki K, Zhao ML, Lopez L, Lee SC, Berman JW, Goldstein H (2003a) Microglia from mice transgenic for a provirus encoding a monocyte-tropic HIV type 1 isolate produce infectious virus and display in vitro and in vivo upregulation of lipopolysaccharide-induced chemokine gene expression. AIDS Res Hum Retroviruses 19:755–765
Wang Z, Pekarskaya O, Bencheikh M, Chao W, Gelbard HA, Ghorpade A, Rothstein JD, Volsky DJ (2003b) Reduced expression of glutamate transporter EAAT2 and impaired glutamate transport in human primary astrocytes exposed to HIV-1 or gp120. Virology 312:60–73
Wang GJ, Chang L, Volkow ND, Telang F, Logan J, Ernst T, Fowler JS (2004) Decreased brain dopaminergic transporters in HIV-associated dementia patients. Brain 127:2452–2458
Weed MR, Steward DJ (2005) Neuropsychopathology in the SIV/macaque model of AIDS. Front Biosci 10:710–727
Weisse CS (1992) Depression and immunocompetence: a review of the literature. Psychol Bull 111:475–489
Weiss JM, Nath A, Major EO, Berman JW (1999) HIV-1 Tat induces monocyte chemoattractant protein-1-mediated monocyte transmigration across a model of the human blood–brain barrier and up-regulates CCR5 expression on human monocytes. J Immunol 163:2953–2959
Wetzel MA, Steele AD, Eisenstein TK, Adler MW, Henderson EE, Rogers TJ (2000) Mu-opioid induction of monocyte chemoattractant protein-1, RANTES, and IFN-gamma-inducible protein-10 expression in human peripheral blood mononuclear cells. J Immunol 165:6519–6524
Williams KC, Corey S, Westmoreland SV, Pauley D, Knight H, deBakker C, Alvarez X, Lackner AA (2001) Perivascular macrophages are the primary cell type productively infected by simian immunodeficiency virus in the brains of macaques: implications for the neuropathogenesis of AIDS. J Exp Med 193:905–915
Woodman SE, Benveniste EN, Nath A, Berman JW (1999) Human immunodeficiency virus type 1 TAT protein induces adhesion molecule expression in astrocytes. J Neurovirol 5:678–684
Yi Y, Lee C, Liu QH, Freedman BD, Collman RG (2004) Chemokine receptor utilization and macrophage signaling by human immunodeficiency virus type 1 gp120: implications for neuropathogenesis. J Neurovirol 10(Suppl 1):91–96
Zhang L, Looney D, Taub D, Chang SL, Way D, Witte MH, Graves MC, Fiala M (1998) Cocaine opens the blood–brain barrier to HIV-1 invasion. J Neurovirol 4:619–626
Zhu HJ, Liu GQ (2004) Glutamate up-regulates P-glycoprotein expression in rat brain microvessel endothelial cells by an NMDA receptor-mediated mechanism. Life Sci 75:1313–1322
Zink MC, Clements JE (2000) A rapid, reproducible model of AIDS and encephalitis in SIV infected macaques demonstrates the role of viral load in CNS disease. NeuroAIDS 3: Online AAAS Science publication http://AID Science.org/NeuroAIDS/
Zink MC, Clements JE (2002) A novel simian immunodeficiency virus model that provides insight into mechanisms of human immunodeficiency virus central nervous system disease. J Neurovirol 8(Suppl 2):42–48
Zink MC, Spelman JP, Robinson RB, Clements JE (1998) SIV infection of macaques—modeling the progression to AIDS dementia. J Neurovirol 4:249–259
Zink MC, Suryanarayana K, Mankowski JL, Shen A, Piatak M Jr, Spelman JP, Carter DL, Adams RJ, Lifson JD, Clements JE (1999) High viral load in the cerebrospinal fluid and brain correlates with severity of simian immunodeficiency virus encephalitis. J Virol 73:10480–10488
Zink MC, Coleman GD, Mankowski JL, Adams RJ, Tarwater PM, Fox K, Clements JE (2001) Increased macrophage chemoattractant protein-1 in cerebrospinal fluid precedes and predicts simian immunodeficiency virus encephalitis. J Infect Dis 184:1015–1021
Zink MC, Uhrlaub J, DeWitt J, Voelker T, Bullock B, Mankowski J, Tarwater P, Clements J, Barber S (2005) Neuroprotective and anti-human immunodeficiency virus activity of minocycline. JAMA 293:2003–2011
Zukowska-Grojec Z, Karwatowska-Prokopczuk E, Rose W, Rone J, Movafagh S, Ji H, Yeh Y, Chen WT, Kleinman HK, Grouzmann E, Grant DS (1998) Neuropeptide Y: a novel angiogenic factor from the sympathetic nerves and endothelium. Circ Res 83:187–195
Acknowledgements
This work was supported by the National Institute of Mental Health grant, MH52974 and MH0702297, National Institutes of Health grant NS11920, NIH Centers for AIDS Research grant AI-051519 (to all authors), and Molecular Neuropathology Training grant NIH NS07098 (to CMB).
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Buckner, C.M., Luers, A.J., Calderon, T.M. et al. Neuroimmunity and the Blood–Brain Barrier: Molecular Regulation of Leukocyte Transmigration and Viral Entry into the Nervous System with a Focus on NeuroAIDS. Jrnl NeuroImmune Pharm 1, 160–181 (2006). https://doi.org/10.1007/s11481-006-9017-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11481-006-9017-3