
Abstract
Urinary metabolites of α-pyrrolidinovalerophenone (α-PVP) in humans were investigated by analyzing urine specimens obtained from abusers. Unambiguous identification and accurate quantification of major metabolites were realized using gas chromatography–mass spectrometry and liquid chromatography-tandem mass spectrometry with newly synthesized authentic standards. Two major metabolic pathways were revealed: (1) the reduction of the β-keto moiety to 1-phenyl-2-(pyrrolidin-1-yl)pentan-1-ol (OH-α-PVP, diastereomers) partly followed by conjugation to its glucuronide, and (2) the oxidation at the 2″-position of the pyrrolidine ring to α-(2″-oxo-pyrrolidino)valerophenone (2″-oxo-α-PVP) via the putative intermediate α-(2″-hydroxypyrrolidino)valerophenone (2″-OH-α-PVP). Of the metabolites retaining the structural characteristics of the parent drug, OH-α-PVP was most abundant in most of the specimens examined.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
α-Pyrrolidinovalerophenone (α-PVP) [1] is one of a new class of designer drugs known as α-pyrrolidinophenones, which also includes pyrovalerone [2, 3], methylenedioxypyrovalerone (MDPV) [3, 4], 4-methyl-α-pyrrolidinopropiophenone (MPPP) [5], and 3,4-methylenedioxy-α-pyrrolidinopropiophenone (MDPPP) [6]. This particular class of drugs provides selective inhibitors of dopamine and norepinephrine transporters with little effect on serotonin trafficking, resulting in central nervous system stimulatory effects [7]. Various analogs of these drugs have appeared on black markets around the world [3, 8]. Such drugs are easily available through Internet websites and local dealers and are often sold as “fragrance powder,” “aroma liquid,” or “herbal smoking mixture,” and labeled “not for human consumption” to circumvent drug abuse legislation. The abuse of α-PVP has increased recently, especially among teenagers and young adults in many countries, including Japan [1, 9–11]. The escalating popularity and potent pharmacological effects of α-PVP will most certainly increase the number of suicides, murders, robberies, and traffic accidents caused by its abuse. In addition, increasing numbers of acute poisonings (i.e., overdoses) have been reported [12]. In response, the authorities in Japan have assigned these drugs as controlled substances under the Narcotics and Psychotropics Control Law in March, 2013. Under this law, both the possession and misuse of α-PVP are strictly prohibited.
Information about the metabolic pathways of α-PVP and the development of analytical methods for its metabolites are indispensable for regulating the abuse or for determining the cause of death and poisoning in forensic or toxicological studies. However, although an in vivo study was performed in rats by Sauer et al. [1], there are no data available regarding α-PVP metabolism in humans.
In this study, gas chromatography-mass spectrometry (GC–MS), validated liquid chromatography–tandem mass spectrometry (LC–MS–MS) procedures, and authentic standards synthesized in our laboratory were used to identify and quantitate α-PVP and several phase I metabolites, including diastereomers, in urine specimens obtained from human abusers. In addition, other metabolites, including phase II metabolites, have been identified from product ion mass spectra and relative retention times obtained from LC–MS–MS experiments.
Materials and methods
Reagents
α-PVP, 1-phenyl-2-(pyrrolidin-1-yl)pentan-1-ol (OH-α-PVP), α-(2″-oxo-pyrrolidino)valerophenone (2″-oxo-α-PVP) were synthesized in our laboratory according to the methods described below. All synthesized standards were confirmed by 1H nuclear magnetic resonance (NMR) spectroscopy, and ensured as >98 % pure based on 1H NMR and LC–MS analysis by the direct flow injection method. Dibenzylamine (DBA), used as internal standard (IS), was obtained from Wako Pure Chemical (Osaka, Japan). Standard stock solutions of all compounds were prepared in methanol and adjusted to appropriate concentrations with distilled water immediately prior to use. β-Glucuronidase/sulfatase (Helix pomatia, Type H-1) was purchased from Sigma-Aldrich (St. Louis, MO, USA). All other chemicals and reagents of analytical grade or quality were obtained from Wako Pure Chemical. Distilled water and HPLC-grade methanol were used throughout the experiments.
Chemical syntheses
α-Pyrrolidinovalerophenone
To a solution of valerophenone (Wako) in dichloromethane, one drop of bromine in dichloromethane was added. This solution was stirred for 5 min for the reaction to initiate. An equimolar amount of bromine solution was added over a period of a further 10 min. The solvent was removed under vacuum to yield 2-bromo-valerophenone.
To a solution of 2-bromo-valerophenone in tetrahydrofuran (THF) was dropwise added pyrrolidine (Wako) in THF. The mixture was stirred at room temperature overnight. The reaction mixture was treated with 10 % HCl aqueous solution to make it acidic and was then washed with diethyl ether. The aqueous layer was then made basic with 10 % sodium carbonate and extracted with ethyl acetate. The organic extract was washed with brine, dried over anhydrous sodium sulfate, and evaporated to give crude α-PVP as pale yellow oil. Finally, 10 % HCl methanol solution was added dropwise to the oil. After removal of the solvent, the crude hydrochloride salt was purified by recrystallization from diethyl ether/2-propanol.
α-(2″-Oxo-pyrrolidino)valerophenone
To a solution of 2-bromo-valerophenone in THF was added dropwise 2-pyrrolidinone (Wako) in 15 % sodium methoxide methanol solution (Wako). The mixture was stirred at room temperature for 48 h. The reaction mixture was treated with 10 % HCl aqueous solution to make it acidic and it was then washed with diethyl ether. The aqueous layer was then made basic with 10 % sodium carbonate and extracted with ethyl acetate. The organic extract was washed with brine, dried over anhydrous sodium sulfate, and evaporated under vacuum. The resultant residue was subjected to preparative LC using silica gel as packing material and an ethyl acetate/n-hexane mixture (1:1, v/v) as eluent to isolate 2″-oxo-α-PVP. The solution was concentrated under vacuum to give 2″-oxo-α-PVP as a pale yellow oil.
1H NMR (400 MHz, CDCl3): δ 8.06–8.01 (m, 2H), 7.59–7.53 (m, 1H), 7.49–7.43 (m, 2H), 5.65 (dd, J = 5.4, 9.4 Hz, 1H), 3.35 (ddd, J = 6.0, 8.4, 9.6 Hz, 1H), 3.26 (ddd, J = 5.6, 8.4, 9.6 Hz, 1H), 2.42 (ddd, J = 7.2, 9.6, 17 Hz, 1H), 2.32 (ddd, J = 6.4, 9.6, 17 Hz, 1H), 2.04–1.81 (m, 3H), 1.68–1.79 (m, 1H), 1.32 (sext, J = 7.6 Hz, 2H), 0.96 (t, J = 7.6 Hz, 3H).
1-Phenyl-2-(pyrrolidin-1-yl)pentan-1-ol
To a solution of α-PVP in ethanol at 60 °C was added sodium tetrahydroborate, and the mixture was stirred at 60 °C for 1 h. The reaction mixture was evaporated under vacuum, and the residue was treated with 10 % HCl aqueous solution to make it acidic before it was washed with diethyl ether. The aqueous layer was then made basic with 10 % sodium carbonate and extracted with ethyl acetate. The organic layer was washed with brine, dried over anhydrous sodium sulfate, and evaporated to give diastereomic OH-α-PVP (referred to OH-α-PVP-1 and OH-α-PVP-2 as defined below) as a pale yellow oil. The product ratio of the diastereomers was determined by NMR spectroscopy.
1H NMR (400 MHz, CDCl3; the compound existed as a mixture of diastereomers, OH-α-PVP-1 denoted by §, OH-α-PVP-2 denoted by *): δ 7.39–7.29 (m, 4H*, 4H§), 7.29–7.20 (m,1H*, 1H§), 4.97 (d, J = 4.0 Hz, 1H*), 4.18 (d, J = 9.2 Hz, 1H§), 2.82–2.70 (m, 4H*, 4H§), 2.66–2.59 (m, 1H§), 2.53 (q, J = 4.0 Hz, 1H*), 1.84–1.75 (m, 4H*, 4H§), 1.52–0.74 (m, 4H*, 4H§), 0.69 (t, J = 7.2 Hz, 3H*, 3H§).
Urine specimens
Urine specimens obtained from α-PVP users were submitted to our laboratory for forensic analysis. The specimens were collected from 19 abusers, and were stored at −20 °C until analysis. Drug-free urine specimens used for validation experiments were obtained from healthy volunteers.
Sample preparation for GC–MS
A 100-μl aliquot of a urine sample was adjusted to pH 9 with a carbonate buffer (1 M, pH 9.5) and saturated with NaCl. To the solution, 500 μl of a chloroform/2-propanol (3:1, v/v) was added and the mixture was vortex-mixed for 1 min. After centrifuging at 7,000 g for 10 min, the organic layer was transferred to a stoppered glass test tube. After addition of 50 μl of acetic acid, it was evaporated to dryness under a nitrogen stream at 40 °C. The residue was trimethylsilylated at 70 °C for 30 min after reconstitution in 100 μl of N-methyl-N-(trimethylsilyl)trifluoroacetamide (MSTFA), and a 1-μl aliquot of the reaction mixture was injected into the GC–MS system.
Sample preparation for LC–MS–MS
A 1-ml aliquot of urine was adjusted to pH 5 with 1 M acetic acid and incubated at 37 °C for 5 h with β-glucuronidase/sulfatase (15,000/750 U/ml urine, respectively). To aliquots of prehydrolysis urine and posthydrolysis urine (100 μl each), 100 μl of DBA aqueous solution (0.1 μg/ml) was added as IS. After mixing briefly, 600 μl of methanol was added to each solution, and each mixture was vortex-mixed for 1 min, and centrifuged at 7,000 g for 10 min. The supernatant fraction was transferred to a stoppered glass test tube and evaporated to dryness under a nitrogen stream at 50 °C. Each residue was dissolved in 100 μl of distilled water. After filtration through a 0.22-μm membrane filter, a 5-μl aliquot of each was used for LC–MS–MS analysis.
Nuclear magnetic resonance spectroscopy
All NMR spectra of the synthetic compounds were acquired on a JNM-ECS 400 FT NMR system (JEOL Resonance, Akishima, Japan) in CDCl3 using tetramethylsilane as the internal standard.
GC–MS conditions
A GCMS-QP2010 Ultra gas chromatograph–mass spectrometer (Shimadzu, Kyoto, Japan) was operated in the electron ionization (EI) mode using a DB-5MS fused-silica capillary column (30 m × 0.25 mm i.d., film thickness 0.25 μm; Agilent, Santa Clara, CA, USA). Injections were made automatically in the splitless mode at an injection port temperature of 250 °C. The carrier gas was high-purity helium at a flow rate of 3.0 ml/min. The column temperature was initially held at 60 °C for 1 min, and increased to 300 °C at 10 °C/min. The ionization energy and interface temperature were set at 70 eV and 250 °C, respectively.
LC–MS–MS conditions
Analysis was performed on a Prominence Series UFLC system (Shimadzu) linked to an API 3200 QTRAP hybrid triple quadrupole linear ion-trap mass spectrometer (AB Sciex, Concord, ON, Canada) equipped with an electrospray ionization (ESI) interface. LC separation was carried out using an L-column2 ODS semi-micro column (150 × 1.5 mm i.d., 5 μm particles; Chemicals Evaluation and Research Institute, Tokyo, Japan). The analytes were chromatographed by linear gradient elution with (A) 10 mM ammonium formate buffer (pH 5) and (B) methanol at a flow rate of 0.1 ml/min at a column temperature of 40 °C. A gradient was applied starting from 95 % A/5 % B, linearly changed to 5 % A/95 % B over the 20 min, and held for 5 min. ESI–MS–MS was conducted in the positive ion mode, and the protonated molecules were used as precursor ions. Analyses were performed in the enhanced product ion scan (EPI) mode for identification, and selected reaction monitoring (SRM) mode for quantitation. Nitrogen was used as nebulizer and collision gas, and the declustering potential (DP) and collision energy (CE) were set at 30 V and 25 eV, respectively, for EPI. The SRM parameters for each compound were set as shown in Table 1.
Results and discussion
Synthesis of α-PVP and its putative metabolites
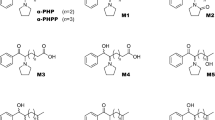
A previous in vivo study in rats by Sauer et al. [1] is the only available study on the metabolic pathways and metabolite analysis of α-PVP. Our group, however, reported on the metabolism of other cathinone-derivative drugs in humans and rats [13–16]. Based on these findings and preliminary experimental GC–MS and LC–MS–MS results, reduction of the ketone moiety and oxidation at the 2″ position of the pyrrolidine ring were most likely as the principal metabolic pathways of α-PVP. In the present study, therefore, OH-α-PVP and 2″-oxo-α-PVP were synthesised in our laboratory and used as standards for the metabolites that retain the structural characteristics of the parent drug α-PVP. The reductive metabolite OH-α-PVP has two chiral carbon atoms and would be excreted in human urine as a mixture of diastereomers. It was therefore prepared as a diastereomeric mixture according to the methods described above. The product ratio (OH-α-PVP-1/OH-α-PVP-2) was 1.5 as calculated from the peak areas of H§ and H* obtained from NMR spectra. This ratio allows each of the diastereomers in urine to be quantitated separately.
Identification of α-PVP and its metabolites in urine by GC–MS
Figure 1 shows the total ion chromatogram (TIC), extracted mass chromatograms, and electron ionisation (EI) mass spectra obtained from the urine specimen of an α-PVP abuser and a mixture of the synthesized authentic standards. The free bases of OH-α-PVP and 2″-oxo-α-PVP were detected in the abuser’s urine together with the parent α-PVP. The diastereomic metabolite, OH-α-PVP, yielded two peaks [(b-i) and (b-ii) in Fig. 1a]. No significant differences were observed between the mass spectra of the two diastereomers (data not shown). Regarding the reduction of β-keto moieties in cathinones, Meyer et al. [17] reported that 4-methylmethcathinone was reduced to only one diastereomer, because of steric hindrance effects in the enzymatic reaction. In contrast, our previous study on 3,4-dimethylmethcathinone (DMMC) revealed that racemic DMMC was metabolized to two diastereomers [14]. The identification of both diastereomers of OH-α-PVP in the present study agrees with the results of the latter study.

Total ion chromatogram (TIC) and extracted mass chromatograms obtained from an authentic standard mixture (1 μg/ml each) and a urine specimen of an α-PVP abuser (subject 2) without derivatization (a) and with trimethylsilyl (TMS) derivatization (b), and the electron ionization mass spectra of α-PVP and its metabolites with/without TMS derivatization (c) all obtained by gas chromatography-mass spectrometry. Peaks: a α-PVP, b-i OH-α-PVP-1, b-ii OH-α-PVP-2, c 2″-oxo-α-PVP. Mass spectra: a α-PVP, b OH-α-PVP-1, c 2″-oxo-α-PVP, d TMS derivative of OH-α-PVP-1. The numbers in parentheses represent magnification ratios
Trimethylsilyl (TMS) derivatization with MSTFA is a common pretreatment that enhances the sensitivity of GC–MS analyses of alcohols and phenols. The TMS derivative of OH-α-PVP gave tenfold enhancement in sensitivity as compared with nonderivatized free base, but resulted in significantly lower resolution between the peaks of the two OH-α-PVP diastereomers, as shown in Fig. 1b. The TMS derivatization would be disadvantageous for accurate quantitation of the individual diastereomers. Several studies [14, 18] reported that trifluoroacetic derivatives of β-hydroxylated metabolites of cathinones isomerize in the GC inlet, although TMS derivatization under such circumstances has never been discussed. These reports suggest that GC–MS analyses may not be satisfactorily reproducible or reliable for quantitative measurements of OH-α-PVP diastereomers. To obtain more reliable quantitative results, simultaneous analyses of α-PVP and its metabolites were performed on an LC–MS–MS system.
Quantitation of α-PVP and its metabolites by LC–MS–MS
To simultaneously measure levels of α-PVP and its metabolites in urine, a sensitive and reliable LC–MS–MS procedure was established. A linear gradient elution on a C18 semi-micro column was optimized to yield clear separation of all four analytes, including the two diastereomers, as shown in Fig. 2a, b.

Extracted mass chromatograms obtained from an authentic standard mixture (1 μg/ml each) (a) and a urine specimen of an α-PVP abuser (b), and product ion mass spectra of α-PVP and its identified and putative metabolites (c) all obtained by liquid chromatography–tandem mass spectrometry. Peaks: a α-PVP, b-i OH-α-PVP-1, b-ii OH-α-PVP-2, c 2″-oxo-α-PVP, d-i and d-ii 2″-OH-α-PVP, e-i, e-ii, e-iii and e-iv OH-α-PVP-glucuronides, IS internal standard. Numbers in parentheses represent magnification ratios. Each protonated molecule was selected as a precursor ion
Qualitative analyses by MS–MS were performed in the EPI mode, and CE was set at 25 eV to obtain abundant product ions. Figure 2c shows product ion mass spectra of α-PVP and its metabolites obtained from the urine of an α-PVP abuser. A slight but significant difference was observed between the product ion mass spectra of the diastereomers [peaks (b-i) and (b-ii)]. Relatively intense ion peaks at m/z 72 and 91 allowed the discrimination of OH-α-PVP-1 from OH-α-PVP-2, although detailed structural information was not available. The mass spectra obtained from the urine specimen agreed with those obtained with the authentic standards.
Quantitative analyses by LC–MS–MS were performed in the SRM mode. Table 2 summarizes the validation data obtained by analyzing samples containing various amounts of analyte spiked into drug-free urine. Pretreating the urine specimens with methanol to precipitate proteins provided high recoveries not less than 90 %. The LC–MS-MS technique exhibited a high degree of linearity throughout the concentration range from 10 to 10,000 ng/ml. Samples containing analyte levels above the linear calibration range could be successfully quantitated by diluting with drug-free urine prior to analysis. Accuracy and precision were <10 % for all compounds at all of the concentrations examined.
The concentrations of α-PVP and its three metabolites were quantitated in unknown urine samples using the above validated LC–MS–MS procedure and calibration curves. Excretion profiles were obtained, and the results are summarized in Table 3. Unchanged α-PVP and its three metabolites were detected in all of the samples. In 14 of the 19 specimens, OH-α-PVP-1 was most abundant among the identified metabolites that retain the structural characteristics of the parent drug. In the remaining five specimens, 2″-oxo-α-PVP was the most abundant metabolite. This suggests that OH-α-PVP would be the preferable marker to prove α-PVP use by urinalysis. Sauer et al. [1] identified 2″-oxo-α-PVP as one of the main metabolites in rat urine based on EI mass spectra. The present study shows that both 2″-oxo-α-PVP and OH-α-PVP can be considered as primary metabolites in human urine. The differences in the urinary metabolites detected in our present study and those found in previous studies can be attributed to interspecies variation.
This study is the first to report the separation and quantitation of OH-α-PVP stereoisomers. The concentration ratio of OH-α-PVP-1 to OH-α-PVP-2 (OH-α-PVP-1/OH-α-PVP-2) was estimated as 8.7–59.5 (n = 16 with a mean of 41.0 ± 11.8). This significant difference in urinary concentrations can be attributed to the stereospecific affinity of R- and S-α-PVP to the enzyme and/or to the stereoselective reduction of the keto moiety, likely catalyzed by carbonyl reductase [19, 20]. However, detailed properties of the enzymes involved in the reduction and conjugation of α-PVP are not clear. Future studies are required to fully elucidate the metabolism of α-PVP.
Previous studies [14–16] indicated an increase in urinary levels of the metabolites of cathinone-derived drugs after enzymatic hydrolysis of conjugated metabolites. In the present study, concentrations of OH-α-PVP in urine (subjects 1 and 2) were slightly higher following enzymatic hydrolysis (Table 3). However, the exact rate of conjugation was unknown, because the conjugates of OH-α-PVP were not completely cleaved by enzymatic hydrolysis (data not shown).
Investigation of other metabolites in α-PVP abusers’ urine
Urine specimens were analyzed by LC–MS–MS prior to enzymatic hydrolysis to survey the other metabolites present, including conjugated metabolites. The putative metabolites were identified from their product ion spectra and relative retention times. The putative oxidative metabolites of the pyrrolidine ring appear as a pair of diastereomers [peaks (d-i) and (d-ii), 2″-hydroxy form] detected at retention times of 18 and 19 min, as shown in Fig. 2b. No significant differences were observed between the product ion mass spectra of the diastereomers (d-i) and (d-ii) (data for d-ii not shown). This putative metabolite reveals the following metabolic steps: hydroxylation at the 2″ position of the pyrrolidine ring followed by dehydrogenation to 2″-oxo-α-PVP. This assumption is supported by a previous study [21], detailing the metabolism of nicotine, which possesses a similar pyrrolidine ring.
In addition, four putative conjugated metabolites, whose peak intensities disappeared or decreased after hydrolysis, were detected. No significant differences were observed in the product ion mass spectra of the four peaks, (e-i), (e-ii), (e-iii), and (e-iv), as shown in Fig. 2b, c. The spectra are characterized by ions produced through the neutral loss of a mass of 176 (a glucuronyl group) to produce a protonated molecule of OH-α-PVP (m/z 234) and its characteristic substructural ions (m/z 216 and 173). These data indicate that the four detected peaks correspond to diastereomic OH-α-PVP glucuronides. To discriminate aglycones of the four diastereomers, these peaks were further analyzed by MS–MS using an in-source CID technique. Product ion spectra were obtained by selecting each ion at m/z 234, corresponding to the protonated molecule of OH-α-PVP, as the precursor ion. The mass spectra obtained from peaks (e-i) and (e-iii), and peaks (e-ii) and (e-iv) were consistent with those of OH-α-PVP-1 and OH-α-PVP-2, respectively, suggesting that peaks (i) and (iii) correspond to OH-α-PVP-1-glucuronides and that peaks (ii) and (iv) correspond to OH-α-PVP-2-glucuronides.
In the 1980s, Brenneisen et al. [22] reported on the metabolism of α-aminopropiophenone (cathinone) in khat leaves and concluded that (S)-cathinone and (R)-cathinone are metabolized to 1R,2S-(−)-norephedrine and 1R,2R-(+)-norpseudoephedrine, respectively, by a stereospecific 1R keto reduction. However, the detection of OH-α-PVP glucuronides in the urine of α-PVP abusers indicates that OH-α-PVP exists in both R- and S-configurations. Differences in stereoisomeric metabolism are likely due to steric hindrance by the aliphatic side chain and pyrrolidine ring of α-PVP, leading to the disappearance of stereospecificity in the keto reduction. In addition, the high intensity of peak (e-iv), which corresponds to OH-α-PVP-2-glucuronide, demonstrates that enantioselectivity plays a role in O-glucuronidation in the metabolism of α-PVP.
Proposed metabolic pathways
The identification and quantitation of urinary metabolites in the present study suggest the principal metabolic pathways of α-PVP shown in Fig. 3. The metabolism of α-PVP includes reduction of the ketone moiety to OH-α-PVP and oxidation at the 2″ position of the pyrrolidine ring to 2″-oxo-α-PVP. In addition, putative metabolites suggest the following metabolic steps: hydroxylation at the 2″ position of the pyrrolidine ring to form 2″-OH-α-PVP as the intermediate to 2″-oxo-α-PVP, and a reduction to diastereomic OH-α-PVP followed by partial conjugation to the respective glucuronide.

Proposed major metabolic pathways for α-PVP in humans
A previous study on the metabolism of α-PVP [1] has suggested other metabolic pathways, including a ring-opening reaction to form an aliphatic aldehyde and further oxidation to a carboxylic acid, degradation of the pyrrolidine ring to primary amines, and hydroxylation of the phenyl ring. These pathways were not confirmed in the present study.
Conclusions
α-PVP and its three metabolites including diastereomers were unequivocally identified and quantitated in α-PVP abusers’ urine specimens using newly synthesized authentic standards. Our study revealed two metabolic pathways of considerable quantitative significance for α-PVP and three characteristic metabolites in humans. The first pathway leads, via reduction of β-keto moiety, to diastereomic OH-α-PVP partly followed by conjugation to the respective glucuronide. The second pathway leads, via hydroxylation on position 2″ of the pyrrolidine ring, to 2″-OH-α-PVP followed by dehydrogenation to 2″-oxo-α-PVP. These findings will contribute not only to developing a reliable analytical procedure for unequivocal proof of the intake of newly encountered designer drugs of similar structures but also to estimating their metabolic pathways.
References
Sauer S, Peters FT, Haas C, Meyer MR, Fritschi G, Maurer HH (2009) New designer drug α-pyrrolidinovalerophenone (PVP): studies on its metabolism and toxicological detection in rat urine using gas chromatographic/mass spectrometric techniques. J Mass Spectrom 44:952–964
Shin H-S, Shin Y-SO, Lee S, Park B–B (1996) Detection and identification of pyrovalerone and its hydroxylated metabolite in the rat. J Anal Toxicol 20:568–572
Jane MP, Lewis SN (2012) The toxicology of bath salts: a review of synthetic cathinones. J Med Toxicol 8:33–42
Meyer MR, Du P, Schuster F, Maurer HH (2010) Studies on the metabolism of the α-pyrrolidinophenone designer drug methylenedioxy-pyrovalerone (MDPV) in rat and human urine and human liver microsomes using GC–MS and LC-high resolution MS and its detectability in urine by GC–MS. J Mass Spectrom 45:1426–1442
Springer D, Fritschi G, Maurer HH (2002) Metabolism and toxicological detection of the new designer drug 4′-methyl-alpha-pyrrolidinopropiophenone in urine using gas chromatography-mass spectrometry. J Chromatogr B 773:25–33
Springer D, Fritschi G, Maurer HH (2003) Metabolism of the new designer drug 3′,4′-methylenedioxy-alpha-pyrrolidinopropiophenone studied in urine using gas chromatography–mass spectrometry. J Chromatogr B 793:377–388
Meltzer PC, Butler D, Deschamps JR, Madras BK (2006) 1-(4-Methylphenyl)-2-pyrrolidin-1-yl-pentan-1-one (pyrovalerone) analogs: a promising class of monoamine uptake inhibitors. J Med Chem 49:1420–1432
Ammann D, McLaren JM, Gerostamoulos D, Beyer J (2012) Detection and quantification of new designer drugs in human blood: part 2—designer cathinones. J Anal Toxicol 36:381–389
Kikura-Hanajiri R, Uchiyama N, Kawamura M, Goda Y (2013) Changes in the prevalence of synthetic cannabinoids and cathinone derivatives in Japan until early 2012. Forensic Toxicol 31:44–53
Namera A, Nakamoto A, Saito T, Nagao M (2011) Colorimetric detection and chromatographic analyses of designer drugs in biological materials: a comprehensive review. Forensic Toxicol 29:1–24
Namera A, Urabe S, Saito T, Torikoshi-Hatano A, Shiraishi H, Arima Y, Nagao M (2013) A fatal case of 3,4-methylenedioxypyrovalerone poisoning: coexistence of α-pyrrolidinobutiophenone and α-pyrrolidinovalerophenone in blood and/or hair. Forensic Toxicol 31:338–343
Saito T, Namera A, Osawa M, Aoki H, Inokuchi S (2013) SPME–GC–MS analysis of α-pyrrolidinovalerophenone in blood in a fatal poisoning case 31:328–332
Kamata HT, Shima N, Zaitsu K, Kamata T, Miki A, Nishikawa M, Katagi M, Tsuchihashi H (2006) Metabolism of the recently encountered designer drug, methylone, in humans and rats. Xenobiotica 36:709–723
Zaitsu K, Katagi M, Kamata HT, Kamata T, Shima N, Miki A, Tsuchihashi H, Mori Y (2009) Determination of the metabolites of the new designer drugs bk-MBDB and bk-MDEA in human urine. Forensic Sci Int 188:131–139
Zaitsu K, Katagi M, Tatsuno M, Sato T, Tsuchihashi H, Suzuki K (2011) Recently abused β-keto derivates of 3,4-methylenedioxyphenylalkylamines: a review of their metabolisms and toxicological analysis. Forensic Toxicol 29:73–84
Shima N, Katagi M, Kamata H, Matsuta S, Nakanishi K, Zaitsu K, Kamata T, Nishioka H, Miki A, Tatsuno M, Sato T, Tsuchihashi H, Suzuki K (2013) Urinary excretion and metabolism of the newly encountered designer drug 3,4-dimethylmethcathinone in humans. Forensic Toxicol 31:101–112
Meyer MR, Wilhelm J, Peters FT, Maurer HH (2010) Beta-keto amphetamines: studies on the metabolism of the designer drug mephedrone and toxicological detection of mephedrone, butylone, and methylone in urine using gas chromatography-mass spectrometry. Anal Bioanal Chem 397:1225–1233
Morita M, Ando H (1983) Analysis of methamphetamine and its metabolites in urine from a habitual user of the stimulant (in Japanese with English abstract). Eisei Kagaku 29:318–322
Imamura Y, Kojima Y, Higuchi T, Akita H, Oishi T, Otagiri M (1989) Stereoselective reduction of acetohexamide in cytosol of rabbit liver. J Pharmacobiodyn 12:731–735
Kobana K, Watanabe K, Kimura T, Matsunaga T, Kondo S, Yamamoto I (2000) A carbonyl reductase-catalyzing reduction of N 3-phenacyluridine in rabbit liver. Biol Pharm Bull 23:917–921
Hukkanen J, Jacob P III, Benowitz NL (2005) Metabolism and disposition kinetics of nicotine. Pharmacol Rev 57:79–115
Brenneisen R, Geisshüsler S, Schorno X (1986) Metabolism of cathinone to (−)-norephedrine and (−)-norpseudoephedrine. J Pharm Pharmacol 38:298–300
Acknowledgments
The authors would like to thank Prof. Dr. Iwamura (Matsuyama University) for his valuable suggestions in synthesizing the standards.
Conflict of interest
There are no financial or other relations that could lead to a conflict of interest.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
Shima, N., Katagi, M., Kamata, H. et al. Metabolism of the newly encountered designer drug α-pyrrolidinovalerophenone in humans: identification and quantitation of urinary metabolites. Forensic Toxicol 32, 59–67 (2014). https://doi.org/10.1007/s11419-013-0202-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11419-013-0202-9