
Abstract
Cliniatines A–C (1–3), three new Amaryllidaceae alkaloids, consisting of 2,6-dimetylpyridine and lycorine-type and/or galanthamine-type were isolated from Clivia miniata (Lindl.) Bosse. The structures and absolute configurations of 1–3 were elucidated based on spectroscopic data and chemical correlation. Cliniatines A–C showed moderate inhibitory activity against acetylcholinesterase.
Graphic abstract

Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Clivia miniata (Lindl.) Bosse. is an evergreen plant that is distributed in southern Africa. The rhizome of the plant had been known as a remedy for snakebite, or associated pain. [1, 2]
Plants of the genus Clivia comprise about 4 species, [1] which were reported to be rich sources of Amaryllidaceae alkaloids such as lycorine [3], clivonine [4], clivacetine [5], clivimine [6], and galanthamine [7], among which galanthamine is used in the treatment of Alzheimer's disease due to its selective reversible and competitive inhibitory activity against Acetylcholinesterase (AChE). [8, 9] In our search for biogenetically interesting intermediates and new alkaloids with a novel skeleton from medicinal plants, [10,11,12,13,14,15] two new dimeric and a monomeric alkaloid with 2,6-dimethylpyridine unit, cliniatines A–C (1–3) were isolated from the whole plants of C. miniata (Fig. 1). Here we reported the isolation and structure elucidation of 1–3, and its inhibitory activity against AChE.

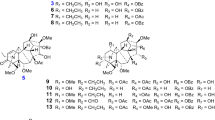
Structures of 1–3
Results and discussion
Cliniatine A (1) was shown to have the molecular formula C41H43N3O10 by HRESIMS [m/z 738.3027, (M + H)+, ∆ + 0.0 mmu]. The IR absorptions implied the presence of NH and/or OH (3340 cm−1) and carbonyl (1720 cm−1) functionalities. The analysis of 1H and 13C NMR data (Table 1) and HSQC spectrum of 1 revealed 41 carbon signals due to 7 sp3 methines, 10 sp3 methylenes, 3 methyls, 1 sp3 quaternary carbon, 5 sp2 methines, and 15 sp2 quaternary carbons. Among them, 4 sp3 methines (δC 69.4; δH 5.77, δC 78.9; δH 4.65, δC 88.9; δH 4.42, and δC 70.9; δH 5.38) and 1 sp3 methylene (δC 104.3; δH 6.14 and 6.15) were attributed to those attached to an oxygen atom. Also, 1 sp3 methine (δC 70.2; δH 4.23), 3 sp3 methylenes (δC 56.2; δH 3.25 and 3.92, δC 46.8; δH 3.58 and 3.64, and δC 51.7; δH 4.26 and 4.39), and 1 methyl (δC 43.9; δH 3.23) were considered to be connected to the nitrogen atoms.
The gross structure of 1 was elucidated by analysis of 2D NMR data including the 1H–1H COSY, HMQC, and HMBC spectra in CD3OD (Fig. 2).

Selected 2D NMR correlations for cliniatine A (1)
The 1H–1H COSY spectra revealed the presence of four partial structures, a (C-2 ~ C5, C3a, C-5a, and C-11b ~ C-11c), b (C-4a′ and C-5′ ~ C-8′), c (C-9′ ~ C-10′), and d (C-1′ ~ C-2′), as shown in Fig. 2.
In unit A, the connectivity of C-2, C-11c, and C-12 through a nitrogen atom was revealed by the HMBC correlations of H3-12/C-2 and C-11c. The HMBC cross-peaks of H2-13/C-9 and C-10, H-11/C-7a and C-9, and H-8/C-9, C-10, and C-11a indicated the presence of the 1,3-benzodioxole (C-7a, C-8 ~ C-11, C-13, C-11a) moiety. In addition, the HMBC correlations of H-11/C-11b and H-8/C-7 indicated the linkage of C-11a ~ C-11b and C-7a ~ C-7, respectively. Finally, the connectivity between C-5a and C-7 through oxygen atom was deduced from its chemical shifts (CH-5a: δC 78.9; δH 4.65 and C-7: 164.9). Thus, unit A was assigned as clivonine [4].
In unit B, the presence of a cyclohexane ring (C-4a′, C-5′ ~ C-8′, and C-8a′) was elucidated by the HMBC correlations of H-5′b and H-8′/C-8a′ and H-4a′/C-8′. HMBC cross-peaks of H-9′/C-14′, H-10′/C-8a′, H-10′/C-12′, and H-12′/C-14′ indicated the presence of dehydroazepane ring (C-8a′, C-9′ ~ C-10′, C-12′ ~ C-14′, and N). Furthermore, the HMBC correlations of H-1′/C-3′ and C-14′ and H-2′/C-13′ and C-3a′ implied the presence of a phenol moiety (C-1′ ~ C-3′, C-3a′, and C-13′ ~ C-14′). Those HMBC correlations data and the chemical shift of C-3a′ (δC 147.4) and CH-4a′ (δC 88.9; δH 4.42) revealed that unit B was O-demethyl form of N-demethyllycoramine [16].
The remaining of unit C was elucidated as 2,6-dimethylpyridine with two ester functionalities by chemical shifts of literature [17] and HMBC correlations. The HMBC correlation of H-5/C-8″ and the chemical shift characteristic of CH-6′ (δC 70.9; δH 5.38) established the connection between C-5 and C-8″ also C-6′ and C-9″ through an ester bond, respectively.
Thus, the gross structure of cliniatine A was assigned as 1.
The relative stereochemistry of 1 was elucidated by NOESY correlations and 3 J coupling constants.
In unit A, the NOESY correlation of H-3a/H-11c suggested that H-3a and H-11c were oriented to the same side (Fig. 3). In addition, the coupling constants of 3JH-11b/H-11c (12.6 Hz) and 3JH-11b/H-5a (12.3 Hz) indicated the anti-relationship of H-11b/H-11c and H-11b/H-5a, respectively. Furthermore, the gauche-relationship between H-5 and H-5a was elucidated by the 3 J coupling constant of H-5/H-5a (2.8 Hz). Thus, the relative configurations of unit A were the same as those of clivonine [4].

Selected NOESY correlations and 3 J coupling constants for unit A of cliniatine A (1)
On the other hand, in unit B (Fig. 4), the coupling pattern of both H-4a′ and H-6′ was broad singlet. This indicated that both H-4a′ and H-6′ oriented equatorially in the cyclohexane ring (C-4a′, C-5′ ~ C-8′, and C-8a′). Then the NOESY correlation of H-4a′/H-9′a suggested that H-4a′ and C-9′ were oriented to the same side. Thus, the relative configuration of unit B was identified to be the same as O-demethyl form of N-demethyllycoramine [16].

Selected NOESY correlations and 3 J coupling constants for unit B of cliniatine A (1)
Cliniatine B {2, [α] -28 (c 1.0, MeOH) showed the pseudomolecular ion peak at m/z 776 (M + Na)+ and the molecular formula, C41H43N3O11, was established by HRESIMS [m/z 776.2797, (M + Na)+, ∆ + 0.2 mmu]. The IR absorptions implied the presence of NH and/or OH (3350 cm−1) and carbonyl (1720 cm−1) functionalities. The 1H and 13C NMR (Table 2) spectra suggested that 2 had a similar structure as that of 1, except for the presence of a hydroxy group at C-9′ (δC 72.8; δH 4.10). The α-configuration of H-9′ was elucidated by NOESY correlation of H-4a′/H-9′ (Fig. 5).

Selected NOESY correlation for cliniatine B (3)
The HRESIMS data [m/z 509.1925, (M + H)+, ∆ + 0.1 mmu] of cliniatine C (3) established the molecular formula to be C27H28N2O8, which was smaller than cliniatine A (1) by a C14H15NO2 unit. The 1H and 13C NMR data (Table 3) of 3 were analogous to those of 1, although 1H and 13C signals of N-demethyllycoramine moiety observed for 1 were absent for 3.
Detailed analyses of 2D NMR spectra of 3 indicated that cliniatine C possessed methyl ester instead of N-demethyllycoramine moiety of 1 (Fig. 6). The relative stereochemistry of clivonine moiety of 3 was the same as 1.

Selected 2D NMR correlations for cliniatine C (3)
The CD spectra (Fig. 7) of 1–3 showed a similar CD curve to that of clivonine [18] [λmax 233 (∆ε + 16.9), 252 (− 1.46), 269 (+ 0.90), and 314 (− 2.65) nm]. The absolute stereochemistry of clivonine has been established by chemical correlation [19,20,21,22,23,24]. Therefore, the absolute configurations of clivonine moiety of 1–3 were assigned as 3aR,5S,5aR,11bS,11cR.

CD spectra of cliniatines A–C (1–3) in MeOH
To elucidate the absolute configurations of N-demethyllycoramine moiety, 2 was hydrolyzed by LiAlH4 condition. However, the reaction gave a complex mixture.
Therefore, the absolute configuration was elucidated by comparing the CD data of N-demethyllycoramine, which was isolated in this study, and lycoramine [7] synthesized from commercially available (–)–galanthamine. The CD spectrum of N-demethyllycoramine in MeOH showed a similar CD curve to that of lycoramine (see supporting information S22). Considering that N-demethyllycoramine and cliniatines A and B those were isolated from this study having similar structures, the absolute configuration of 1 and 2 were considered as 4a′S,6′S,8a′S and 4a′S,6′S,8a′S,9′R, respectively.
A plausible biogenetic pathway for cliniatines A–C is proposed as shown in Fig. 8. It was considered that both galanthamine and clivonine moieties of cliniatine A were generated from nobelladine, and 2,6-dimethylpyridine-3,5-dicarboxylate binding to them was biosynthesized from four acetic acid, ammonia, and formaldehyde. [17]

Plausible biogenetic pathway for cliniatines A–C (1–3)
The isolated compounds were tested for inhibitory activity against AChE. [25] As can be seen in Table 3, 1 inhibited AChE with an IC50 value of 5.7 μM, which was comparable to that of galanthamine (Table 3).
Experimental section
General Experimental Procedures. Optical rotations were measured with a JASCO P-1030 polarimeter. IR spectra were obtained by a JASCO FT/IR-230 using Zn/Se cell. CD spectra were obtained using a JASCO J-820 spectropolarimeter. 1D and 2D spectra were recorded on a JEOL ECZ600 and Bruker AV400 spectrometer. Chemical shifts (ppm) were referenced to the residual solvent peaks (δH 7.26 and δC 77.0 for CDCl3 and δH 3.31 and δC 49.0 for CD3OD). Positive mode ESITOFMS was obtained on a Waters Xevo G2-XS QTof LC/MS spectrometer using a sample dissolved in MeOH. Column chromatography was performed using silica gel (230–400 mesh; Merck KGaA, Darmstadt, Germany), amino silica gel (NH-DM1020; Fuji Silysia Chemical Ltd., Aichi, Japan), and ODS HPLC (CAPCELL PAK C18 MG II; Shiseido, Tokyo, Japan).
Material. Clivia miniata was collected at Akita, Japan in 2015. The botanical identification was made by Dr. Yusuke Hirasawa, Faculty of Pharmaceutical Sciences, Hoshi University. The voucher specimen (Herbarium No. HS31) was deposited at the Herbarium of the Faculty of Pharmaceutical Sciences, Hoshi University, Tokyo, Japan.
Extraction and isolation. The whole plants of Clivia miniata (481 g) collected in Akita were extracted with MeOH at rt, and the extract (93 g) was partitioned between EtOAc and 3% tartaric acid. Water-soluble materials, which were adjusted at pH 10 with sat. Na2CO3 aq., were extracted with CHCl3. CHCl3-soluble materials (1050 mg) were subjected to an amino SiO2 column (hexane/EtOAc, 1:0 → 0:1, and then CHCl3/MeOH, 1:0 → 0:1) to give nine fractions (I–IX).
The fraction VIII was further separated by a SiO2 column (CHCl3/MeOH, 4:1 → 0:1) and, then ODS HPLC (25% CH3CN aq/0.1% TFA) to afford cliniatines A (1, 2.7 mg, 0.0006%), B (2, 8.2 mg, 0.002%), C (3, 3.3 mg, 0.0007%), and lycorine (10.6 mg, 0.002%). The fraction VI was subjected to a SiO2 column to give clivimine (280 mg, 0.06%) and N-demethyllycoramine (5.6 mg, 0.001%). The fraction II was purified by SiO2 column (CHCl3/MeOH, 10:1 → 0:1) to afford clivonine (30.5 mg, 0.006%).
Cliniatine A (1): colorless amorphous solid; [α] − 30 (c 1.0, MeOH); IR (Zn-Se) λmax 3340, 2920, 1720, 1260, and 1030 cm−1; UV (MeOH) λmax 306 (log ε 3.57), 273 (3.84), 227 (4.41), and 206 (4.41) nm; CD (MeOH) λmax 225 (∆ε + 6.18), 240 (− 1.73), 271 (+ 1.50), 289 (− 0.70) nm; ESIMS m/z 738 (M + H)+; HRESIMS m/z 738.3027 [calcd for C41H44N3O10 (M + H)+].
Cliniatine B (2): colorless amorphous solid; [α] − 28 (c 1.0, MeOH); IR (Zn-Se) λmax 3350, 2960, 2930, 1720, and 1560 cm−1; UV (MeOH) λmax 306 (log ε 3.49), 273 (3.74), and 205 (4.62) nm; CD (MeOH) λmax 208 (∆ε − 11.27), 229 (+ 10.00), 241 (− 3.79), 270 (+ 3.09), 294 (− 2.03) nm; ESIMS m/z 776 (M + Na)+; HRESIMS m/z 776.2797 [calcd for C41H43N3O11Na (M + Na)+].
Cliniatine C (3): pale yellow amorphous solid; [α] + 4 (c 1.0, MeOH); IR (Zn–Se) λmax 3440, 2950, 1710, 1670, and 1030 cm−1; UV (MeOH) λmax 306 (log ε 3.44), 273 (3.57), 228 (4.13), and 204 (4.24) nm; CD (MeOH) λmax 209 (∆ε − 4.13), 230 (+ 5.30), 246 (− 1.83), 270 (+ 1.26), 293 (− 1.11) nm; ESIMS m/z 509 (M + H)+; HRESIMS m/z 509.1925 [calcd for C27H29N2O8 (M + H)+].
Hydrolysis of cliniatine B (2). To a solution of 2 (0.5 mg) in THF (100 μl) was added LiAlH4. The mixture was allowed to stand at 80 °C for 12 h, and then concentrated under reduced pressure. The residue was dissolved in CHCl3 and washed with Na2CO3 aq. After evaporation of solvent, complex mixture was obtained.
Hydrogenation of galanthamine. Galanthamine (20.7 mg) was dissolved in MeOH and hydrogenated over 10% Pd/C (17 mg) for 19 h. The catalyst was removed by filtration, and the filtrate was evaporated to give a residue, which was separated by SiO2 (CHCl3/MeOH, 10:1 → 0:1) to give lycoramine (7.0 mg) as an colorless amorphous solid; 1H NMR (400 MHz, CDCl3) δ 6.56 (1H, d, 8.4), 6.61 (1H, d, 8.4), 4.33 (1H, t, 3.2), 4.05 (1H, m), 3.95 (1H, d, 15.0), 3.82 (3H, s), 3.58 (1H, d, 15.1), 3.17 (1H, dd, 13.4, 13.4), 3.00 (1H, m), 2.45 (1H, brd, 16.0), 2.33 (3H, s); ESIMS m/z 290.
References
Crouch NR, Mulholland DA, Pohl TL, Ndlovu ES (2003) The ethnobotany and chemistry of the genus Clivia (Amaryllidaceae). Afr J Bot 69:144–147
Musara C, Aladejana EB, Aladejana AE (2021) Clivia miniata (Lindl.) Bosse, (Amaryllidaceae): Botany, medicinal uses, phytochemistry and pharmacological properties. J Appl Pharm Sci 11(2):012–018
Govindachari TR, Thyagarajan BS (1954) Structure of lycorine. Chem Ind 1:374–375
Briggs CK, Highet PF, Highet RJ, Wildman WC (1956) Alkaloids of the Amaryllidaceae. VII. Alkaloids containing the hemiacetal of lactone group1. J Am Chem Soc 78:2899–2904
Kobayashi S, Ishikawa H, Sasakawa E, Kihara M, Shingu T, Kato A (1980) Isolation of clivacetine from Clivia miniata Regel. (Amaryllidaceae). Chem Pharm Bull 28:1827–1831
Boit HG, Mehlis B (1961) Structure of clivimine and clivonine. Sci Nat 48:603
Kobayashi S, Shingu T, Uyeo S (1956) Structure of galanthamine and lycoramine. Chem Ind 1(11):177–178
Bartolucci C, Perola E, Pilger C, Fels G, Lamba D (2001) Three-dimensional structure of a complex of galanthamine (nivalin) with acetylcholinesterase from Torpedo californica: implications for the design of new anti-Alzheimer drugs. Proteins 42:182–191
Berkov S, Codina C, Viladomat F, Bastida J (2008) N-Alkylated galanthamine derivatives: potent acetylcholinesterase inhibitors from Leucojum aestivum. Bioorg Med Chem Lett 18:2263–2266
Hirasawa Y, Dai X, Deguchi J, Hatano S, Sasaki T, Ohtsuka R, Nugroho AE, Kaneda T, Morita H (2019) New vasorelaxant indole alkaloids, taberniacins A and B, from Tabernaemontana divaricata. J Nat Med 73:533–540
Amelia P, Nugroho AE, Hirasawa Y, Kaneda T, Tougan T, Horii T, Morita H (2019) Indole alkaloids from Tabernaemontana macrocarpa Jack. J Nat Med 73:820–825
Tang Y, Nugroho AE, Hirasawa Y, Tougan T, Horii T, Hadi AHA, Morita H (2019) Leucophyllinines A and B, bisindole alkaloids from Leuconotis eugeniifolia. J Nat Med 73:533–540
Hirasawa Y, Mitsui C, Uchiyama N, Hakamatsuka T, Morita H (2018) Hupercumines A and B, Lycopodium alkaloids from Huperzia cunninghamioides, inhibiting acetylcholinesterase. Org Lett 20:1384–1387
Nugroho AE, Zhang W, Hirasawa Y, Tang Y, Wong CP, Kaneda T, Hadi AHA, Morita H (2018) Bisleuconothines B-D, modified eburnane–aspidosperma bisindole alkaloids from Leuconotis griffithii. J Nat Prod 81:2600–2604
Nugroho AE, Inoue D, Wong CP, Hirasawa Y, Kaneda T, Shirota O, Hadi AHA, Morita H (2018) Reinereins A and B, new onocerane triterpenoids from Reinwardtiodendron cinereum. J Nat Med 72:588–592
Kihara M, Koike T, Imakura Y, Kida K, Shingu T, Kobayashi S (1987) Alkaloidal Constituents of Hymenocallis rotata HERB. (Amaryllidaceae). Chem Pharm Bull 35:1070–1075
Kobayashi S, Imakura Y, Ishikawa H, Sasakawa E (1980) Biogenetic-type synthesis of clivimine from clivacetine. Heterocycles 14:751–754
Wagner J, Pham HL, Döpke W (1996) Alkaloids from Hippeastrum equestre Herb. -5. Circular dichroism studies. Tetrahedron 52:6591–6600
Mehlis B (1965) Structure of clivimine and clivonine. Sci Nat 52(2):33–34
Döpke W, Bienert M (1967) Structures and stereochemistry of clivonine and clivimine. Tetrahedron Lett 8:451–457
Kitagawa T, Uyeo S, Yokoyama N (1959) Stereochemistry of lycorenine, homolycorine, pluviine, and their hydrogenation products. J Chem Soc 1959:3741–3751
Shiro M, Sato T, Koyama H (1966) Crystal structure of dihydrolycorine hydrobromide. Chem Ind 1(28):1229
Jeffs PW, Hansen JF, Döpke W, Bienert M (1971) The structure and stereochemistyr of clivonine. Tetrahedron 27:5065–5079
Schnoes HK, Smith DH, Burlingame AL, Jeffs PW, Döpke W (1968) Mass spectra of amaryllidaceae alkaloids: the lycorenine series. Tetrahedron 24:2825–2837
Ellman GL, Courtney KD, Andres V Jr, Feather-Stone RM (1961) A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 7:88–95
Acknowledgements
This work was partly supported by JSPS KAKENHI Grant Number JP19K07152, Japan.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Hirasawa, Y., Tanaka, T., Hirasawa, S. et al. Cliniatines A–C, new Amaryllidaceae alkaloids from Clivia miniata, inhibiting Acetylcholinesterase. J Nat Med 76, 171–177 (2022). https://doi.org/10.1007/s11418-021-01570-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11418-021-01570-6