
Abstract
Six new pyrrole 2-carbaldehyde derived alkaloids, dahurines A–F (1–6), along with five known ones (7–11) and butyl 2-pyrrolidone-5-carboxylate (12) were isolated from the roots of Angelica dahurica. Their structures were determined by extensive spectroscopic and spectrometric data (1D and 2D NMR, IR, and HRESIMS) and calculated electronic circular dichroism (ECD) methods. Although compounds 7 and 8 have been chemically synthesized, they were obtained from natural materials for the first time. Compounds 2, 3, 4, 10, and 11 exhibited acetylcholinesterase inhibitory activity with IC50 values in the range of 47.5–52.5 μM.
Graphic abstract
Pyrrole 2-carbaldehyde derived alkaloids from the roots of Angelica dahurica

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Angelica dahurica is a perennial medicinal plant belonging to the family Apiaceae. The roots of A. dahurica are clinically used as an analgesic agent for the treatment of migraine headache and toothache in Chinese medicine, and the promising therapeutic effects have attracted great interests from scientific researchers, leading to publication of a mass of documents concerning the pharmacological activities and chemical constituents of A. dahurica [1]. It is now generally believed that the major chemical constituents of A. dahurica roots are coumarins, and many of these coumarins have been demonstrated as possessing significant anti-inflammatory, anti-tumor, and acetylcholinesterase (AChE) inhibitory activities [2,3,4,5,6,7,8]. Our previous investigation on the coumarins in the roots of A. dahurica by LC–MS revealed that dimeric furanocoumarins might occur in the roots of A. dahurica in trace amounts but very complex structures, and further LC–MS-guided separation and purification of the targeted fractions led to the determination of 10 potentially anti-inflammatory dimeric furanocoumarins [9, 10]. Notably, Lu and coworkers reported the elucidation of one pyrrole 2-carbaldehyde alkaloid named as desmodimine from the roots of A. dahurica [11]. Given that alkaloids are sparsely reported from A. dahurica, and natural pyrrole 2-carbaldehyde alkaloids exhibit interesting bioactivities such as hepatoprotective, immunostimulatory, and AChE inhibitory activities [12], further investigation on the chemical constituents of the remnant fractions of A. dahurica were performed, which resulted in the identification of six new pyrrole 2-carbaldehyde alkaloids, dahurines A–F (1–6), along with five known ones (7–11) and a known pyrrolidone derivative (12). Herein, the isolation and structural elucidation of the isolated compounds and an evaluation of their inhibitory effects on AChE are described.
Results and discussion
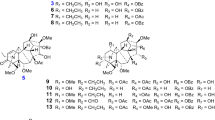
The n-BuOH-soluble fraction of the EtOH extract of the roots of A. dahurica was subjected to repeated silica gel, Sephadex LH-20, and RP-C18 gel column chromatography, followed by semipreparative RP-HPLC, to afford six new pyrrole 2-carbaldehyde derived alkaloids (1–6; Fig. 1), along with five known ones (7–11) and butyl 2-pyrrolidone-5-carboxylate (12).

Structures of compounds 1–8 from the roots of A. dahurica
Compound 1 was obtained as a pale yellow powder: \([\alpha ]_{\text{D}}^{21}\) + 6.0 (c 0.1, MeOH). Its molecular formula was assigned as C17H20N2O6 by the presence of a [M + H]+ ion peak at m/z 349.1480 in the HRESIMS spectrum, which was supported by the 13C NMR data (Table 1). The IR spectrum exhibited the absorption bands of ester carbonyl (1781 cm−1) and aldehyde (1663 cm−1) groups. The maximum UV absorption was observed at 292 nm, which is characteristic for pyrrole-2-aldehyde [13]. The 1H and 13C NMR spectra of 1 showed the presence of a 5-hydroxymethyl-pyrrole 2-carbaldehyde moiety [δH 9.48 (1H, s, H-7), 7.05 (1H, d, J = 4.0 Hz, H-3), 6.47 (1H, d, J = 4.0 Hz, H-4), 5.36 (1H, d, J = 13.5 Hz, H-6a), 5.14 (1H, d, J = 13.5 Hz, H-6b); δC 179.3, 57.6, 137.1, 113.9, 126.0, 132.7], and a 5-oxopyrrolidine-2-carboxylic acid moiety [δH 4.22 (1H, m, H-2′), 2.43 (1H, m, H-3′a), 2.19 (1H, m, H-3′b), 2.34 (2H, m, H2-4′); δC 177.8, 171.5, 55.4, 29.1, 24.9]. The remaining NMR signals [δH 4.71 (1H, d, J = 11.5 Hz, H-8), δH 4.30 (1H, m, H-11), 2.74 (1H, m, H-12), 1.63 (3H, d, J = 6.0 Hz, H3-14), 1.16 (3H, d, J = 6.5 Hz, H3-13); δC 171.2, 80.7, 63.3, 43.6, 18.6, 15.1] suggested the presence of a 4,5-dimethyldihydrofuran-2-one moiety in 1, which was further confirmed by the 1H–1H COSY correlations (Fig. 2) of H-8/H-12, H-12/H-11, H-12/H3-13, and H-11/H3-14, and the HMBC correlations (Fig. 2) of H-8/C-9, C-12, C-13; H3-13/C-11, C-12; and H3-14/C-11, C-12. Additionally, the HMBC correlations of H2-6/C-1′ and H-8/C-2, C-5 demonstrated that the moieties of 5-oxopyrrolidine-2-carboxylic acid and 4,5-dimethyldihydrofuran-2-one were located at C-6 and N-1, respectively. The relative configuration of 1 was determined by the NOE correlations of H3-13/H-8, H-11 (Fig. 3). The absolute configuration of 1 was determined by calculated electronic circular dichroism (ECD) method. Given that the relative configuration of C-2′ could not be solved by NOESY experiment, the ECD spectra of (8R, 11S, 12R, 2′S)-1, (8R, 11S, 12R, 2′R)-1, (8S, 11R, 12S, 2′R)-1, and (8S, 11R, 12S, 2′S)-1 were calculated using time-dependent density functional theory (TDDFT) at the B3LYP/6-31G level with the CPCM model. As shown in Fig. 4, the calculated spectrum of (8R, 11S, 12R, 2′S)-1 is in good agreement with the experimental spectrum. Accordingly, the absolute configuration of 1 was defined as (8R, 11S, 12R, 2′S)-1, named as dahurine A.

Selected COSY and HMBC correlations of compounds 1–6

Selected NOESY correlations of compounds 1–4

Experimental and calculated ECD spectra of compounds 1, 5, and 6 (in MeOH)
Compound 2 was obtained as a pale yellow powder: \([\alpha ]_{\text{D}}^{21}\) + 48.0 (c 0.1, MeOH). Its molecular formula was identified as C16H23NO4 by the presence of a [M + H]+ ion peak at m/z 294.1721 in the HRESIMS spectrum, which was supported by the 13C NMR data (Table 1). Comparison of the NMR data of 2 with those of 1 revealed that the 5-oxopyrrolidine-2-carboxylic acid group in 1 was replaced by an n-butoxy group, which was deduced from the protons resonating at δH 3.42 (2H, m, H2-1′), 1.54 (2H, m, H2-2′), 1.33 (2H, m, H2-3′), and 0.89 (3H, t, J = 7.5 Hz, H3-4′), and the carbons resonating at δC 70.1 (C-1′), 31.5 (C-2′), 19.3 (C-3′), and 14.8 (C-4′). The group of n-butoxy was determined to be linked at C-6 by the HMBC correlations between H2-6 and C-1′. The planar structure of 2 was further confirmed by the HMBC correlations (Fig. 2) of H-3/C-2, C-7, C-3; H-4/C-5, C-6; H2-6/C-5, C-1′; H-7/C-2; H-8/C-2, C-5, C-9, C-12, C-13; H-12/C-8, C-11; H3-13/C-11, C-12; and H3-14/C-11, C-12. The relative configuration of 2 was determined by the NOE correlations (Fig. 3) of H-8/H-11, H3-13; H-11/H3-13; and H-12/H3-14. Given that the experimental CD spectrum of 2 and the calculated ECD spectra of (8R, 11S, 12R)-2 are superimposed (Fig. 5), the structure of 2 was determined as shown in Fig. 1, named as dahurine B.

Experimental and calculated ECD spectra of compounds 2, 3, 4, 7, and 8 (in MeOH). The calculated ECD spectra of compounds 4, 7 and 8 were not presented due to the high similarity of these spectra to that of 2
Compound 3 was obtained as a pale yellow powder: \([\alpha ]_{\text{D}}^{21}\) − 74.0 (c 0.1, MeOH), with a molecular formula of C16H23NO4 determined by HRESIMS and NMR spectroscopic data (Table 1). Comparison of the NMR data of 3 with those of 2 revealed that compound 3 shared a similar skeleton with that of 2. Unambiguous assignment of the protons and carbons of 3 by 2D NMR experiments allowed establishing the planar structure of 3, which was completely the same to that of 2. However, the quite different retention time on the HPLC column (Fig. S47) suggested that 3 is a stereoisomer of 2. In the NOESY spectrum of 3, the NOE correlations (Fig. 3) of H-8/H-12, H-12/H3-14, and H-11/H3-13 suggested that the orientation of H-8 in 3 is opposite to that in 2. The absolute configuration of 3 was assigned as (8S,11S,12R)-3 by comparison of its experimental and calculated ECD spectra (Fig. 5). Therefore, the structure of 3 was elucidated as shown in Fig. 1, and named as dahurine C.
Compound 4 was isolated as a pale yellow, amorphous powder: \([\alpha ]_{\text{D}}^{21}\) + 18.0 (c 0.1, MeOH). Its molecular formula was assigned as C12H15NO3 by the presence of an [M + Na]+ ion peak at m/z 244.0919 in the HRESIMS spectrum. Comparison of the 1H and 13C NMR data of 4 with those of 2 revealed the absence of an n-butoxy group and the significantly up-fielded chemical shift of C-6 (δC 13.0, ΔδC − 43.5) in 4, suggesting that the –CH2OCH2CH2CH2CH3 moiety in 2 was replaced by a methyl group [δH 2.33 (3H, s, H3-6), δC 13.0]. The deduction was further confirmed by the HMBC correlations between H3-6 and C-4/C-5. The relative configuration of 4 was determined by the NOE correlations of H-8/H3-13 and H-11/H3-13 (Fig. 3). Given that the experimental spectrum of 4 and the calculated ECD spectrum of (8R, 11S, 12R)-4 are superimposed (Fig. 5), the structure of 4 was established as shown in Fig. 1, and named as dahurine D.
Compound 5 was obtained as a pale yellow powder: \([\alpha ]_{\text{D}}^{21}\) + 2.0 (c 0.1, MeOH). Its molecular formula was assigned as C23H31NO4 by the presence of a [M + H]+ ion peak at m/z 386.2323 in the HRESIMS spectrum. The IR spectrum exhibited the absorption bands of ester carbonyl (1780 cm−1), aldehyde (1659 cm−1), and phenyl (1541 and 1487 cm−1) groups. The 1H NMR spectrum of 5 indicated the characteristic signals due to a 5-hydroxymethyl-pyrrole-2-carbaldehyde moiety [δH 9.50 (1H, s, H-7), 6.99 (1H, d, J = 4.0 Hz, H-3), and δH 6.05 (1H, d, J = 4.0 Hz, H-4), and 3.88 (1H, br s, H-6a), δH 3.53 (1H, br s, H-6b)], and signals contributing to a phenylalanine moiety [δH 7.16 (3H, overlapped, H-13, H-14, H-15), 6.84 (2H, overlapped, H-12, H-16), 5.26 (1H, br s, H-8), 3.74 (1H, dd, J = 14.0, 2.5 Hz, H-10a), and 3.32 (1H, dd, J = 14.0, 2.5 Hz, H-10b)]. In addition, signals contributing to two n-butoxy groups were also observed in the 1H NMR spectrum of 5. Comparison of the NMR data of 5 with those of methyl (2R)-[2-formyl-5-(methoxymethyl)-1H-pyrrol-1-yl]-3-(phenyl) propanoate, a pyrrole 2-carbaldehyde derived alkaloid previously isolated from Lycium chinense [14], revealed that the two methyl groups in the known compound were replaced by two n-butoxy groups. Further analysis of the 1D and 2D NMR spectra of 5 led to the establishment of the planar structure of 5. The CD spectrum of 5 indicated negative Cotton effects at 308 nm (Δεmax −8.3), which is similar to that of the known analogue aforementioned [14], resulting in the assignment of an 8R configuration in 5. In addition, the experimental CD spectrum of 5 is highly similar to the calculated ECD spectrum of (8R)-5 (Fig. 5). Thus, the structure of 5 was elucidated as shown in Fig. 1, and named as dahurine E.
Compound 6 was isolated as a pale yellow powder: \([\alpha ]_{\text{D}}^{21}\) + 2.0 (c 0.1, MeOH). Its molecular formula was assigned as C17H27NO4 by the presence of a [M + Na]+ ion peak at m/z 332.1852 in the HRESIMS spectrum. The NMR data of 6 were similar to those of 5, except for the replacement of the benzyl group in 5 by a methyl group. The aforementioned structure of 6 was further confirmed by the key HMBC correlations of H3-10/C-8, C-9, and the 1H–1H COSY correlations of H-3/H-4, H3-10/H-8, H2-1′/H2-2′/H2-3′/H3-4′, and H2-1″/H2-2″/H2-3″/H3-4″. Akin to the CD spectrum of 5, negative Cotton effects at 309 nm were also observed in the CD spectrum of 6, and the experimental CD spectrum of 6 was in good agreement with the calculated spectrum of (8R)-6. Accordingly, the structure of 6 was determined as shown in Fig. 1, and named as dahurine F.
By comparison of their spectroscopic data (Table S1) with those reported, and using calculated ECD method (Fig. 5), compounds 7 and 8 were identified as (8R,11S,12R)-funebral and (8R,11S,12R)-3,4-dihydro-3-amino-4,5-dimethylfuran-2[5H]-one-2-formyl pyrrole, which have been chemically synthesized by Dong and coworkers [15]. Notably, this is the first time that compounds 7 and 8 were isolated from natural materials. In addition, by comparison of their NMR and specific rotation data with those reported in literature, compounds 9–12 were identified as 4″-butyl-2-formyl-5-(hydroxymethyl)-1H-pyrrole-1-butanoic acid (9) [16], butyl 2-formyl-5-butoxymethyl-1H-pyrrole-1-butanoate (10) [17], hemerocallisamine II (11) [18], and butyl 2-pyrrolidone-5-carboxylate (12) [19], respectively.
Compounds 1–12 were evaluated for their inhibitory effects on AChE using Ellman’s method, using huperzine A as a positive control (IC50 = 0.6 ± 0.1 μM). Compounds 2, 3, 4, 10, and 11 exhibited inhibition of AChE with IC50 values of 52.0 ± 0.5, 48.2 ± 0.1, 47.5 ± 0.2, 50.4 ± 0.6, and 52.5 ± 3.4 μM, respectively. Compounds 1, 5–9, and 12 were inactive (< 50% inhibition at 100 μM).
Experimental
General procedures
Optical rotations were obtained on a Rudolph Autopol IV automatic polarimeter (NJ, USA). IR spectra were recorded on a Thermo Nicolet Nexus 470 FT-IR spectrophotometer (MA, USA) with KBr pellets. UV spectra were obtained using a Shimadzu UV-2450 spectrophotometer (Tokyo, Japan). NMR spectra were recorded on a Varian INOVA-500 spectrometer (CA, USA) operating at 500 MHz for 1H NMR and 125 MHz for 13C NMR. HRESIMS was recorded on an LCMS-IT-TOF system, fitted with a Prominence UFLC system and an ESI interface (Shimadzu, Kyoto, Japan). Silica gel (200–300 mesh, Qingdao Marine Chemical Inc., Qingdao, China), LiChroprep RP-C18 gel (40–63 μm, Merck, Germany), D101 macroporous adsorption resin (Qingdao Marine Chemical Inc., Qingdao, China), and Sephadex LH-20 (Pharmacia) were used for open-column chromatography (CC). HPLC was performed on a Shimadzu LC-20AT pump system (Shimadzu Corporation, Tokyo, Japan), equipped with a SPD-M20A photodiode array detector monitoring at 254 nm. A semi-preparative HPLC column (YMC-Pack C18, 250 × 10 mm, 5 μm) was utilized for compound separation and purification. TLC was performed using GF254 plates (Qingdao Marine Chemical Inc., Qingdao, China).
Plant material
The roots of A. dahurica (Fisch. ex Hoffm.) Benth. et Hook. were collected in Bozhou, Anhui province, People’s Republic of China, in December 2013, and plant authentication was performed by one of the authors (P.F. Tu). A voucher specimen (SPSHI-ADB-201312) is deposited in the Modern Research Center for Traditional Chinese Medicine, Beijing University of Chinese Medicine.
Extraction and isolation
The air-dried roots of A. dahurica (29.6 kg) were refluxed with 80% EtOH (3 × 180 L, 2 h each). After removal of the solvent under reduced pressure, the residue (1.5 kg) was suspended in H2O (4 L) and partitioned with petroleum ether (4 × 4 L), EtOAc (4 × 4 L), and n-BuOH (4 × 4 L), successively. The n-BuOH extract (805 g) was subjected to D101 macroporous adsorption resin chromatography and eluted with H2O–EtOH (100:0, 80:20, 20:80, v/v) to yield three fractions (fr. A–C). Fr. C (202 g) was subjected to silica gel chromatography and eluted with a gradient of CH2Cl2–MeOH from 15:1 to 1:1 to give 20 subfractions (subfr. C1–C20). Subfr. C1 (10.1 g) was subjected to an RP-C18 open column and eluted with a stepwise gradient elution using MeOH–H2O to yield 12 fractions (fr. C1A–C1L). Fr. C1E (80 mg) was purified by semipreparative RP-HPLC using 20% aqueous MeCN as the mobile phase to afford 4 (7.5 mg, tR = 13.0 min), 7 (25.0 mg, tR = 18.0 min), 8 (10.0 mg, tR = 19.0 min), and 1 (1.7 mg, tR = 21.5 min). Fr. C1F (90 mg) was purified by semipreparative RP-HPLC using 50% aqueous MeOH as the mobile phase to afford 11 (12.0 mg, tR = 23.0 min), 12 (20.0 mg, tR = 25.0 min), and 10 (8.5 mg, tR = 33.5 min). Fr. C1G (70 mg) was purified by semipreparative RP-HPLC using 70% aqueous MeOH as the mobile phase to afford 2 (2.2 mg, tR = 19.0 min), 3 (5.5 mg, tR = 20.5 min), 9 (5.2 mg, tR = 25.0 min), 6 (2.2 mg, tR = 28.0 min), and 5 (1.7 mg, tR = 37.0 min).
Dahurine A (1): pale yellow powder; \([\alpha ]_{\text{D}}^{21}\) + 6.0 (c 0.1, MeOH); UV (MeOH) λmax (log ε): 292 (3.79); IR (KBr) νmax: 2973, 2932, 2873, 1781, 1663, 1452, 1335, 1190, 1052, 781 cm−1; 1H and 13C NMR data, see Table 1; positive-ion HRESIMS: m/z 349.1380 [M + H]+ (calcd. for C17H21N2O6, 349.1394).
Dahurine B (2): pale yellow powder; \([\alpha ]_{\text{D}}^{21}\) + 48.0 (c 0.1, MeOH); UV (MeOH) λmax (log ε): 291 (3.81); IR (KBr) νmax: 2959, 2931, 2871, 1783, 1664, 1451, 1334, 1189, 1089, 1052, 779 cm−1; 1H and 13C NMR data, see Table 1; positive-ion HRESIMS: m/z 294.1721 [M + H]+ (calcd. for C16H24NO4, 294.1700).
Dahurine C (3): pale yellow powder; \([\alpha ]_{\text{D}}^{21}\) − 74.0 (c 0.1, MeOH); UV (MeOH) λmax (log ε): 293 (3.79); IR (KBr) νmax: 2975, 2934, 2876, 1779, 1658, 1451, 1337, 1309, 1193, 1048, 781 cm−1; 1H and 13C NMR data, see Table 1; positive-ion HRESIMS: m/z 294.1688 [M + H]+ (calcd. for C16H24NO4, 294.1700).
Dahurine D (4): pale yellow powder; \([\alpha ]_{\text{D}}^{21}\) + 18.0 (c 0.1, MeOH); UV (MeOH) λmax (log ε): 292 (3.78); IR (KBr) νmax: 2974, 2933, 2876, 1779, 1659, 1451, 1337, 1310, 1194, 1049, 781 cm−1; 1H and 13C NMR data, see Table 1; positive-ion HRESIMS: m/z 244.0919 [M + Na]+ (calcd. for C12H15NO3Na, 244.0944).
Dahurine E (5): pale yellow powder; \([\alpha ]_{\text{D}}^{21}\) + 2.0 (c 0.1, MeOH); UV (MeOH) λmax (log ε): 292 (3.79); IR (KBr) νmax: 2974, 2933, 2876, 1780, 1659, 1541, 1451, 1310, 1241, 1087, 1049, 781 cm−1; 1H and 13C NMR data, see Table 2; positive-ion HRESIMS: m/z 386.2323 [M + H]+ (calcd. for C23H32NO4, 386.2326).
Dahurine F (6): pale yellow powder; \([\alpha ]_{\text{D}}^{21}\) + 2.0 (c 0.1, MeOH); UV (MeOH) λmax (log ε): 292 (3.69); IR (KBr) νmax: 2976, 2931, 2849, 1782, 1658, 1472, 1413, 1188, 1050, 934, 763 cm−1; 1H and 13C NMR data, see Table 2; positive-ion HRESIMS: m/z 332.1852 [M + Na]+ (calcd. for C17H27NO4Na, 332.1832).
Biological assays
AChE inhibitory activity was determined via the method of Mukherjee by a 96-well microplate reader based on Ellman’s method [20, 21]. Briefly, 100 μL of 0.1 M PBS buffer (pH 8.0), 20 μL of AChE solution (50 mU/mL in buffer), and 20 μL of sample diluted in 0.2% DMSO buffer solution were added to a microplate and incubated at 37 °C for 20 min. Then, 40 μL of 0.6 mM 5,5′-dithiobis(2-nitrobenzoic acid) (DTNB) was added. The reaction was initiated by adding 20 μL of 1.2 mM acetylthiocholine iodide (ATCh). After incubating the reaction solution at 37 °C for 30 min, the reaction was quenched by the addition of 4% sodium dodecyl sulfate (SDS) solution. The optical densities were immediately measured in a 96-well plate reader at 405 nm. The experiments were performed in triplicate. The percentage inhibition was evaluated using the equation I (%) = [1 − (Asample − Abackground)/Ablank] × 100%, where Asample is the absorbance of each test compound, and Abackground is the absorbance of the background without enzyme. The IC50 values were determined using GraphPad Prism 5 software from experiments performed (GraphPad Software, Inc., San Diego, CA, USA). Huperzine A was used as a positive control (IC50 = 0.6 ± 0.1 μM).
ECD calculations
The relative configurations of compounds 1–4, 7, and 8 were initially established on the basis of their NOESY spectra and submitted to random conformational analysis with the MMFF94s force field and using the SYBYL-X 2.0 software package. The conformers were further optimized by using the time-dependent density functional theory (TDDFT) method at the B3LYP/6-31G(d) level, and the frequency was calculated at the same level of theory. The stable conformers without imaginary frequencies were subjected to ECD calculation by the TDDFT method at the B3LYP/6-31 + G(d) level with the CPCM model in methanol solution. ECD spectra of different conformers were simulated using SpecDis v1.51 with a half-bandwidth of 0.16–0.4 eV, and the final ECD spectra were obtained according to the Boltzmann-calculated contribution of each conformer after UV correction. The calculated ECD spectra were compared with the experimental data. All calculations were performed with the Gaussian 09 program package [22, 23].
References
Chinese Pharmacopoeia Commission (2015) Pharmacopoeia of the People’s Republic of China, vol I. Chemical Industry Press, Beijing, pp 105–106
Kwon YS, Shin SJ, Kim MJ, Kim CM (2002) A new coumarin from the stem of Angelica dahurica. Arch Pharm Res 25:53–56
Kwon YS, Kobayashi A, Kajiyama SI, Kawazu K, Kanzaki H, Kim CM (1997) Antimicrobial constituents of Angelica dahurica roots. Phytochemistry 44:887–889
Thuong PT, Hung TM, Ngoc TM, Ha DT, Min BS, Kwack SJ, Kang TS, Choi JS, Bae KH (2010) Antioxidant activities of coumarins from Korean medicinal plants and their structure–activity relationships. Phytother Res 24:101–106
Thanh PN, Jin WY, Song GY, Bae K, Kang SS (2004) Cytotoxic coumarins from the root of Angelica dahurica. Arch Pharm Res 27:1211–1215
Baek NI, Ahn EM, Kim HY, Park YD (2000) Furanocoumarins from the root of Angelica dahurica. Arch Pharm Res 23:467–470
Liu DP, Luo Q, Wang GH, Xu Y, Zhang XK, Chen QC, Chen HF (2011) Furocoumarin derivatives from radix Angelicae dahuricae and their effects on RXRα transcriptional regulation. Molecules 16:6339–6448
Seo WD, Kim JY, Ryu HW, Kim JH, Han SI, Ra JE, Seo KH, Jang KC, Lee JH (2013) Identification and characterisation of coumarins from the roots of Angelica dahurica and their inhibitory effects against cholinesterase. J Funct Foods 5:1421–1431
Yang WQ, Song YL, Zhu ZX, Su C, Zhang X, Wang J, Shi SP, Tu PF (2015) Anti-inflammatory dimericfuranocoumarins from the roots of Angelica dahurica. Fitoterapia 105:187–193
Yang WQ, Zhu ZX, Song YL, Qi BW, Wang J, Su C, Tu PF, Shi SP (2017) Dimeric furanocoumarins from the roots of Angelica dahurica. Nat Prod Res 31:870–877
Lu J, Jin L, Jin YS, Chen HS (2007) Chemical constituents in roots of Angelica dahurica var. formosana. Acad J Second Mil Med Univ 28:294–298
Wood JM, Furkert DP, Brimble MA (2018) 2-Formylpyrrole natural products: origin, structural diversity, bioactivity and synthesis. Nat Prod Rep. https://doi.org/10.1039/c8np00051d
Kim SB, Chang BY, Jo YH, Lee SH, Han SB, Hwang BY, Kim SY, Lee MK (2013) Macrophage activating activity of pyrrole alkaloids from Morus alba fruits. J Ethnopharmacol 145:393–396
Youn UJ, Lee JY, Kil YS, Han AR, Chae CH, Ryu SY, Seo EK (2016) Identification of new pyrrole alkaloids from the fruits of Lycium chinense. Arch Pharm Res 39:321–327
Dong Y, Pai NN, Ablaza SL, Yu SX, Bolvig S, Forsyth DA, Quesne PWL (1999) Quararibea metabolites. 4. Total synthesis and conformational studies of (±)-funebrine and (±)-funebral. J Org Chem 64:2657–2666
Yao CP, Zou ZX, Zhang Y, Li J, Cheng F, Xu PS, Zhou G, Li XM, Xu KP, Tan GS (2018) New adenine analogues and a pyrrole alkaloid from Selaginella delicatula. Nat Prod Res. https://doi.org/10.1080/14786419.2018.1482892
Fang CW, Chen JJ, Liu SJ (2010) Studies on the chemical constituents of rhizoma of Coniogramme japonica. J Chin Med Mater 33:557–559
Matsumoto T, Nakamura S, Ohta T, Fujimoto K, Yoshikawa M, Ogawa K, Matsuda H (2015) A rare glutamine derivative from the flower buds of daylily. Org Lett 16:3076–3078
Park MR, Kim YC, Lee S, Kim IS (2009) Identification of an ISR-related metabolite produced by rhizobacterium Klebsiella oxytoca C1036 active against soft-rot disease pathogen in tobacco. Pest Manag Sci 65:1114–1117
Wang MZ, Sun MW, Hao HL, Lu CH (2015) Avertoxins A–D, prenyl asteltoxin derivatives from Aspergillus versicolor Y10, an endophytic fungus of Huperzia serrata. J Nat Prod 78:3067–3070
Ying YM, Shan WG, Zhan ZJ (2015) Biotransformation of Huperzine A by a fungal endophyte of Huperzia serrata furnished sesquiterpenoid–alkaloid hybrids. J Nat Prod 77:2054–2059
Qi BW, Liu X, Mo T, Zhu ZX, Li J, Wang J, Shi XP, Zeng KW, Wang XH, Tu PF, Abe I, Shi SP (2017) 3,5-Dimethylorsellinic acid derived meroterpenoids from Penicillium chrysogenum MT-12, an endophytic fungus isolated from Huperzia serrata. J Nat Prod 80:2699–2707
Qi BW, Liu X, Mo T, Li SS, Wang J, Shi SP, Xiao XH, Zhu ZX, Zhao YF, Jin HW, Tu PF, Shi SP (2017) Nitric oxide inhibitory polyketides from Penicillium chrysogenum MT-12, an endophytic fungus isolated from Huperzia serrata. Fitoterapia 123:35–43
Acknowledgements
This work was partially supported by the National Natural Science Foundation of China (no. 81573312).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Qi, B., Yang, W., Ding, N. et al. Pyrrole 2-carbaldehyde derived alkaloids from the roots of Angelica dahurica. J Nat Med 73, 769–776 (2019). https://doi.org/10.1007/s11418-019-01328-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11418-019-01328-1