
Abstract
As average lifespan of humans increases in western countries, cardiac diseases become the first cause of death. Aging is among the most important risk factors that increase susceptibility for developing cardiovascular diseases. The heart has very aerobic metabolism, and is highly dependent on mitochondrial function, since mitochondria generate more than 90 % of the intracellular ATP consumed by cardiomyocytes. In the last few decades, several investigations have supported the relevance of mitochondria and oxidative stress both in heart aging and in the development of cardiac diseases such as heart failure, cardiac hypertrophy, and diabetic cardiomyopathy. In the current review, we compile different studies corroborating this role. Increased mitochondria DNA instability, impaired bioenergetic efficiency, enhanced apoptosis, and inflammation processes are some of the events related to mitochondria that occur in aging heart, leading to reduced cellular survival and cardiac dysfunction. Knowing the mitochondrial mechanisms involved in the aging process will provide a better understanding of them and allow finding approaches to more efficiently improve this process.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Background
According to different organizations such as the American Heart Association and the World Health Organization (WHO), cardiovascular diseases are becoming the first cause of death in western countries (Heidenreich et al. 2011). The morbidity due to these sorts of diseases is even higher than those generated by neurodegenerative disorders, and it is believed that the percentages will increase in the near future. In 2012, ischemic heart disease was the main cause of premature mortality in high-income countries worldwide, and it is expected that more than 23 million people will die from ischemic heart disease in 2030. By that year, the proportion of total deaths worldwide due to cardiovascular diseases within the elderly population (>70 years) will be 40 % (World health statistics 2014. World Health Organization).
Although long-term exposure to risk factors, such as those related to lifestyle like diet and physical inactivity, plays a major role in the etiopathogenesis of cardiac disorders, aging itself is considered to be the major determinant for developing cardiac diseases. Moreover, other age-related diseases such as dyslipidemia and diabetes mellitus enhance the deleterious effects of aging on the cardiovascular system. Life expectancy at birth has increased by 6 years worldwide in the last 3 decades. As previously mentioned, such an increase has a direct consequence on there being a higher rate of rate of age-related diseases, in particular cardiovascular ones. More importantly, the increase in average lifespan is expected to keep rising over the next 20 years, when approximately 20 % of the population will be 65 years old or older.
The heart is primarily a postmitotic tissue and exhibits highly aerobic metabolism. These features implicate two consequences for normal organ function: dependence on healthy mitochondria and on healthy cells (Judge and Leeuwenburgh 2007). Studying bioenergetics of aged cardiomyocytes has been an important research area for many years. Mitochondria play a fundamental role in the survival and function of cardiomyocytes and are critical for the high demand of energy in the myocardium. In physiological states, 20–30 % of the cell volume of cardiomyocytes is occupied by mitochondria, but the numbers can increase with enhanced myocardial energy requirements. Primary energy production in myocardial tissue occurs in the form of adenosine triphosphate (ATP) by mitochondria to bridge the metabolism of nutrients and oxidative respiration. The heart consumes the equivalent of 6 kg of ATP per day, which is mainly generated through mitochondrial oxidative phosphorylation from catabolism of lipids and carbohydrates and used for various biological events (Ren et al. 2010). The energy pool of the heart includes ATP (≈5 μmol/g wet weight) and phosphocreatine (PCr; ≈8 μmol/g wet weight), with the latter serving as an ATP transport and buffer system (Beer et al. 2002). In the mitochondria, the high-energy phosphate bond in ATP can be transferred to creatine by mitochondrial creatine kinase to form PCr. With a lower molecular weight than ATP, PCr can easily diffuse through the mitochondrial membrane into the cytosol. Here, it can be used to generate ATP from adenosine diphosphate (ADP) through reactions catalyzed by the cytosolic creatine kinase (Neubauer 2007). There is a fine balance between nuclear and mitochondrial gene expression, which governs the assembly of mitochondrial respiratory complexes. Especially under conditions of exercise, mitochondrial biogenesis is triggered through modulation of the ATP/ADP ratio, activation of adenosine monophosphate-activated protein kinase (AMPK), and consequent expression of the transcriptional factor peroxisomal proliferator activator receptor γ co-activator 1α (PGC-1α) and nuclear respiratory factor-1 (NRF1) (Kim et al. 2008). In turn, these increases in cardiac energy demands enhance gene expression of nuclear and mitochondrial DNA (mtDNA) maximizing the capacity of mitochondria to perform oxidative phosphorylation.
Mitochondrial role in aging
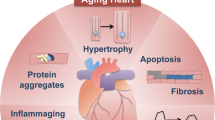
Taking into account that aging is one of the major factors leading to cardiac dysfunction, it is especially important to know the mechanisms underlying the aging process in order to find approaches to improve it. Several factors have been considered important contributors to the aging process, such as mitochondrial dysfunction, genomic instability, and epigenetic alterations among others (Lopez-Otin et al. 2013). One of the main features of aging is the accumulation of genetic damage and increased DNA instability (Beckman and Ames 1999, Lopez-Otin et al. 2013). Due to continuous exposure to both exogenous and endogenous threats, nuclear and mitochondrial DNA integrity are compromised during aging (Garinis et al. 2008, Gredilla et al. 2010). Interestingly, the main sites of reactive oxygen species (ROS) generation, under physiological conditions, are located within the electron transport chain in mitochondria (Barja 1999, Brand 2010). Mitochondrial DNA instability has been considered especially relevant in the aging process, since it leads to mitochondrial dysfunction (Kujoth et al. 2007, Atkinson et al. 2011, Tyurina et al. 2012). Relevance of mtDNA in cellular function was clearly evident after the identification of mitochondrial diseases as a result of either inherited or spontaneous mutations in mtDNA, leading to mitochondrial dysfunction, such as chronic progressive external ophthalmoplegia syndrome or NADH dehydrogenase deficiencies, both mitochondrial myopathies. In addition, different premature aging syndromes are related to increased DNA instability, and several of them are accompanied by cardiovascular aging (Minamino and Komuro 2008, Jeppesen et al. 2011). Moreover, the generation of a knock-in mouse expressing defective mitochondrial DNA polymerase (PolG mouse) has provided the first clear support that accumulation of somatic mtDNA mutations causes a variety of aging phenotypes in mammals (Trifunovic et al. 2004, Kujoth et al. 2005). One of the main consequences of mtDNA instability is mitochondrial dysfunction which, as mentioned above, is thought to significantly contribute to the aging process. Mitochondria are not only the critical organelle for the generation of metabolic energy in eukaryotic cells via oxidative phosphorylation but they also participate in an important number of cellular functions including apoptosis, Ca2+ homeostasis, and redox signaling among others (Tyler, 1991). Consequently, impaired mitochondrial function leads to loss of cellular homeostasis, and hence to cellular dysfunction (Fig. 1).

Several factors contribute to heart aging and higher susceptibility to cardiac diseases (see text for details and references). Increased mtDNA instability, enhanced apoptotic processes, and loss of homeostasis (e.g., Ca2+ buffering) in cardiomyocytes lead to impaired bioenergetics and reduced survival. Moreover, release of cytokines from cardiac fibroblasts and different growing factors, such as TGFβ, play a critical role in survival decline of cardiomyocytes with aging, in cardiac remodeling and in the development of fibrosis, which contribute to cardiac hypertrophy. In addition, important positive feedback between oxidative stress and inflammatory processes takes place. All these events will ultimately lead to cardiac dysfunction
As organisms age, the efficiency of the mitochondrial respiratory chain has been reported to decrease in different tissues, diminishing ATP generation (Drew and Leeuwenburgh 2004, Bratic and Trifunovic 2010, Green et al. 2011). As already mentioned, one of the main factors causing mitochondrial dysfunction and defective bioenergetics is the accumulation of mtDNA mutations. However, other mechanisms also contribute to the age-related loss of mitochondrial function such as oxidation of mitochondrial proteins, destabilization of electron transport chain complexes, alterations in the lipid composition of mitochondrial membranes, and defective quality control by mitophagy (Lopez-Otin et al. 2013). Importantly, mitochondrial function is not only affected by mtDNA instability; since most of the mitochondrial proteins are encoded by nDNA, increased nDNA instability also contributes to impairment of mitochondrial function (Fang et al. 2016).
Mitochondrial dysfunction not only affects bioenergetics but it is also known to influence apoptotic events, which contribute to the decline of tissue functionality (Kujoth et al. 2006, Tower 2015). The effect of mitochondrial malfunction on programed cell death pathways differs depending on cell types and tissues. Age-related enhancement in apoptosis has been related to cell loss in several tissues, including the skeletal muscle, heart, and brain leading to dysfunctional conditions such as sarcopenia, cardiac dysfunction, and neurodegenerative diseases (Tower 2015).
Metabolic changes in aged heart
The main metabolic pathway used by the heart is long-chain fatty acid β-oxidation and utilizes 70 to 90 % of cardiac ATP (Modrego et al. 2013). The remaining 10 to 30 % comes from the oxidation of glucose and lactate, as well as small amounts of ketone bodies and certain amino acids (Doenst et al. 2013). Use of fatty acids or glucose substrate may directly inhibit the use of the others (the Randle cycle). Substrate selection and interaction between glucose and fatty acids used in the heart have been considered highly relevant in cardiac disease, since complete metabolism of glucose is more oxygen efficient than that of fatty acid (Opie and Knuuti 2009). When it comes to mitochondria in the aged heart, levels of respiratory proteins and other key proteins involved in mitochondrial metabolism decline including those in fatty acid metabolism. On the contrary, glucose metabolic pathways as well as extracellular structural proteins increase significantly with age (Dai et al. 2014). Thus, increased expression of glycolytic proteins, together with a decline in fatty acid oxidation, would be indicating a metabolic remodeling with age similar to heart failure in younger individuals (Dai et al. 2012). Due to the high energetic demand of the heart, age-related defects in mitochondrial bioenergetics have been commonly related to normal cardiac aging (Bratic and Larsson 2013, Tocchi et al. 2015). For instance, it has been proposed that heart failure (HF) is associated with a reactive hyperadrenergic state that increases circulating plasma free fatty acids, which leads to impaired glucose metabolism and insulin resistance (Opie and Knuuti 2009). Also, aging-associated pathophysiological changes in metabolism like diabetes crucially contribute to the generation of oxidized proteins and advanced glycation end products (Fulop et al. 2007). Age-dependent cardiac alterations related to metabolic changes will be expanded upon throughout this review.
Cardiac remodeling in aging heart
Myocardial remodeling during aging is related to changes in the amount and organization of extracellular matrix components (Kwak 2013). Proper collagen arrangement prevents excessive stretch and damage and preserves heart function (de Souza 2002). One of the most recurrent and well-characterized alterations associated with cardiac aging is increased collagen deposition within cardiomyocytes and around blood vessels (Gazoti Debessa et al. 2001, Horn et al. 2012).
Aging increases the rate of ventricular collagen turnover and deposition by fibroblasts (Mendes et al. 2012). Age-related fibrosis is characterized by increased collagen content, decreased collagen solubility, and increased collagen cross-linking (Thomas et al. 2001). In addition to collagen deposition, modifications and cross-linking of both collagen and elastin contribute to the development of vascular calcification, which increases with aging (Atkinson 2008). Related extracellular matrix modifications participate in the development of valve sclerosis, which is present in 30 to 80 % of elderly individuals. Indeed, deposition of collagen fibers or fibrosis is associated with stiffer ventricles and diastolic dysfunction in the senescent heart. A recent study has shown that metalloproteinase-mediated degradation of elastin contributed to valve mineralization and calcification by inducing calcium deposition onto fragmented elastin in humans (Nassimiha et al. 2001).
One of the most common consequences of cardiac remodeling in aged heart is enhanced left ventricular hypertrophy (LVH) evidenced by the thickening of ventricular walls. Clinical trials such as The Framingham Heart Study and the Baltimore Longitudinal Study on Aging have demonstrated an age-dependent increase in LVH in healthy adults free of hypertension, a known risk factor for LVH (Lakatta and Levy 2003). With age, several cardiac hypertrophic mechanisms are enhanced such as mechanical load and reduced cardiomyocytes as well as the previously described higher interstitial fibrosis.
Cardiomyocyte hypertrophy undergoes several deleterious processes in aging heart. Progressive inhibition of autophagy is attributed, at least, in part to intralysosomal accumulation of lipofuscin (De Meyer et al. 2010). In this scenario, crosslinked polymeric lipofuscin cannot be degraded by lysosomal hydrolases and might accumulate as lipofuscin-loaded lysosomes at the expense of active autolysosomes (Rajawat et al. 2009). Exacerbated autophagy stimulates further accumulation of damaged mitochondria, usually deficient in ATP production and, induces increased amounts of ROS, while oxidative modified cytosolic proteins form large indigestible aggregates. These events enhance lipofuscingenesis and sensitize cardiomyocytes to undergo apoptosis. Progression of these changes seems to result in enhanced oxidative stress, decreased ATP production, and collapse of cellular catabolic mechanisms, which eventually are incompatible with cardiomyocyte viability (Terman et al. 2010).
On the other hand, changes in single myocytes may also reach adaptive limits as in calcium handling. Intracellular calcium plays a critical role in modulating cardiac function. Calcium-induced calcium release regulates myocardial contractility through activation of ion channels, activation of ryanodine receptor, and activity of sodium/calcium exchanger. Re-sequestration of Ca2+ in the sarcoplasmic reticulum by the activity of sarcoplasmic endoplasmic reticulum Ca2+–ATPase 2a (SERCA 2a) triggers myocardial relaxation. In senescent myocardium, the active diastolic relaxation properties of the myocytes are highly modified causing delayed ventricular relaxation due to impaired Ca2+ cycling/handling (Lakatta 2003, Upadhya et al. 2015). In recent studies, it has been shown that the increased oxidative stress observed in aging heart leads to oxidative damage of the sarcoplasmic reticulum SERCA pump, thus decreasing its Ca2+-sequestering activity and prolonging diastolic relaxation (Upadhya et al. 2015).
Moreover, in a study conducted in female Fischer rats, a commonly used animal model of aging-associated heart failure (Boluyt et al. 2004), prolonged cellular contractions in older animals have been described as being caused by slower decay of the intracellular Ca2+ transients and increased expression of slower β-isoform of myosin heavy chain molecules (Carnes et al. 2004, Campbell et al. 2013). Progression of these modifications will adversely affect other electrical activity and contractile power output. In the mentioned study, authors showed that the shift in transmural properties is caused by age- and region-dependent changes in Ca2+ transient morphology and altered Ca2+-relaxation coupling at the level of the myofilaments. Decreased phosphorylation of troponin I, which contributed to the myofilament level effect, was also described (Boluyt et al. 2004).
Oxidative stress and DNA instability in cardiac mitochondria
Since they were initially postulated as major determinants of lifespan (Harman 1972), a large body of evidence has supported the role of mitochondria and oxidative stress in the aging process. Free radicals are generated in different cellular compartments (i.e., mitochondria, peroxisomes) and by multiple enzymes, such as NADPH oxidase or xanthine oxidase. However, mitochondrial ROS production is considered the most important source of cellular ROS in healthy tissues, since the main free radical generator, the electron transport chain is located in the inner mitochondrial membrane (Barja 1999). Mitochondrial free radicals are continuously generated during oxidative phosphorylation and ATP generation. For several years, ROS have been primarily investigated due to their damage-promoting effects. However, they also play an important role as signaling molecules, being involved in different physiological responses that control cellular homeostasis (Shadel and Horvath 2015) In fact, various researchers have started questioning the role of mitochondrial free radicals in the aging process, considering that some ROS may actually act as pro-longevity signaling molecules. These studies have been mainly performed in animal models of aging like Caenorhabditis elegans (Dillin et al. 2002, Schulz et al. 2007, Ristow and Zarse 2010) and Drosophila melanogaster (Sanz 2016, Scialo et al. 2016). Furthermore, the absence of effect on lifespan when antioxidants are overexpressed or depleted in mouse models has also been considered contrary to the theoretical idea of oxidative stress and mitochondrial free radicals as main determinants of the rate of aging by some authors (Jang et al. 2009, Zhang et al. 2009). Consequently, the debate about the role of free radicals in the aging process has increased in the last few years (Gruber et al. 2008, Barja 2013, Rugarli and Trifunovic 2015, Speakman et al. 2015). Meanwhile, new aspects of mitochondrial function have come out suggesting that, even if mitochondrial free radicals are not as critical as thought in the aging process, mitochondria themselves would play a central role after all (Gonzalez-Freire et al. 2015).
Taking into account that aging is a multifactorial process (Lopez-Otin et al. 2013) and despite the reports questioning mitochondrial free radicals as main determinants of aging, different studies in animal models and observations in humans suggest that mitochondrial function and oxidative stress are important factors contributing to aging (Barja 2013, 2014). Free radicals like ·OH and OONO− can react and damage all biological components of the cell. Among the different mitochondrial molecules attacked by ROS, mtDNA is the most critical one for mitochondrial function (Gredilla and Barja 2005). Free radicals generate a wide number of mtDNA lesions, including oxidized DNA bases, abasic sites, and single- and double-strand breaks (DSBs), the latter being some of the most harmful DNA lesions. Many of these ROS-induced DNA lesions show mutagenic or cytotoxic effects due to mispair of bases, which may give rise to mutations upon DNA replication (Grollman and Moriya 1993, Kavli et al. 2007). DNA damage accumulation may lead to blockage of DNA replication and transcription, contributing to genomic instability. In order to maintain genomic integrity and stability, DNA repair pathways have evolved and the particular pathway employed depends, in part, upon the type of DNA damage that is being repaired (Gredilla et al. 2010). DSBs are repaired by recombination-based processes, which are rarely error-free, hence leading to sequence deletions of various lengths (Krishnan et al. 2008). Mitochondria contain a very efficient DNA repair pathway for simpler lesions such as alkylation or oxidation products caused by ROS, the base excision repair (BER) pathway (Seeberg et al. 1995). Moreover, mismatch and SSB repair have been described to take place in mitochondria (Gredilla et al. 2010, Marin-Garcia 2016). Despite recent studies on recombination repair in mitochondria (Ling et al. 2013, Sen et al. 2016), our knowledge of the actual mechanism of DNA recombination in mammalian mitochondria is still limited (Liu and Demple 2010, Chen 2013).
Although some reports suggest that cardiomyocytes can re-enter the cell cycle under certain circumstances (Leri et al. 2000, Beltrami et al. 2001, Walsh et al. 2010), the proliferative and hence, regenerative potential of adult mammalian cardiomyocytes is quite limited. Due mostly to their postmitotic nature, accumulation of mtDNA damage in cardiomyocytes is especially relevant, since it highly compromises their functionality. Similarly to other tissues, the main sites of ROS generation in mitochondria of cardiomyocytes are located within the electron transport chain. Under physiological conditions, a variable amount of oxygen is converted into superoxide anion (O2 ·−) due to the leaking of electrons mainly from complexes I and III (Barja 1999, St-Pierre et al. 2002). Superoxide dismutases convert O2 ·− into H2O2, which will be further neutralized into O2 and H2O. Moreover, H2O2 can be converted into the highly reactive hydroxyl radical (OH−) through Fenton and Haber-Weiss reactions in the presence of iron. Interestingly, mitochondrial iron content increases with aging in different tissues, including myocardium, which may exacerbate the generation of OH− and hence oxidative damage in later life (Xu et al. 2010, Mallikarjun et al. 2014). The continuous production of O2 ·− in mitochondria leads to increased levels of oxidative damage to mitochondrial proteins, lipids, and DNA in aged organisms. The frequency of mtDNA point mutations and deletions is approximately 3-fold higher in the heart of aged mice when compared to young animals (Dai and Rabinovitch 2009). Important support of mtDNA mutations as a critical factor inducing heart dysfunction and aging has been provided by animal models with increased mtDNA instability. An early mouse model where mitochondrial transcription factor A was disrupted specifically in the heart and muscle reproduces important pathophysiological features of mtDNA mutation disorders, including respiratory chain deficiency caused by impaired mtDNA expression and accumulation of morphologically abnormal mitochondria (Wang et al. 1999). Moreover, these alterations were associated with increased in vivo apoptosis (Wang et al. 2001). Interestingly, this tissue-specific knockout shows normal respiratory chain function in the heart at birth, but the development of dilated cardiomyopathy in the postnatal period (Wang et al. 1999). More recently, a new animal model has attracted attention, the PolG mouse (Trifunovic et al. 2004, Kujoth et al. 2005). These mice accumulate a high load of mtDNA point mutations and deletions in all tissues (Vermulst et al. 2008, Edgar et al. 2009) and are characterized by an early aging phenotype. Cardiac mitochondria from PolG mice also show elevated levels of protein carbonyls. As a result, heart mitochondria of PolG mice have abnormal ETC activity with depressed activity in different complexes (Trifunovic et al. 2004). It has also been proposed that the elevated accumulation of random point mutations in mtDNA may affect the proper assembly of supercomplexes, leading to reduced ATP production and bioenergetic impairment (Edgar et al. 2009). Moreover, these mice show increased apoptosis in cardiac tissue and premature age-related changes, cardiac hypertrophy, and reduced systolic and diastolic function (Dai et al. 2009, Dai et al. 2010). Interestingly, it has been reported that accumulation of protein carbonyls and mtDNA deletions in PolG mice is partially reduced to control levels after overexpression of catalase targeted at mitochondria, with cardiac function being partially rescued (Dai et al. 2009, Dai et al. 2010). Similarly, when PolG mice are subjected to endurance exercise, premature age-related changes, both in the skeletal muscle and heart, are prevented (Safdar et al. 2011). The effect of endurance exercise seems to be mediated by induction of mitochondrial biogenesis, prevention of mtDNA mutations, increased mitochondrial oxidative capacity and respiratory chain assembly and reduction in apoptosis (Safdar et al. 2011). Very recently, a new animal model has been generated showing accelerated accumulation of mtDNA deletions in the myocardium (Baris et al. 2015). Like PolG animals, these mice accumulate randomly distributed cardiomyocytes with compromised mitochondrial function, which seems to promote premature cardiac arrhythmia.
Involvement of mitochondrial dysfunction in aged heart
During aging, physiological processes decline progressively, decreasing homeostasis control and increasing morbidity. Moreover, the incidence of age-related diseases exponentially increases during aging. Although all tissues are affected during aging, those containing postmitotic cells like the brain and heart are considered to be especially affected (Miquel et al. 1980). Cardiac aging implies several changes in the physiology and biochemistry of the heart and associated vessels. Morphologically, the heart undergoes thickening of the left ventricle and hypertrophy of the left ventricle and interventricular septum. There is stiffening, scarring, and calcification of aortic valve leaflets and aortic sclerosis. Electrical activity on the myocardium is also affected in aged heart by mitral annular calcification and apoptotic reduction of the sinoatrial and atrioventricular nodes’ pacemaker cells along with deposition of collagen, adipose tissue, and amyloid (Karavidas et al. 2010, Maruyama 2012).
Cardiac hypertrophy
Cardiomyocytes can be damaged in different ways such as by ischemia, presence of toxic substances, microorganisms, etc. derived by an increased load on the heart. These pathological situations lead to inadequate contraction or loss of cardiomyocytes (e.g., infarction), where the compensatory response of the heart is manifested by the remodeling that cardiac hypertrophy undergoes (Pangonyte et al. 2008). During hypertrophy, protein synthesis intensifies, new sarcomeres are produced, cardiomyocytes become thicker and longer, the thickness of ventricular wall grows, and cardiac contraction increases. This response temporarily eliminates, or at least reduces, the hemodynamic overload of the heart (Weber et al. 1995, Swynghedauw 1999). Cardiac hypertrophy has traditionally been seen as an adaptive response. However, many epidemiological studies have demonstrated that LVH is associated with a significantly increased risk of diastolic dysfunction, HF, and malignant arrhythmias (Frey and Olson 2003a, Martin-Fernandez et al. 2009). It is known that pathological cardiac hypertrophy is associated with depletion of energy reserves manifested as maintained ATP levels and a reduction of the energy reserve compound, PCr (Liao et al. 1996, Tian et al. 1997). PCr/ATP ratio decreases and significant decreases of ATP are observed as compensated hypertrophy advances to overt HF, (Ingwall 2009). For instance, LVH in animal models of pressure overload is blunted when treated with antioxidants (Seddon et al. 2007). In cultured cardiomyocytes, hypertrophy induced by angiotensin II, endothelin 1, norepinephrine, TNF-α, or mechanical stress is associated with increased levels of oxidative stress and ROS-mediated activation of several intracellular signaling pathways, including mitogen activated protein kinases and nuclear factor κB (Dutta et al. 2012).
Heart failure
Cardiac hypertrophy has traditionally been seen as an adaptive response. However, many epidemiological studies have demonstrated that LVH is associated with a significantly increased risk of HF and malignant arrhythmias (Koren et al. 1991, Gutstein et al. 2001, Frey and Olson 2003b). HF is a growing public health problem, mainly because of the increased lifespan of the population and an elevated prevalence in the elderly. In developing countries, around 2 % of adults suffer from HF; the prevalence is found to be increased to approximately 6–10 % over the age of 65 (McMurray and Pfeffer 2005).
The mechanisms of HF are complex and multifactorial. HF manifests as a reduction in velocity of myocardial relaxation, as well as decreasing myocardial compliance (Ren and Bode 2000, Finck and Kelly 2007). It is well known that HF is associated with mitochondrial dysfunction, which is associated with increased oxidative stress, making mitochondria-targeted reactive oxygen species arousing an attractive therapeutic strategy. Mitochondrial dysfunction is not only involved in the pathogenesis of LVH but also in the transition from compensated LVH to HF (Abel and Doenst 2011). Decreases in energy production due to reductions in mitochondrial respiration, increased oxidative stress, and defective contractile and intracellular Ca2+ regulatory proteins contribute to selective cardiac dysfunction (Martin-Fernandez et al. 2009). In addition, alterations in Ca2+ influx and myofilament function contribute to the cardiomyopathic alterations. Mitochondria in endothelial cells are thought to play an important role in cellular signaling as sensors for local oxygen concentration and regulators of nitric oxide (NO) production thus, playing an important role in coronary disease (Davidson and Duchen 2007). In addition, increased oxidative stress directly impacts cardiomyocyte structure and function by activating signaling pathways involved in myocardial remodeling and failure (Ide et al. 1999). This evidence suggests a pathogenic link between enhanced ROS production, mitochondrial dysfunction, and the development of HF.
In contrast to the well-identified diastolic dysfunction at rest, systolic function as measured by ejection fraction is preserved with age. Heart failure with preserved ejection fraction (HFpEF) displays no cardiac dilation, yet is characterized by high filling pressures and lung congestion, dyspnea, and intolerance to effort (Conceicao et al. 2016). It has been observed to become more common with aging, and this number is expected to increase as projected by the increase in life expectancy and by population growth projections (Dhingra et al. 2014), particularly older women, where 90 % of new HF cases are HFpEF (Gottdiener et al. 2000). Originally considered to be predominantly caused by diastolic dysfunction, more recent insights indicate that HFpEF in the elderly is characterized by a broad range of cardiac and non-cardiac abnormalities and reduced reserve capacity in multiple organ systems. HFpEF is driven by inherent age-related changes, multiple, concomitant comorbidities and HFpEF itself, which is like a systemic disorder (Dhingra et al. 2014). In comparison with heart failure with reduced ejection fraction (HFrEF), the overall prognosis of HFpEF patients is similar to those with HFrEF, with a higher degree of repeat hospitalizations in the former group. The development of effective therapeutic strategies for HFpEF has stimulated the establishment of proper animal models. For instance, aortic banding and systemic hypertension animal models have been broadly used, since hypertension is a major contributor to HFpEF (Conceicao et al. 2016). The Dahl salt-sensitive rat is characterized by hypersensitivity to sodium intake and represents the most published HFpEF animal model. When feeding animals a high-salt diet (8 % NaCl) from the age of 7 weeks, Dahl/SS rats develop renal failure, fast-developing hypertension (>175 mmHg) and LVH, falling into HFpEF between 12 and 19 weeks of age (Doi et al. 2000). Moderate transverse-aortic constriction imposed at an early age triggers concentric LVH accompanied by compensated chamber performance, with marked diastolic filling abnormalities. These abnormalities become progressively more exaggerated at 12 and 18 weeks, thus representing a good model for studying HFpEF.
Diabetic cardiomyopathy
In obese people and individuals with diabetes mellitus, cardiac dysfunction, independent of macro- and microvascular disease, is considered a consequence of diabetic cardiomyopathy (Kolwicz et al. 2013). Diabetic cardiomyopathy is defined as the presence of left ventricular dysfunction beyond that which can be accounted for by arterial hypertension, coronary artery disease, or evidence of any other structural cardiac disease in individuals with diabetes (Westermeier et al. 2015).
Rates of diabetes increase with advancing age with a particular rise in type 2 diabetes. The global prevalence of diabetes among adults aged 60 years and older is 19 %—approximately 135 million people—and accounts for 35 % of all cases of diabetes in adults (L’Heveder and Nolan 2013). The number of health conditions increases with advancing age. Thus, in addition to diabetes, four out of five adults aged 65–74 years and older are diagnosed with at least one comorbid condition. Rates of coronary heart disease, stroke, congestive heart failure, hypertension, neuropathy, visual impairment, and arthritis are significantly higher among older adults with diabetes compared to similar age adults without diabetes (Kalyani et al. 2010).
We can dissociate between systemic and cardiac insulin resistance. Cardiac insulin resistance is defined as ablated insulin signaling or insulin-stimulated glucose uptake in the heart in the absence of systemic insulin resistance or known risk factors for coronary heart disease such as obesity, hyperglycemia, hyperinsulinemia, hypercholesterolemia, and hypertension (Dei Cas et al. 2015). Insulin resistance alone has severe adverse effects on cardiac dysfunction. In fact, the onset of hyperglycemia and diabetes is often preceded by several years of IR. Previous studies carried out in transgenic mice with cardiomyocyte-specific deletion of IR (CIRKO) or insulin receptor substrate (CIRSKO) have explored the causal association between insulin resistance and cardiac dysfunction. CIRKO and CIRSKO mice showed reduced insulin-stimulated glucose uptake and also impairment in cardiac function. In glucose transporter 4 (GLUT4) KO mice also showed cardiac dysfunction development implicating insulin resistance as a contributing factor in the development of cardiac dysfunction (McQueen et al. 2005, Sena et al. 2009, Domenighetti et al. 2010).
Insulin resistance in presence of diabetes is highly associated with HF. Approximately 24 % of HF patients overall and 40 % of hospitalized HF patients have diabetes mellitus and in the next few decades, it is expected to grow exponentially with the aging of population (Aroor et al. 2012). Insulin resistance not only induces cardiac negative effects but also increases the risk of type 2 diabetes in both aged men and women which predispose to coronary heart disease in the elderly (Kannel 2002, Wilson and Kannel 2002). Many mechanisms underlying insulin resistance-induced cardiac dysfunction with age have been proposed. For instance, the development of endothelial dysfunction in response to insulin and decreased telomere length of leukocytes (Demissie et al. 2006), which plays an important role in the development of age-related hypertension (Li et al. 2009), are highly accepted theories.
Since mitochondria are the major source of ATP for meeting the energy demands of the heart, mitochondrial dysfunction might be an underlying cause of metabolic disorders and insulin-resistance-associated heart disease (Boudina et al. 2009). Under normal physiological conditions, the heart uses energy from substrates (FA and carbohydrates) based on metabolic demand and availability (Stanley et al. 2005); however, in the setting of insulin resistance, the myocardium’s ability to use glucose as an energy source is reduced (Ashrafian et al. 2007, Opie and Knuuti 2009). Pathophysiology of diabetic cardiomyopathy becomes highly influenced by the change in substrate preference (Battiprolu et al. 2012). Mitochondria generate more than 90 % of the intracellular ATP consumed by the heart (Piquereau et al. 2013). Thus, it has been proposed that mitochondrial dysfunction could be a causative factor in metabolic disorders and insulin resistance-associated heart diseases (Chen and Knowlton 2011). An intracellular accumulation of toxic metabolic intermediates in diabetic cardiomyopathy has been shown. These intermediates, such as long-chain acyl-CoA and acylcarnitine, affect mitochondrial ATP/ADP ratio, leading to diminished mitochondrial metabolic function (Lopaschuk and Spafford 1989).
Concluding remarks
Cardiac diseases are associated with profound changes in cardiac metabolism. Metabolic remodeling in HF is characterized by decreased cardiac energy production, which may result from progressive impairments in substrate use and mitochondrial biogenesis and function. Alterations in mitochondrial biogenesis as well as mitochondrial content and function in cardiomyocytes contribute to provoking and aggravating a heterogeneous group of cardiac diseases. The maintenance of a healthy pool of mitochondria and the removal of damaged organelles are vital for the preservation of cardiomyocyte function and viability. Although we are, at present, far from understanding the processes that determine the aging of heart, and although the relevance of the mentioned factors differs between individuals, it is clear that a large number of different processes work together in the pacing of the aging process. To accomplish this challenging task, several critical issues need to be addressed. The mechanisms responsible for the loss of protection in the aged heart include alterations in gene/protein expression, signal transduction cascades, and mitochondrial function (e.g., ROS formation, respiration). It is mandatory to identify specific pathways or substrates of dysfunctional mitochondria, the derangements of which are primarily involved in heart senescence. Taken together, these findings stress the importance of mitochondrial ROS, mtDNA mutation accumulation and mitochondrial function in cardiomyocyte function and cardiac aging. Understanding fundamental mechanisms that dictate the pace of aging could lead to significant advances in both preventative and therapeutic treatments of cardiac diseases.
References
Abel ED, Doenst T (2011) Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy. Cardiovasc Res 90:234–242
Aroor AR, Mandavia CH, Sowers JR (2012) Insulin resistance and heart failure: molecular mechanisms. Heart Fail Clin 8:609–617
Ashrafian H, Frenneaux MP, Opie LH (2007) Metabolic mechanisms in heart failure. Circulation 116:434–448
Atkinson J (2008) Age-related medial elastocalcinosis in arteries: mechanisms, animal models, and physiological consequences. J Appl Physiol (1985) 105:1643–1651
Atkinson J, Kapralov AA, Yanamala N, Tyurina YY, Amoscato AA, Pearce L, Peterson J, Huang Z, Jiang J, Samhan-Arias AK, Maeda A, Feng W, Wasserloos K, Belikova NA, Tyurin VA, Wang H, Fletcher J, Wang Y, Vlasova II, Klein-Seetharaman J, Stoyanovsky DA, Bayir H, Pitt BR, Epperly MW, Greenberger JS, Kagan VE (2011) A mitochondria-targeted inhibitor of cytochrome c peroxidase mitigates radiation-induced death. Nat Commun 2:497
Baris OR, Ederer S, Neuhaus JF, von Kleist-Retzow JC, Wunderlich CM, Pal M, Wunderlich FT, Peeva V, Zsurka G, Kunz WS, Hickethier T, Bunck AC, Stockigt F, Schrickel JW, Wiesner RJ (2015) Mosaic deficiency in mitochondrial oxidative metabolism promotes cardiac arrhythmia during aging. Cell Metab 21:667–677
Barja G (1999) Mitochondrial oxygen radical generation and leak: sites of production in states 4 and 3, organ specificity, and relation to aging and longevity. J Bioenerg Biomembr 31:347–366
Barja G (2013) Updating the mitochondrial free radical theory of aging: an integrated view, key aspects, and confounding concepts. Antioxid Redox Signal 19:1420–1445
Barja G (2014) The mitochondrial free radical theory of aging. Prog Mol Biol Transl Sci 127:1–27
Battiprolu PK, Hojayev B, Jiang N, Wang ZV, Luo X, Iglewski M, Shelton JM, Gerard RD, Rothermel BA, Gillette TG, Lavandero S, Hill JA (2012) Metabolic stress-induced activation of FoxO1 triggers diabetic cardiomyopathy in mice. J Clin Invest 122:1109–1118
Beckman KB, Ames BN (1999) Endogenous oxidative damage of mtDNA. Mutat Res 424:51–58
Beer M, Seyfarth T, Sandstede J, Landschutz W, Lipke C, Kostler H, von Kienlin M, Harre K, Hahn D, Neubauer S (2002) Absolute concentrations of high-energy phosphate metabolites in normal, hypertrophied, and failing human myocardium measured noninvasively with P-31-SLOOP magnetic resonance spectroscopy. J Am Coll Cardiol 40:1267–1274
Beltrami AP, Urbanek K, Kajstura J, Yan SM, Finato N, Bussani R, Nadal-Ginard B, Silvestri F, Leri A, Beltrami CA, Anversa P (2001) Evidence that human cardiac myocytes divide after myocardial infarction. N Engl J Med 344:1750–1757
Boluyt MO, Converso K, Hwang HS, Mikkor A, Russell MW (2004) Echocardiographic assessment of age-associated changes in systolic and diastolic function of the female F344 rat heart. J Appl Physiol (1985) 96:822–828
Boudina S, Bugger H, Sena S, O’Neill BT, Zaha VG, Ilkun O, Wright JJ, Mazumder PK, Palfreyman E, Tidwell TJ, Theobald H, Khalimonchuk O, Wayment B, Sheng X, Rodnick KJ, Centini R, Chen D, Litwin SE, Weimer BE, Abel ED (2009) Contribution of impaired myocardial insulin signaling to mitochondrial dysfunction and oxidative stress in the heart. Circulation 119:1272–1283
Brand MD (2010) The sites and topology of mitochondrial superoxide production. Exp Gerontol 45:466–472
Bratic A, Larsson NG (2013) The role of mitochondria in aging. J Clin Invest 123:951–957
Bratic I, Trifunovic A (2010) Mitochondrial energy metabolism and ageing. Biochim Biophys Acta 1797:961–967
Campbell SG, Haynes P, Kelsey Snapp W, Nava KE, Campbell KS (2013) Altered ventricular torsion and transmural patterns of myocyte relaxation precede heart failure in aging F344 rats. Am J Physiol Heart Circ Physiol 305:H676–H686
Carnes CA, Geisbuhler TP, Reiser PJ (2004) Age-dependent changes in contraction and regional myocardial myosin heavy chain isoform expression in rats. J Appl Physiol (1985) 97:446–453
Conceicao G, Heinonen I, Lourenco AP, Duncker DJ, Falcao-Pires I (2016) Animal models of heart failure with preserved ejection fraction. Neth Heart J 24:275–286
Chen L, Knowlton AA (2011) Mitochondrial dynamics in heart failure. Congest Heart Fail 17:257–261
Chen XJ (2013) Mechanism of homologous recombination and implications for aging-related deletions in mitochondrial DNA. Microbiol Mol Biol Rev 77:476–496
Dai DF, Chen T, Wanagat J, Laflamme M, Marcinek DJ, Emond MJ, Ngo CP, Prolla TA, Rabinovitch PS (2010) Age-dependent cardiomyopathy in mitochondrial mutator mice is attenuated by overexpression of catalase targeted to mitochondria. Aging Cell 9:536–544
Dai DF, Hsieh EJ, Liu Y, Chen T, Beyer RP, Chin MT, MacCoss MJ, Rabinovitch PS (2012) Mitochondrial proteome remodelling in pressure overload-induced heart failure: the role of mitochondrial oxidative stress. Cardiovasc Res 93:79–88
Dai DF, Karunadharma PP, Chiao YA, Basisty N, Crispin D, Hsieh EJ, Chen T, Gu H, Djukovic D, Raftery D, Beyer RP, MacCoss MJ, Rabinovitch PS (2014) Altered proteome turnover and remodeling by short-term caloric restriction or rapamycin rejuvenate the aging heart. Aging Cell 13:529–539
Dai DF, Rabinovitch PS (2009) Cardiac aging in mice and humans: the role of mitochondrial oxidative stress. Trends in cardiovascular medicine 19:213–220
Dai DF, Santana LF, Vermulst M, Tomazela DM, Emond MJ, MacCoss MJ, Gollahon K, Martin GM, Loeb LA, Ladiges WC, Rabinovitch PS (2009) Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation 119:2789–2797
Davidson SM, Duchen MR (2007) Endothelial mitochondria: contributing to vascular function and disease. Circ Res 100:1128–1141
De Meyer GR, De Keulenaer GW, Martinet W (2010) Role of autophagy in heart failure associated with aging. Heart Fail Rev 15:423–430
de Souza RR (2002) Aging of myocardial collagen. Biogerontology 3:325–335
Dei Cas A, Khan SS, Butler J, Mentz RJ, Bonow RO, Avogaro A, Tschoepe D, Doehner W, Greene SJ, Senni M, Gheorghiade M, Fonarow GC (2015) Impact of diabetes on epidemiology, treatment, and outcomes of patients with heart failure. JACC Heart Fail 3:136–145
Demissie S, Levy D, Benjamin EJ, Cupples LA, Gardner JP, Herbert A, Kimura M, Larson MG, Meigs JB, Keaney JF, Aviv A (2006) Insulin resistance, oxidative stress, hypertension, and leukocyte telomere length in men from the Framingham Heart Study. Aging Cell 5:325–330
Dhingra A, Garg A, Kaur S, Chopra S, Batra JS, Pandey A, Chaanine AH, Agarwal SK (2014) Epidemiology of heart failure with preserved ejection fraction. Curr Heart Fail Rep 11:354–365
Dillin A, Hsu AL, Arantes-Oliveira N, Lehrer-Graiwer J, Hsin H, Fraser AG, Kamath RS, Ahringer J, Kenyon C (2002) Rates of behavior and aging specified by mitochondrial function during development. Science 298:2398–2401
Doenst T, Nguyen TD, Abel ED (2013) Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res 113:709–724
Doi R, Masuyama T, Yamamoto K, Doi Y, Mano T, Sakata Y, Ono K, Kuzuya T, Hirota S, Koyama T, Miwa T, Hori M (2000) Development of different phenotypes of hypertensive heart failure: systolic versus diastolic failure in Dahl salt-sensitive rats. J Hypertens 18:111–120
Domenighetti AA, Danes VR, Curl CL, Favaloro JM, Proietto J, Delbridge LM (2010) Targeted GLUT-4 deficiency in the heart induces cardiomyocyte hypertrophy and impaired contractility linked with Ca(2+) and proton flux dysregulation. J Mol Cell Cardiol 48:663–672
Drew B, Leeuwenburgh C (2004) Ageing and subcellular distribution of mitochondria: role of mitochondrial DNA deletions and energy production. Acta Physiol Scand 182:333–341
Dutta D, Calvani R, Bernabei R, Leeuwenburgh C, Marzetti E (2012) Contribution of impaired mitochondrial autophagy to cardiac aging mechanisms and therapeutic opportunities. Circ Res 110:1125–1138
Edgar D, Shabalina I, Camara Y, Wredenberg A, Calvaruso MA, Nijtmans L, Nedergaard J, Cannon B, Larsson NG, Trifunovic A (2009) Random point mutations with major effects on protein-coding genes are the driving force behind premature aging in mtDNA mutator mice. Cell Metab 10:131–138
Fang EF, Scheibye-Knudsen M, Chua KF, Mattson MP, Croteau DL, Bohr VA (2016) Nuclear DNA damage signalling to mitochondria in ageing. Nat Rev Mol Cell Biol 17:308–321
Finck BN, Kelly DP (2007) Peroxisome proliferator-activated receptor gamma coactivator-1 (PGC-1) regulatory cascade in cardiac physiology and disease. Circulation 115:2540–2548
Frey N, Olson EN (2003a) Cardiac hypertrophy: the good, the bad and the ugly. Annu Rev Physiol 65:45–79
Frey N, Olson EN (2003b) Cardiac hypertrophy: the good, the bad, and the ugly. Annu Rev Physiol 65:45–79
Fulop N, Mason MM, Dutta K, Wang P, Davidoff AJ, Marchase RB, Chatham JC (2007) Impact of type 2 diabetes and aging on cardiomyocyte function and O-linked N-acetylglucosamine levels in the heart. Am J Physiol Cell Physiol 292:C1370–C1378
Garinis GA, van der Horst GT, Vijg J, Hoeijmakers JH (2008) DNA damage and ageing: new-age ideas for an age-old problem. Nat Cell Biol 10:1241–1247
Gazoti Debessa CR, Mesiano Maifrino LB, Rodrigues de Souza R (2001) Age related changes of the collagen network of the human heart. Mech Ageing Dev 122:1049–1058
Gonzalez-Freire M, de Cabo R, Bernier M, Sollott SJ, Fabbri E, Navas P, Ferrucci L (2015) Reconsidering the role of mitochondria in aging. J Gerontol A Biol Sci Med Sci 70:1334–1342
Gottdiener JS, Arnold AM, Aurigemma GP, Polak JF, Tracy RP, Kitzman DW, Gardin JM, Rutledge JE, Boineau RC (2000) Predictors of congestive heart failure in the elderly: the Cardiovascular Health Study. J Am Coll Cardiol 35:1628–1637
Gredilla R, Barja G (2005) Minireview: the role of oxidative stress in relation to caloric restriction and longevity. Endocrinology 146:3713–3717
Gredilla R, Bohr VA, Stevnsner T (2010) Mitochondrial DNA repair and association with aging—an update. Exp Gerontol 45:478–488
Green DR, Galluzzi L, Kroemer G (2011) Mitochondria and the autophagy-inflammation-cell death axis in organismal aging. Science 333:1109–1112
Grollman AP, Moriya M (1993) Mutagenesis by 8-oxoguanine: an enemy within. Trends Genet 9:246–249
Gruber J, Schaffer S, Halliwell B (2008) The mitochondrial free radical theory of ageing—where do we stand? Front Biosci 13:6554–6579
Gutstein DE, Morley GE, Fishman GI (2001) Conditional gene targeting of connexin43: exploring the consequences of gap junction remodeling in the heart. Cell Commun Adhes 8:345–348
Harman D (1972) The biologic clock: the mitochondria? J Am Geriatr Soc 20:145–147
Heidenreich PA, Trogdon JG, Khavjou OA, Butler J, Dracup K, Ezekowitz MD, Finkelstein EA, Hong Y, Johnston SC, Khera A, Lloyd-Jones DM, Nelson SA, Nichol G, Orenstein D, Wilson PW, Woo YJ, American Heart Association Advocacy Coordinating C, Stroke C, Council on Cardiovascular R, Intervention, Council on Clinical C, Council on E, Prevention, Council on A, Thrombosis, Vascular B, Council on C, Critical C, Perioperative, Resuscitation, Council on Cardiovascular N, Council on the Kidney in Cardiovascular D, Council on Cardiovascular S, Anesthesia, Interdisciplinary Council on Quality of C, Outcomes R (2011) Forecasting the future of cardiovascular disease in the United States: a policy statement from the American Heart Association. Circulation 123:933–944
Horn MA, Graham HK, Richards MA, Clarke JD, Greensmith DJ, Briston SJ, Hall MC, Dibb KM, Trafford AW (2012) Age-related divergent remodeling of the cardiac extracellular matrix in heart failure: collagen accumulation in the young and loss in the aged. J Mol Cell Cardiol 53:82–90
Ide T, Tsutsui H, Kinugawa S, Utsumi H, Kang D, Hattori N, Uchida K, Arimura K, Egashira K, Takeshita A (1999) Mitochondrial electron transport complex I is a potential source of oxygen free radicals in the failing myocardium. Circ Res 85:357–363
Ingwall JS (2009) Energy metabolism in heart failure and remodelling. Cardiovasc Res 81:412–419
Jang YC, Perez VI, Song W, Lustgarten MS, Salmon AB, Mele J, Qi W, Liu Y, Liang H, Chaudhuri A, Ikeno Y, Epstein CJ, Van Remmen H, Richardson A (2009) Overexpression of Mn superoxide dismutase does not increase life span in mice. J Gerontol A Biol Sci Med Sci 64:1114–1125
Jeppesen DK, Bohr VA, Stevnsner T (2011) DNA repair deficiency in neurodegeneration. Prog Neurobiol 94:166–200
Judge S, Leeuwenburgh C (2007) Cardiac mitochondrial bioenergetics, oxidative stress, and aging. Am J Physiol Cell Physiol 292:C1983–C1992
Kalyani RR, Saudek CD, Brancati FL, Selvin E (2010) Association of diabetes, comorbidities, and A1C with functional disability in older adults: results from the National Health and Nutrition Examination Survey (NHANES), 1999-2006. Diabetes Care 33:1055–1060
Kannel WB (2002) Coronary heart disease risk factors in the elderly. Am J Geriatr Cardiol 11:101–107
Karavidas A, Lazaros G, Tsiachris D, Pyrgakis V (2010) Aging and the cardiovascular system. Hell J Cardiol 51:421–427
Kavli B, Otterlei M, Slupphaug G, Krokan HE (2007) Uracil in DNA—general mutagen, but normal intermediate in acquired immunity. DNA Repair (Amst) 6:505–516
Kim JA, Wei YZ, Sowers JR (2008) Role of mitochondrial dysfunction in insulin resistance. Circ Res 102:401–414
Kolwicz SC Jr, Purohit S, Tian R (2013) Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res 113:603–616
Koren MJ, Devereux RB, Casale PN, Savage DD, Laragh JH (1991) Relation of left ventricular mass and geometry to morbidity and mortality in uncomplicated essential hypertension. Ann Intern Med 114:345–352
Krishnan KJ, Reeve AK, Samuels DC, Chinnery PF, Blackwood JK, Taylor RW, Wanrooij S, Spelbrink JN, Lightowlers RN, Turnbull DM (2008) What causes mitochondrial DNA deletions in human cells? Nat Genet 40:275–279
Kujoth GC, Bradshaw PC, Haroon S, Prolla TA (2007) The role of mitochondrial DNA mutations in mammalian aging. PLoS Genet 3:e24
Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA (2005) Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309:481–484
Kujoth GC, Leeuwenburgh C, Prolla TA (2006) Mitochondrial DNA mutations and apoptosis in mammalian aging. Cancer Res 66:7386–7389
Kwak HB (2013) Aging, exercise, and extracellular matrix in the heart. Journal of exercise rehabilitation 9:338–347
L’Heveder R, Nolan T (2013) International diabetes federation. Diabetes Res Clin Pract 101:349–351
Lakatta EG (2003) Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part III: cellular and molecular clues to heart and arterial aging. Circulation 107:490–497
Lakatta EG, Levy D (2003) Arterial and cardiac aging: major shareholders in cardiovascular disease enterprises: part II: the aging heart in health: links to heart disease. Circulation 107:346–354
Leri A, Malhotra A, Liew CC, Kajstura J, Anversa P (2000) Telomerase activity in rat cardiac myocytes is age and gender dependent. J Mol Cell Cardiol 32:385–390
Li QX, Xiong ZY, BP H, Tian ZJ, Zhang HF, Gou WY, Wang HC, Gao F, Zhang QJ (2009) Aging-associated insulin resistance predisposes to hypertension and its reversal by exercise: the role of vascular vasorelaxation to insulin. Basic Res Cardiol 104:269–284
Liao RL, Nascimben L, Friedrich J, Gwathmey JK, Ingwall JS (1996) Decreased energy reserve in an animal model of dilated cardiomyopathy relationship to contractile performance. Circ Res 78:893–902
Ling F, Hori A, Yoshitani A, Niu R, Yoshida M, Shibata T (2013) Din7 and Mhr1 expression levels regulate double-strand-break-induced replication and recombination of mtDNA at ori5 in yeast. Nucleic Acids Res 41:5799–5816
Liu P, Demple B (2010) DNA repair in mammalian mitochondria: much more than we thought? Environ Mol Mutagen 51:417–426
Lopaschuk GD, Spafford M (1989) Response of isolated working hearts to fatty acids and carnitine palmitoyltransferase I inhibition during reduction of coronary flow in acutely and chronically diabetic rats. Circ Res 65:378–387
Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G (2013) The hallmarks of aging. Cell 153:1194–1217
Mallikarjun V, Sriram A, Scialo F, Sanz A (2014) The interplay between mitochondrial protein and iron homeostasis and its possible role in ageing. Exp Gerontol 56:123–134
Marin-Garcia J (2016) Mitochondrial DNA repair: a novel therapeutic target for heart failure. Heart Fail Rev.
Martin-Fernandez B, Miana M, De las Heras N, Ruiz-Hurtado G, Fernandez-Velasco M, Bas M, Ballesteros S, Lahera V, Cachofeiro V, Delgado C (2009) Cardiac L-type calcium current is increased in a model of hyperaldosteronism in the rat. Exp Physiol 94:675–683
Maruyama Y (2012) Aging and arterial-cardiac interactions in the elderly. Int J Cardiol 155:14–19
McMurray JJV, Pfeffer MA (2005) Heart failure. Lancet 365:1877–1889
McQueen AP, Zhang D, Hu P, Swenson L, Yang Y, Zaha VG, Hoffman JL, Yun UJ, Chakrabarti G, Wang Z, Albertine KH, Abel ED, Litwin SE (2005) Contractile dysfunction in hypertrophied hearts with deficient insulin receptor signaling: possible role of reduced capillary density. J Mol Cell Cardiol 39:882–892
Mendes AB, Ferro M, Rodrigues B, Souza MR, Araujo RC, Souza RR (2012) Quantification of left ventricular myocardial collagen system in children, young adults, and the elderly. Medicina 72:216–220
Minamino T, Komuro I (2008) Vascular aging: insights from studies on cellular senescence, stem cell aging, and progeroid syndromes. Nat Clin Pract Cardiovasc Med 5:637–648
Miquel J, Economos AC, Fleming J, JE J Jr (1980) Mitochondrial role in cell aging. Exp Gerontol 15:575–591
Modrego J, de las Heras N, Zamorano-Leon JJ, Mateos-Caceres PJ, Martin-Fernandez B, Valero-Munoz M, Lahera V, Lopez-Farre AJ (2013) Changes in cardiac energy metabolic pathways in overweighed rats fed a high-fat diet. Eur J Nutr 52:847–856
Nassimiha D, Aronow WS, Ahn C, Goldman ME (2001) Association of coronary risk factors with progression of valvular aortic stenosis in older persons. Am J Cardiol 87:1313–1314
Neubauer S (2007) Mechanisms of disease—the failing heart—an engine out of fuel. New Engl J Med 356:1140–1151
Opie LH, Knuuti J (2009) The adrenergic-fatty acid load in heart failure. J Am Coll Cardiol 54:1637–1646
Pangonyte D, Stalioraityte E, Ziuraitiene R, Kazlauskaite D, Palubinskiene J, Balnyte I (2008) Cardiomyocyte remodeling in ischemic heart disease. Med Lith 44:848–854
Piquereau J, Caffin F, Novotova M, Lemaire C, Veksler V, Garnier A, Ventura-Clapier R, Joubert F (2013) Mitochondrial dynamics in the adult cardiomyocytes: which roles for a highly specialized cell? Front Physiol 4:102
Rajawat YS, Hilioti Z, Bossis I (2009) Aging: central role for autophagy and the lysosomal degradative system. Ageing Res Rev 8:199–213
Ren J, Bode AM (2000) Altered cardiac excitation-contraction coupling in ventricular myocytes from spontaneously diabetic BB rats. Am J Physiol-Heart C 279:H238–H244
Ren J, Pulakat L, Whaley-Connell A, Sowers JR (2010) Mitochondrial biogenesis in the metabolic syndrome and cardiovascular disease. J Mol Med 88:993–1001
Ristow M, Zarse K (2010) How increased oxidative stress promotes longevity and metabolic health: the concept of mitochondrial hormesis (mitohormesis). Exp Gerontol 45:410–418
Rugarli E, Trifunovic A (2015) Is mitochondrial free radical theory of aging getting old? Biochim Biophys Acta 1847:1345–1346
Safdar A, Bourgeois JM, Ogborn DI, Little JP, Hettinga BP, Akhtar M, Thompson JE, Melov S, Mocellin NJ, Kujoth GC, Prolla TA, Tarnopolsky MA (2011) Endurance exercise rescues progeroid aging and induces systemic mitochondrial rejuvenation in mtDNA mutator mice. Proc Natl Acad Sci U S A 108:4135–4140
Sanz A (2016) Mitochondrial reactive oxygen species: do they extend or shorten animal lifespan? Biochim Biophys Acta.
Scialo F, Sriram A, Fernandez-Ayala D, Gubina N, Lohmus M, Nelson G, Logan A, Cooper HM, Navas P, Enriquez JA, Murphy MP, Sanz A (2016) Mitochondrial ROS produced via reverse electron transport extend animal lifespan. Cell Metab 23:725–734
Schulz TJ, Zarse K, Voigt A, Urban N, Birringer M, Ristow M (2007) Glucose restriction extends Caenorhabditis elegans life span by inducing mitochondrial respiration and increasing oxidative stress. Cell Metab 6:280–293
Seddon M, Looi YH, Shah AM (2007) Oxidative stress and redox signalling in cardiac hypertrophy and heart failure. Heart 93:903–907
Seeberg E, Eide L, Bjoras M (1995) The base excision repair pathway. Trends Biochem Sci 20:391–397
Sen D, Patel G, Patel SS (2016) Homologous DNA strand exchange activity of the human mitochondrial DNA helicase TWINKLE. Nucleic Acids Res.
Sena S, Hu P, Zhang D, Wang X, Wayment B, Olsen C, Avelar E, Abel ED, Litwin SE (2009) Impaired insulin signaling accelerates cardiac mitochondrial dysfunction after myocardial infarction. J Mol Cell Cardiol 46:910–918
Shadel GS, Horvath TL (2015) Mitochondrial ROS signaling in organismal homeostasis. Cell 163:560–569
Speakman JR, Blount JD, Bronikowski AM, Buffenstein R, Isaksson C, Kirkwood TB, Monaghan P, Ozanne SE, Beaulieu M, Briga M, Carr SK, Christensen LL, Cocheme HM, Cram DL, Dantzer B, Harper JM, Jurk D, King A, Noguera JC, Salin K, Sild E, Simons MJ, Smith S, Stier A, Tobler M, Vitikainen E, Peaker M, Selman C (2015) Oxidative stress and life histories: unresolved issues and current needs. Ecol Evol 5:5745–5757
St-Pierre J, Buckingham JA, Roebuck SJ, Brand MD (2002) Topology of superoxide production from different sites in the mitochondrial electron transport chain. J Biol Chem 277:44784–44790
Stanley WC, Recchia FA, Lopaschuk GD (2005) Myocardial substrate metabolism in the normal and failing heart. Physiol Rev 85:1093–1129
Swynghedauw B (1999) Molecular mechanisms of myocardial remodeling. Physiol Rev 79:215–262
Terman A, Kurz T, Navratil M, Arriaga EA, Brunk UT (2010) Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial-lysosomal axis theory of aging. Antioxid Redox Signal 12:503–535
Thomas DP, Cotter TA, Li X, McCormick RJ, Gosselin LE (2001) Exercise training attenuates aging-associated increases in collagen and collagen crosslinking of the left but not the right ventricle in the rat. Eur J Appl Physiol 85:164–169
Tian R, Nascimben L, Ingwall JS, Lorell BH (1997) Failure to maintain a low ADP concentration impairs diastolic function in hypertrophied rat hearts. Circulation 96:1313–1319
Tocchi A, Quarles EK, Basisty N, Gitari L, Rabinovitch PS (2015) Mitochondrial dysfunction in cardiac aging. Biochim Biophys Acta 1847:1424–1433
Tower J (2015) Programmed cell death in aging. Ageing research reviews.
Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly YM, Gidlof S, Oldfors A, Wibom R, Tornell J, Jacobs HT, Larsson NG (2004) Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429:417–423
Tyler D (1991) The mitochondrion in health and disease. Wiley-VCH, Weinheim
Tyurina YY, Tungekar MA, Jung MY, Tyurin VA, Greenberger JS, Stoyanovsky DA, Kagan VE (2012) Mitochondria targeting of non-peroxidizable triphenylphosphonium conjugated oleic acid protects mouse embryonic cells against apoptosis: role of cardiolipin remodeling. FEBS Lett 586:235–241
Upadhya B, Taffet GE, Cheng CP, Kitzman DW (2015) Heart failure with preserved ejection fraction in the elderly: scope of the problem. J Mol Cell Cardiol 83:73–87
Vermulst M, Wanagat J, Kujoth GC, Bielas JH, Rabinovitch PS, Prolla TA, Loeb LA (2008) DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat Genet 40:392–394
Walsh S, Ponten A, Fleischmann BK, Jovinge S (2010) Cardiomyocyte cell cycle control and growth estimation in vivo—an analysis based on cardiomyocyte nuclei. Cardiovasc Res 86:365–373
Wang J, Silva JP, Gustafsson CM, Rustin P, Larsson NG (2001) Increased in vivo apoptosis in cells lacking mitochondrial DNA gene expression. Proc Natl Acad Sci U S A 98:4038–4043
Wang J, Wilhelmsson H, Graff C, Li H, Oldfors A, Rustin P, Bruning JC, Kahn CR, Clayton DA, Barsh GS, Thoren P, Larsson NG (1999) Dilated cardiomyopathy and atrioventricular conduction blocks induced by heart-specific inactivation of mitochondrial DNA gene expression. Nat Genet 21:133–137
Weber KT, Sun Y, Katwa LC, Cleutjens JPM, Zhou GP (1995) Connective-tissue and repair in the heart—potential regulatory mechanisms. Ann N Y Acad Sci 752:286–299
Westermeier F, Navarro-Marquez M, Lopez-Crisosto C, Bravo-Sagua R, Quiroga C, Bustamante M, Verdejo HE, Zalaquett R, Ibacache M, Parra V, Castro PF, Rothermel BA, Hill JA, Lavandero S (2015) Defective insulin signaling and mitochondrial dynamics in diabetic cardiomyopathy. Biochim Biophys Acta 1853:1113–1118
Wilson PW, Kannel WB (2002) Obesity, diabetes, and risk of cardiovascular disease in the elderly. Am J Geriatr Cardiol 11:119–123 125
World Health Statistics (2014) Global Health Observatory data. World Health Organization (WHO). http://www.who.int/gho/publications/world_health_statistics/2014/en/ Accessed 10 Dec 2015.
Xu J, Marzetti E, Seo AY, Kim JS, Prolla TA, Leeuwenburgh C (2010) The emerging role of iron dyshomeostasis in the mitochondrial decay of aging. Mech Ageing Dev 131:487–493
Zhang Y, Ikeno Y, Qi W, Chaudhuri A, Li Y, Bokov A, Thorpe SR, Baynes JW, Epstein C, Richardson A, Van Remmen H (2009) Mice deficient in both Mn superoxide dismutase and glutathione peroxidase-1 have increased oxidative damage and a greater incidence of pathology but no reduction in longevity. J Gerontol A Biol Sci Med Sci 64:1212–1220
Acknowledgments
Authors’ contributions
Both authors contributed equally to this paper.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Source of funding
Grant from the Complutense University/Community of Madrid to RG (CCG10-UCM/SAL 4798).
About this article

Cite this article
Martín-Fernández, B., Gredilla, R. Mitochondria and oxidative stress in heart aging. AGE 38, 225–238 (2016). https://doi.org/10.1007/s11357-016-9933-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11357-016-9933-y