
Abstract
Neurodegenerative disorders are typically sporadic in nature in addition to usually influenced through an extensive range of environmental factors, lifestyle, and genetic elements. Latest observations have hypothesized that exposure of environmental factors may increase the prospective risk of Alzheimer’s diseases (AD). However, the role of environmental factors as a possible dangerous issue has extended importance concerned in AD pathology, although actual etiology of the disorder is still not yet clear. Thus, the aim of this review is to highlight the possible correlation between environmental factors and AD, based on the present literature view. Environmental risk factors might play an important role in decelerating or accelerating AD progression. Among well-known environmental risk factors, prolonged exposure to several heavy metals, for example, aluminum, arsenic, cadmium, lead, and mercury; particulate air, and some pesticides as well as metal-containing nanoparticles have been participated to cause AD. These heavy metals have the capacity to enhance amyloid β (Aβ) peptide along with tau phosphorylation, initiating amyloid/senile plaques, as well as neurofibrillary tangle formation; therefore, neuronal cell death has been observed. Furthermore, particulate air, pesticides, and heavy metal exposure have been recommended to lead AD susceptibility and phenotypic diversity though epigenetic mechanisms. Therefore, this review deliberates recent findings detailing the mechanisms for a better understanding the relationship between AD and environmental risk factors along with their mechanisms of action on the brain functions.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Alzheimer’s disease (AD) is an extremely progressive as well as fatal neurodegenerative disorder related to aging (Sorgdrager et al. 2019). Clinical and pathological appearance of AD comprises memory impairment and a slow struggle while performing normal regular activities (Castro-Chavira et al. 2015). However, a minor proportion of cases characterized as early-onset AD (EOAD) involves in disease manifestation earlier than age of 60 years (Wingo et al. 2019). EOAD cases are recognized to extremely penetrant genetic mutations of presenilin 1 (PSEN1) in chromosome 14, amyloid precursor protein (APP) in chromosome 21, and presenilin 2 (PSEN2) in chromosome 1 (Bertram 2009; Dai et al. 2018). These mutations lead to the addition of Aβ plaques observed during these mutations which is the pathological symbol of AD progression. Accumulated investigations have recognized numerous nongenetic threat elements known as late-onset AD (LOAD), comprising smoking (Li et al. 2011; Rusanen et al. 2011), hypercholesterolemia (Li et al. 2011), obesity (Anstey et al. 2011), diabetes (Li et al. 2011), hypertension (Li et al. 2011; Sharp et al. 2011), head trauma (Plassman et al. 2000), stroke (Savva et al. 2010), and depression (Mourao et al. 2016). However, there are many protecting factors that decrease the possibility of rising LOAD or interruption of beginning of LOAD which comprise social engagement (Boal et al. 2018), physical activity (Park et al. 2019; Schlosser Covell et al. 2015), mental activity (Fratiglioni and Wang 2007), education (Fratiglioni and Wang 2007; Lindsay et al. 2002), non-steroidal anti-inflammatory drug (NSAID) use (Wichmann et al. 2016), coffee consumption (Larsson and Orsini 2018), moderate alcohol drinking (Wong et al. 2016; Xu et al. 2017), and past vaccinations (Verreault et al. 2001). Most important environmental exposures related through LOAD contain pesticides (Yan et al. 2016), electromagnetic field (Jalilian et al. 2018), solvents (Huang et al. 2018), particulate matter in air pollution (Kilian and Kitazawa 2018), lead (Bakulski et al. 2012), iron (Huat et al. 2019), mercury (Bjorklund et al. 2019) [41], zinc (Chin-Chan et al. 2015), copper (Yao et al. 2018), and aluminum (Liang 2018).
Currently, it is well-studied that these environmental risk factors may play a crucial role in increasing or slowing neurodegenerative disease onset as well as progression. It has been well known that neurodegenerative disorder etiology is multifactorial, and moreover, it is mentioned that prospective external elements comprising chemical exposures as well as lifestyle are connected through the risk assessment of these diseases (Gomez-Gomez and Zapico 2019). Although the enormous cases of AD population are detected in aging people, so far the introduction to risk elements arisen years or decades earlier to diagnosis (Fratiglioni et al. 2004). The valuation of long-lasting exposures is problematic to implement in retrospective investigations to assist them through the improvement of the disease. Therefore, additional investigation for superior description of exposure as well as identification of initial particular biomarkers for the identification and diagnosis of these diseases is urgently needed. To consider environmental risk factors that actually cause harm the nervous system over the mechanisms of epigenetic regulation, following in neurodegenerative disorders in future life. In this review, we concisely describe the effects of numerous environmental factors such as heavy metal, pesticides, particulate air, and nanoparticles on important neurodegenerative disorder Alzheimer’s disease.
Potential role of environmental risk factors in Alzheimer’s disease
AD is well known as a complex neurodegenerative disease with augmented quantities of intracellular neurofibrillary tangles as well as extracellular neuritic plaques, which is connected to genetic variables and lifestyle and is considered as a progressive and irreversible disease in elderly (Huber et al. 2018). EOAD and LOAD are two known forms of AD (Baillon et al. 2019). EOAD is associated with mutations in particular genes of presenilin (PSEN) and amyloid precursor protein (APP), where both are linked to synthesis of amyloid-beta (Aβ) (Mendez 2017). The EOAD starts before 65 years of age, which is 5% of all. The LOAD is the well-known recognized of AD average 95% in the entire cases and is caused by some genetic risk factors such as polymorphisms in apolipoprotein E (ApoE), ApoE neuronal receptor (SORL1), as well as glycogen synthase kinase 3 beta (GSK3β). It has been found that numerous environmental risk as well as genetic factors are responsible in the pathogenesis of LOAD; general damage in clearance of Aβ is perhaps a main provider to AD development (Mawuenyega et al. 2010). However, genetically, ε4 allele of APOE gene is the robust risk element for LOAD pathogenesis (Bu 2009; Corder et al. 1993).
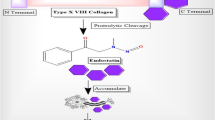
Environmental risk factors have been considered as a key causal factory of the progression and onset of AD. There are two hypotheses for AD development such as (a) increase production of the Aβ leading to formation of neurofibrillary tangles (NFTs) and (b) hyperphosphorylation of tau protein which promotes deposition as NFTs (Mezzaroba et al. 2019; Uddin et al. 2018). It is established that Aβ deposition promote memory loss and AD (Rahman and Rhim 2017; Zenaro et al. 2017). During aging, Aβ is formed by the proteolytic cleavage of APP by the pathway of amyloidogenic. APP is not only produced by β- as well as γ-secretase but also follow the pathway of non-amyloidogenic mechanism (Takahashi et al. 2017; Wirths et al. 2001). The augmented levels of brain Aβ in LOAD patients could be induced by APP expression, activation of amyloidogenic signaling, and inhibition of the non-amyloidogenic signaling. The increase of beta-site APP cleaving enzyme 1 (BACE1) mediates high levels of Aβ in brain (Fig. 1) (Coimbra et al. 2018; Uddin et al. 2019). Conversely, the decline in a desintegrin in addition to metalloproteinase domain-containing protein 10 (ADAM10) activity could promote increasing production of Aβ (Ferrari et al. 2014). Additionally, increased production of Aβ might be mediated by mutations in PSEN 1 or 2 (Piaceri et al. 2013).

The non-amyloidogenic or non-amyloid pathway cleavages APP via α-secretase to produce two fragments C83, an 83 amino acid intracellular C-terminal fragment, and extracellular sAPPα, soluble amyloid precursor protein α. C83 fragment cleave through γ-secretase to yield a P3 peptide short fragment as well as CTF, C terminal fragment. P3 peptide is irrelevant to pathology. The amyloidogenic or amyloid pathway accumulates neurotoxic Aβ. β-Secretase releases extracellular sAPPβ, large N-terminal soluble amyloid precursor protein β and C99, C terminal intracellular fragment. Following cleavage of C99 fragment through γ-secretase produces the Aβ peptide
The aggregation of the microtubule (MT)-associated protein tau could cause neurofibrillary lesions leading to AD. Tau phosphorylation causes MT stabilization, and it has elevated amount of serine as well as threonine residues; thus, it is considered as a substrate of numerous kinases (Dansokho and Heneka 2018). The unusual deposition of tau leads to lesions involved in AD pathogenesis (Jouanne et al. 2017). Tau is hyperphosphorylated which is prominent to aggregation, depolymerization of MTs, and axonal transport disruption under pathological conditions (Jouanne et al. 2017). It is anticipated that repeat domains (RDs) of the MT-binding domain (MBD) are obligatory for aggregation, and for the development of tau filament (Okuda et al. 2015).
Maintenances of AD homeostasis by environmental factors
As the environmental factors including high-fat diet, biogenic metals, heavy metals, and pesticides interrupt Aβ homeostasis pathways, they could trigger AD development. Intake of antioxidants and regular exercise can avert AD progression (Feng and Wang 2012). Evidence shown that numerous single antioxidant such as β-carotene, vitamin C, and vitamin E have been experienced in diverse AD model treatment (Li et al. 2012a). Various environmental stimuli are regarded as oxidative agents. Oxidative stress, high polyunsaturated fatty acids, low antioxidants, and higher enzymatic activities are harmful for brain (Lobo et al. 2010). The environmental factors which stimulate Aβ plaque accumulation and tau hyperphosphorylation causes AD progressions which are presented in Figs. 2 and 3. In culture cells, treatment of Aβ mediates H2O2-induced neurotoxicity, while occurrence of antioxidants inhibits the toxicity (Qi et al. 2018). Various factors produce reactive oxygen species (ROS) while the mechanism responsible for free radicals production by Aβ in AD is unclear. High concentration of Fe3+ in NFTs and Aβ-aggregates increase levels of H2O2, and advanced glycation end products (AGE) in neurodegeneration (Smith et al. 1997). Further, activated microglia is a foundation of NO and O2 in senile plaques (Denis 2013), which forms the peroxinitrite radical (ONOO−) (Smith et al. 1997). Inflammatory stimuli play critical role in pathogenesis of AD (Alam et al. 2016). Astrocyte and microglia are the primary cells concern in the inflammatory process in brain. It is known that Aβ chemotaxis of microglia and amyloid fibril phagocytosis enhance the pro-inflammatory cytokines and ROS, leading to neuronal loss (Wang et al. 2015). Astrocytes degrade Aβ plaques, and thus, it is postulated that astrocytes and microglia activation is a result of aggregation of Aβ (Son et al. 2015). Non-steroidal anti-inflammatory drugs (NSAIDs) decrease the levels of Aβ which support the involvement of inflammation in AD (Miguel-Alvarez et al. 2015).

AD development through diverse mechanisms related with environmental factors. Environmental factors such as several metals (Al, As, Pb, Cd, and Hg), nanoparticles (NPs), pesticides, and diet fat has the possibilities to effect on late-onset Alzheimer’s disease (LOAD). Due to the multiple cellular mechanisms stimulates by these factor ultimately generate amyloid plaque that ends in Aβ senile plaque formation

Environmental factors influence tau hyperphosphorylation in AD. Tau protein become hyperphosphorylated via the stimulation of several factors such as Pb, Hg, Al, Cd, NPs, As, and pesticides. Detailed mechanisms are explained in the text
Exposure of metals in AD epidemiology
Several environmental factors comprising heavy metals, nanoparticles, and pesticides are responsible to stimulate AD and their effects are summarized in Fig. 4. Here, we also describe each individual factor that has particular effects on AD pathology.

Several environmental factors and their pathophysiological effects on AD. Environmental stimuli can distress several additional effects on AD. These might be involved increasing ROS, APP, and Aβ production, as well as impairments of spatial memory and cognition function in AD pathology
Exposure to heavy metals on AD progression
Heavy metal poisoning is the addition of heavy metals such as lead, mercury, copper zinc, cadmium, iron, chromium, manganese, and arsenic. Nonetheless, these metals store in the body in adequate concentrations to cause poisoning effects. Heavy metal poisoning may happen because of air or water pollution, improperly coated food containers, industrial exposure, medicines, foods, and ingestion of lead-based paints. The most important effects of heavy metal exposure on AD progressions are summarized in the following.
Exposure of lead
Lead (Pb) is a neurotoxic metal, but its involvement or direct link with AD development is not known. Pb could exert detrimental effects on intelligence, cognitive functions, speed processing, memory, and motor functions (Zhang et al. 2016). Studies on the level of bone Pb suggest that earlier Pb exposure can deteriorate post cognitive performance (Dorsey et al. 2006). Recently, a study found that there is no involvement in the levels of serum Pb in AD pathology (Ventriglia et al. 2015). The involvement of Pb in AD determined in rats at 1–20 days age examined by drinking water of 200 ppm Pb. After the neonatal exposure of Pb, there is an augment in APP mRNA levels in late life, but not expose as adults’ rats (Basha et al. 2005). A young age Macaca fascicularis, non-human primates, has been exposed to Pb (1.5 mg/kg/day) shows an enhance quantity of amyloid plaque formation at old age. APP and BACE1 might be associated with the increased Aβ levels (Wu et al. 2008). These effects are practical when Pb (5–100 μM/48 h) exposed to differentiated SH-SY5Y cells (Bihaqi and Zawia 2012). Differentiated SH-SY5Y cells enhance Aβ secretion and APP expression, decrease mRNA and protein levels of neprilysin or neutral endopeptidase (NEP), a Aβ degrading enzyme, signifying that both the synthesis and degradation of Aβ are modulated by Pb (Fig. 2) (Reuben 2018). It is shown that there is no significant alteration by 50 μM Pb in NEP expression but an augmentation in APP levels in differentiated SH-SY5Y cells (Chin-Chan et al. 2015). In addition, Pb enhances Aβ through decreasing clearance of Aβ in the brain (Bihaqi 2019). Moreover, it has been found that 27 mg/kg, i.p. of Pb exposure increased the levels of Aβ in the cortex as well as hippocampus of acute APP transgenic mice (V717F) (Gu et al. 2011). Pb also can interrupt the brain Aβ export leading to its accumulation and formation of plaques (Behl et al. 2010).
Exposure of mercury
Mercury (Hg) is another heavy metal which is known to cause neurotoxicity. Hg has facilitated deterioration of the brain development and promotion of cognitive and motor dysfunction (Johansson et al. 2007). Hg is involved with alterations of memory loss and cognitive function in adults (Chang et al. 2008). A previous investigation recommended a correlation between Hg exposure and prevalence of AD. Hg is described to enhance Aβ levels in vitro as well as in vivo, and recommended underlying mechanisms are decreasing degradation in brain clearance of the peptide (Olivieri et al. 2000). Exposure of Hg secreted Aβ-42 and Aβ-40 in neuroblastoma cells along with increased level of ROS (Olivieri et al. 2000). Non-cytotoxic concentrations of MeHg enhanced APP escorted by ROS levels and activation of glia (Monnet-Tschudi et al. 2006). However, 10–1000 nM Hg exposure rise APP expression and decrease in Aβ degradation by NEP in rat pheochromocytoma (PC12) cells (Fig. 2) (Song and Choi 2013). Interestingly, exposure of Hg (10 and 20 μM) increased Aβ-42 expression in SH-SY5Y cells while APP expression is unaffected and activity of NEP (Aβ-degrading enzyme) has been reduced (Chin-Chan et al. 2015). On the other hand, Hg has no significant effects on Aβ aggregation while Zn, Fe, and Cu has the uppermost potential (Bangen et al. 2015). In vivo treatment of 20–2000 μg/kg/day/4 weeks of MeHg increases Aβ-42 in male rat hippocampus, while there is no effect on APP and NEP protein levels (Kim et al. 2014; Song and Choi 2013). In the hippocampus, a suppressed low-density lipoprotein receptor-related protein 1 (LRP1) accomplishes endocytic clearance of Aβ peptide, and receptor expression in the brain is positively connected with increased Aβ and decreased cerebrospinal fluid (CSF) levels which indicate a bargained peptide clearance in the brain (Kim et al. 2014).
Exposure of arsenic
Arsenic (As) is another toxic metal responsible for neuronal toxicity associated with negative effect on the development of brain and cognitive function (Tyler and Allan 2014). Role of As exposure on the development of AD has been poorly studied. In mice, treatment with As (20 mg/L drinking water) increased the loss of spatial memory significantly (Ramos-Chavez et al. 2015). A conceivable mechanism for the cognitive as well as memory loss mediated by As (3–15 mg/L in water) exposure might be the dysregulation of the amyloid pathway. Treatment of inorganic and organic form of 5–10 mM/12–24 h As increases the expression of APP and sAPPβ in cholinergic SN56.B5.G4 cells (Zarazua et al. 2011). Overexpression of a mutant form of APP in Tg2576 mice also shows the similar effects in neurons. It is proposed that increasing APP expression is important in dimethyl arsenic acid (DMA)-induced effects (Zarazua et al. 2011). The underlying entire mechanism through which As effects increasing production of Aβ has not yet been investigated, while As exposure is linked with inflammation and oxidative stress in brain, which is similar with report on AD (Gong and O'Bryant 2010).
Exposure of cadmium
The neurotoxicity has been reported to induce by cadmium (Cd), a toxic heavy metal (Zong et al. 2018). Evidences suggest that 2.5 mg/kg/4 days drinking water of Cd exposure increase production of Aβ. In hippocampus and cerebral cortex of APP/PSEN1 AD mice, treatment of Cd increases Aβ-42 production as well as enlarges size and senile plaque formation (Li et al. 2012b). These things have been recognized for a disintegrin and metalloproteinase domain-containing protein 10 (ADAM10) expression, NEP, and sAPPα proteins, signifying that non-amyloidogenic pathway and Aβ degradation are regulated by Cd contact (Li et al. 2012b). The combination of As, Pb, and Cd treatments in male rats shows that metals increase the production of Aβ in hippocampus and cortex, and these were mediated by APP expression and APP-processing enzymes including BACE1 and PSEN (Karri et al. 2016). Although Pb is the most powerful metal to stimulate Aβ, followed by As, and Cd has the minimum outcome, all of them increase the production of APP (Karri et al. 2016). Fascinatingly, all of them show a synergic effect because of having As, the introduction to these metals significantly increases PSEN1, BACE1 Aβ, and APP, suggesting an increase processing of amyloidogenic pathway (Karri et al. 2016). It is found that exposure to As, Pb, and Cd mixture increases levels of malondialdehyde (MDA). It is connected with decrease activity of enzymatic antioxidant, and the initiation of IL-1β and IL-1α in the hippocampus and frontal cortex of rats (Karri et al. 2016). It is concluded that the APP expression was mediated by increased production of ROS-induced IL-1. These results were supported by the fact that APP mRNA has a responsive element for IL-1 in the 5’UTR region (Chin-Chan et al. 2015; Karri et al. 2016).
Exposure of aluminum
Recently, aluminum (Al) is considered as a pollutant component concerned within the etiology of the progressive damage of structure and function of neurons that can cause neuronal cell death and may lead to the elderly disorders like AD; but, there is no reliable proof yet. Al pollution gave first proof of potential neurotoxicity when people exposed to Al during this region is observed to have brain pathological characteristics usually found in AD patients (Colomina and Peris-Sampedro 2017), further progressing to their brain functions (Yang et al. 2019). Shen et al. state a peripheral positive correlation of soil Al levels and the death due to AD in China (Zhang 2018; Zhang et al. 2010), whereas others report no correlation. Investigational evidence seems to be further consistent. It has been stated that continuous oral Al administration from 6 months old to rest of their lives in rats raises APP in cortical and hippocampal tissues, and thereby increases the production of Aβ (Walton and Wang 2009). However, rat cortical neurons treated with Al (50 μM/48 days) results in the buildup of Aβ; further, Al-induced structural alterations of Aβ then increase its collection by creating fibrillary accumulation on the external surface of in vitro neuronal culture. Desferroxamine, a chelator of Al, is able to dissolve the aggregated Aβ (Alghamdi 2018; Hu et al. 2019). It is also found that increased production of Aβ in the hippocampus and cortex (AD animal model), impairment of memory is successfully produced by the co-treatment of Al with D-galactose which contribute to an amplified BACE1 expression and a reduced NEP (Luo et al. 2009). Al (2 mg/kg in diet/9 months) reduces the degradation of Aβ by reducing cathepsin B activity, suggesting the probable link between the amyloidogenic pathway activation and a decrease of the Aβ catabolism (Fig. 2) (Sakamoto et al. 2006). Furthermore, a reduction of LRP1 expression is conjointly determined in the mice co-treated with D-galactose and Al, representing an attainable drop in Aβ clearance (Luo et al. 2009). Diet (2 mg/kg/9 months) of Al-fed transgenic mice (Tg2576) has found to increase Aβ production and proteins related to its anabolism. Therefore, formation of amyloid plaques has reduced by the action of vitamin E (a potent antioxidant), recommending the involvement of oxidative stress induced by Al (Pratico et al. 2002).
Exposure of pesticides
Long-lasting chemical contact and the frequency of dementias as well as AD may be closely connected, but to confirm, we need more comprehensive studies. Based on epidemiological studies, role of pesticides in modifications of cerebral functions as well as AD development has been supported; however, the mechanisms are poorly understood. OCl pesticides like DDE, in addition, its source DDT (1 μM/48 h) treatment stimulate the production of APP on in vitro differentiated SH-SY5Y cells (Richardson et al. 2014). DDT has been reported to amplify Aβ production by elevating BACE1 and APP, further via decreasing the degradation and clearance of Aβ by dropping the activity of Aβ-degrading enzyme, ATP-binding cassette transporter A1, and IDE in H4-AβPPswe (human neuroglioma) cells (Fig. 2) (Li et al. 2015). Six-month acute subcutaneous administration of CPF (50 mg/Kg) (Chlorpyrifos), an OP pesticide linked with oxidative stress, neuronal impairment, and cognitive impairment have significantly amplified Aβ production within the cortex and hippocampus, further enhanced memory damage and lowered motor activity in Tg2576 mice (Fig. 4) (Salazar et al. 2011). But, another study has been reported that 25 mg/kg of CPF treatment in Tg2576 mice shows no change in Aβ production or memory acquisition (Peris-Sampedro et al. 2014). Therefore, further studies are required to clarify the mechanisms of action by which OCl, OP, and different pesticides are coupled to AD pathogenesis.
Paraquat (PQ), a commonly applied chemical herbicide recommended to be linked in AD pathogenesis. In 3 weeks, 10 mg/kg/twice a week of PQ treated wild-type and APP transgenic (Tg2576) mice shows elevated levels of Aβ in transgenic mice that is related to mitochondrial oxidative impairment in neural structure, resulting in diminishing of memory and learning (Li et al. 2017). Remarkably, peroxiredoxin 3 (a potent mitochondrial antioxidant defense enzyme) overexpression shows better cognitive functions and lower Aβ production in PQ-treated APP transgenic mice showing the potency of pro-oxidant xenobiotics like PQ in the development of AD (Souza et al. 2019).
Exposure of nanoparticles
With the increase of NP synthesis for various applications, such as drug delivery tactics for the treatment of AD, it is important to study the possible poisonous effects on proteins associated with the development of AD. Epidemiological studies required to carry out to link between NP exposure and AD development. However, several experimental pieces of evidence demonstrate the possibility of brain damage by NPs. Mice is treated with TiO2-NPs (nasal administration of 2.5–10 mg/kg/90 days) initiated hippocampus neuronal death, oxidative stress, and gliosis (Fig. 4) (Mushtaq et al. 2015). Microarray study reveals a reduced gene expression related to memory and cognition (Ze et al. 2014). Likewise, rats treated with i.p. 0.5 mg/kg/day for 14 days of CuO-NPs have exhibited poorest spatial cognition; in addition, a decrease in electrophysiological endpoints, for example, long-term potentiation, that coordinated with amplified lipid peroxidation product such as 4-hydroxinonenal-HNE, MDA production, and ROS, then decrease antioxidants enzyme levels (An et al. 2012). Brain alterations like the decline in cognitive, motor and sensory functions are reported based on studies on Al NPs, Ag NPs, and Cu NPs administered at several dosages and different methods in mice and rats (Sharma et al. 2009; Sharma and Sharma 2012). Though, another recent study mentioned that treatment of adult mice with NPs Ag did not cause memory loss (Liu et al. 2013). However, silica NP (SiNPs, 10 μg/mL for 24 h) exposure to human SK-N-SH and mouse Neuro-2a neuroblastoma cells in in vitro conditions have reported to raise the intracellular content of Aβ, with amplified APP and reduction of NEP protein levels. Amplified ROS production by SiNPs suggests that these effects may be facilitated by the production of intracellular ROS (Yang et al. 2014). Similarly, Neuro-2a cells treated with 12.5 μg/mL for 24 h silver NPs has been reported to show the Aβ deposition with an amplified APP expression, but reduces LPR1 (or LDLR) and NEP levels; together, the amyloidogenic pathway alteration by AgNPs can induce AD (Huang et al. 2015). Many NPs and its AD pathological effects are summarized in Fig. 5.

Numerous nanoparticles (NPs) and their effects on AD pathogenesis. NPs may distress numerous pathological effects on AD. NPs involves stimulating neuronal cell death, Aβ and APP production, loss of memory and cognition function, enhances P-tau, and improves ROS production in AD pathology
Environmental factors promotes tau phosphorylation in AD
The existing research demonstrated that numerous environmental risk factors are shown to facilitate AD progression over the alterations on tau aggregation as well as phosphorylation (Fig. 3).
Exposure of metals
Both in vitro as well as in vivo studies have recommended that Hg can potentially induce P-tau. MeHg intake by male mice shows an amplified death of neuronal cells in cerebral cortex and extra migrating astrocytes has been observed, along with augmented P-tau levels facilitated by c-jun N-terminal kinase (Fujimura et al. 2009). Inorganic Hg at a dose of 50 μg/dL/30 min has the ability to amplify tau phosphorylation of SH-SY5Y cells via prompting ROS, which is returned when co-treated by the melatonin which possesses antioxidant properties (Olivieri et al. 2000). A study has reported that Hg ions increases the heparin-prompted aggregation in addition to causes a conformational alteration in tau verified by circular dicroism (CD) (Yang et al. 2010). In contrast, by promoting the aggregation of tau protein, Cd seems to show a role in tau hypothesis. It is revealed that Cd(II) stimulates the heparin-mediated accumulation of tau and it causes variations in conformation verified by CD (Jiang et al. 2007). Rats treated with 3–10 mg/kg/day for 4 to 12 weeks for subchronic As shows increase P-tau, recommending that axonal degeneration may cause by the As destabilization and disruption of the cytoskeleton (Vahidnia et al. 2008). A amplified phosphorylation of tau and cyclin-dependent kinase 5, both mRNA and protein levels are also found to increase (Bihaqi and Zawia 2013). Pb exposures to maternal and early postnatal mice significantly amplified P-tau and cognitive damage (Li et al. 2010). Other studies report that tau aggregation is caused by chronic Al exposure and recommend that Al could be bound to P-tau (Shin et al. 2003; Xu et al. 2018). In vivo and in vitro studies report that Al can resist the breakdown of PHFs (Shin et al. 2003), and also can prevent protein phosphatase 2 (PP2) activity, which is required in the dephosphorylation of P-tau (Fig. 3) (Chin-Chan et al. 2015; Yamamoto et al. 1990).
Exposure of pesticides
Some evidences have suggested that tau functionalities can be disrupted by pesticide exposure (Fig. 3) (Yan et al. 2016). A recent study presented that the insecticide carbofuran (carbamate) and deltamethrin (pyethroid) administration to rats caused the death of neuronal cells in the hippocampus and cortex, and thereby, a loss of spatial memory and learning (Fig. 4). These changes may be occurred by lowering synaptic proteins expression that usually involved in memory consolidation. Furthermore, activated kinase p-GSK3β (phosphorylates tau) and elevated P-tau have also detected (Bian et al. 2016; Chen et al. 2012). Also, it has been reported that PQ (10 mg/Kg) treated mice exhibited P-tau elevation in the striatum, mediated by stimulation of p-GSK3β, also, causes α-tubulin hyperacetylation, suggesting for a cytoskeleton transformation (Wills et al. 2012).
Exposure of nanoparticles
NPs on phosphorylation of tau has not been widely investigated. Silica NPs are being used as a drug has been reported to rise Ser262 as well as Ser396 phosphorylation tau sites that is usually observed in AD (Murugadoss et al. 2017). This effect is mediated by kinase GSK3β activation probably facilitate by oxidative stress as ROS is amplified in mouse Neuro-2a and human SK-N-SH cells in response to these NPs (Yang et al. 2014).
Effects of air pollution on AD pathogenesis
Pathological and Clinical examinations on cell culture, animal, and humans studies partially support on air pollution in AD as a risk factor. Polluted air comprises particulate matter (PM) of numerous sizes in conjunction with deleterious compounds for example sulfur oxide species, nitrogen, metals, carbon monoxide, and inorganic compounds. PM encloses ammonium, carbon, sulfates, chlorides, nitrates, and additional biological material along with dust, which is distributed consistent with size (Li et al. 2003). After inhaled, ultrafine as well as fine PM are proficient to cross into bloodstream and taken up through cells causing mitochondrial damage in addition to oxidative stress (Li et al. 2003), which may be capable to enter the brain directly via the olfactory nerve responsible to AD (Block and Calderon-Garciduenas 2009). Furthermore, it has been found that short-term exposure to high intensities of ultra-fine PM can change inflammatory responses in the brain’s (Kleinman et al. 2008), which is highly relevant to develop AD as well as dementia (Heppner et al. 2015). However, exact risk appears to be altered via additional environmental factors in conjunction with genetic predisposition with APOE gene abnormal interrelated to dementia to the effect of air pollution (Cacciottolo et al. 2017; Chen and Schwartz 2009). For example, individuals staying in extremely polluted zones accumulate greater quantities of Aβ42 and tau hyperphosphorylated in the hippocampus as well as olfactory bulb (Calderon-Garciduenas et al. 2004). More precisely, oxidative damage glial cells may increase risk of AD pathogenesis (Dzamba et al. 2016). Similarly, in AD mouse model, introduction to ultrafine PM cause escalation in the predictable quantity of Aβ plaques formation in addition to decrease hippocampus neuron density (Cacciottolo et al. 2017). It has been revealed that PM exposure provokes modifications in inflammatory reactions, dendritic spine density loss, decrease hippocampus (CA1 region) dendrite length, increase BACE and Aβ expression, and more amyloid precursor protein (APP) in mice brains to stimulate AD (Bhatt et al. 2015). Besides, diesel exposure exhaust elements may lead to stimulate inflammatory-mediated cytokines and generate reactive oxygen species (ROS) in rat brain which has been displayed to decline cognitive function (Durga et al. 2015). Therefore, a relation concerning with neuroinflammation as well as exposure of particulate air pollution creates a possible pathway in AD risk.
Epigenetic evidence to develop AD influences by environmental factors
Epigenetic mechanisms are mainly DNA packaging around nucleosomes, histone tails covalent posttranslational modifications, chromatin folding and attachment to the nuclear matrix (Sadakierska-Chudy and Filip 2015), miRNAs, and DNA methylation (Holliday 2006). DNA methylation can stimulate the expression of corresponding genes by adding methyl groups through DNA methyltransferases (DNMTs) to the cytosine bases placed at cytosine-phosphate-guanine (CpG) sites. Central developments such as embryonic development, differentiation of cells to different cell types and aging are regulated by DNA methylation on the corresponding gene’s promoter regions (Bird 2002; Suelves et al. 2016). An increasing number of evidence suggest that the epigenetic fluctuations within the growing embryo may play necessary roles within the vulnerability to illness in future life resulting from the maternal contacts to environmental elements at critical developmental stages. In several animal models, environmental impacts are associated to epigenetic alterations. Epigenetic effects have been perceived through the environmental and nutritional elements (Heijmans et al. 2008), for example, inorganic contaminants like arsenic (Singh and DuMond Jr. 2007), chemicals like pesticides (Andersen et al. 2008) or fungicides (Anway et al. 2005), methyl donors such as folate (Cropley et al. 2006), drugs like cocaine (Novikova et al. 2008), airborne polycyclic aromatic hydrocarbons (Perera et al. 2009), phytoestrogens (Guerrero-Bosagna et al. 2008), and endocrine disruptors like BPA (Dolinoy et al. 2007; Yaoi et al. 2008). It has also been established that behavioral properties on DNA methylation comprising maternal special effects on nursing behavior (Champagne et al. 2006) as well as depression (Oberlander et al. 2008). For that reason, various models of environmental elements have been exposed to alter the epigenetic. This recommends that a brief contact to chemical compounds is possible to memorize even long after the chemical exposure by the action of epigenetic machinery (Vickers 2014; Weaver et al. 2014). Another study has recommended that epigenetic component to cause neurodegenerative diseases is associated with environmental elements (Marques et al. 2011).
Environmental pollutants (Faulk and Dolinoy 2011), aging (Calvanese et al. 2009), psychiatric consequences (Sananbenesi and Fischer 2009), and neurodegeneration (Urdinguio et al. 2009) are epidemiologic risk factors that are associated with epigenetic fluctuations. Earlier life methylation of particular genes can cause DNA damage mediated by oxidative stress. The hypomethylated APP gene stimulates its self-expression, leads to the APP overproduction and elevated Aβ levels, resulting in the facilitation of the production of ROS, DNA damage, and ultimately neuronal loss (Bhat et al. 2015). On the other hand, the DNA repair pathways and gene transcription are disrupted by the hypermethylation. Both types of DNA methylation can influence the expression of genes and imprint vulnerability to DNA damage by oxidative stress in the elderly brain (Zawia et al. 2009). It is recommended that by interfering with the capacity of DNA methylation, Pb fluctuates the AD-related gene expression. A reduction of brain DNMT activity has been reported in aged monkeys developmentally contacted to Pb. Additionally, Pb (0.1 μM) exposure to primary cells have collected from mouse cerebral cortex shows a parallel result on DNMT1 activity after 1 week of 24-h treatment (Masoud et al. 2016). A dormant rise in AD biomarkers expression and a reduce mRNA and protein levels of the DNA methylation enzymes like Dnmt1 as well as Dnmt3a, and MeCP2 has observed in differentiated SH-SY5Y cells in response to Pb exposure (Bihaqi and Zawia 2012). In postmortem brains, unusual methylation of CpG in tau, GSK3β, and APP genes were observed (Coupland et al. 2016). Furthermore, it recommended that reduction of CpG methylation in the APP promoter could be facilitated by the guanine (8-oxdG) oxidation (Zawia et al. 2009); this is because the adjacent cytosine methylation was preventing by guanine oxidation in CpG dinucleotides (Ito and Kuraoka 2015). Cd, another metal that is also involved in the pathology of AD, reported to reduce the Dnmt enzymatic activity in rat liver cell in vitro (Poirier and Vlasova 2002), nonetheless this effect has not assessed in cerebral cells. Subchronic As exposure has reported to change the methylation of the genes that are intricate in neuronal plasticity, comprising protein phosphatase 1 (PP1) and reelin (RELN), which is linked through memory shortages (Martinez et al. 2011). Mice perinatal introduction to permethrin shows brain function alterations that include dopaminergic neurons biomarkers and spatial memory damage at the age of 6 months (Cinzia et al. 2013).
Conclusions
The epidemiological investigations and experimental studies have directed to emphasize the prospective risk to grow AD because of environmental pollutants exposure include heavy metals, NPs as well as toxic pesticides. Remarkably, these environmental pollutants display related toxicity mechanisms to the oxidative stress mediated AD. For instance, oxidative stress induced by accumulating ROS production or deregulating enzymes of antioxidant stimulates Aβ and tau aggregation and formation, respectively. This pollutant overwhelms the degradation methods and induces neuroinflammation, as a result enhances additional oxidative stress leading to hippocampus as well as cerebral cortex neuronal loss in AD. These neurotoxicants induce oxidative stress and stimulate or prevent several signaling pathways which lead to increased or reduced enzymes activity that encourage the addition of toxic materials, Aβ in AD, damaged proteins, and oxidative byproducts in neuronal cells which might be changed epigenetic or genetic regulation. Conversely, absence of particular biomarkers restricts the earlier diagnosis and appropriate treatment in AD. In particular, biomarker identification is of most importance to conclude the previous exposure to the pollutants of environmental factor for a superior and appropriate controlling of AD. In this respect, precise circulating miRNAs have been related to diagnosis of AD pathology; thus, they are favorable for noninvasive biomarkers. Therefore, additional well-intended epidemiological investigations are essential to develop the quality of earlier life to avoid the progress of this neurodegenerative disorder globally.
References
Alam Q, Alam MZ, Mushtaq G, Damanhouri GA, Rasool M, Kamal MA, Haque A (2016) Inflammatory process in Alzheimer’s and Parkinson’s diseases: central role of cytokines. Curr Pharm Design 22:541–548
Alghamdi BSA (2018) Possible prophylactic anti-excitotoxic and anti-oxidant effects of virgin coconut oil on aluminium chloride-induced Alzheimer’s in rat models. J Integr Neurosci 17:593–607
An L, Liu SC, Yang Z, Zhang T (2012) Cognitive impairment in rats induced by nano-CuO and its possible mechanisms. Toxicol Lett 213:220–227
Andersen HR, Schmidt IM, Grandjean P, Jensen TK, Budtz-Jorgensen E, Kjaerstad MB, Baelum J, Nielsen JB, Skakkebaek NE, Main KM (2008) Impaired reproductive development in sons of women occupationally exposed to pesticides during pregnancy. Environ Health Perspect 116:566–572
Anstey KJ, Cherbuin N, Budge M, Young J (2011) Body mass index in midlife and late-life as a risk factor for dementia: a meta-analysis of prospective studies. Obes Rev 12:e426–e437
Anway MD, Cupp AS, Uzumcu M, Skinner MK (2005) Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science 308:1466–1469
Baillon S, Gasper A, Wilson-Morkeh F, Pritchard M, Jesu A, Velayudhan L (2019) Prevalence and severity of neuropsychiatric symptoms in early- versus late-onset Alzheimer’s disease. Am J Alzheimers Dis Other Dement 34(7-8):433–438 1533317519841191
Bakulski KM, Rozek LS, Dolinoy DC, Paulson HL, Hu H (2012) Alzheimer's disease and environmental exposure to lead: the epidemiologic evidence and potential role of epigenetics. Curr Alzheimer Res 9:563–573
Bangen KJ, Nation DA, Delano-Wood L, Weissberger GH, Hansen LA, Galasko DR, Salmon DP, Bondi MW (2015) Aggregate effects of vascular risk factors on cerebrovascular changes in autopsy-confirmed Alzheimer’s disease. Alzheimers Dement 11:394–403
Basha MR, Wei W, Bakheet SA, Benitez N, Siddiqi HK, Ge YW, Lahiri DK, Zawia NH (2005) The fetal basis of amyloidogenesis: exposure to lead and latent overexpression of amyloid precursor protein and beta-amyloid in the aging brain. J Neurosci 25:823–829
Behl M, Zhang Y, Shi Y, Cheng J, Du Y, Zheng W (2010) Lead-induced accumulation of beta-amyloid in the choroid plexus: role of low density lipoprotein receptor protein-1 and protein kinase C. Neurotoxicology 31:524–532
Bertram L (2009) Alzheimer's disease genetics current status and future perspectives. Int Rev Neurobiol 84:167–184
Bhat AH, Dar KB, Anees S, Zargar MA, Masood A, Sofi MA, Ganie SA (2015) Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed Pharmacother 74:101–110
Bhatt DP, Puig KL, Gorr MW, Wold LE, Combs CK (2015) A pilot study to assess effects of long-term inhalation of airborne particulate matter on early Alzheimer-like changes in the mouse brain. PLoS One 10:e0127102
Bian H, Bian W, Lin X, Ma Z, Chen W, Pu Y (2016) RNA interference silencing of glycogen synthase kinase 3beta inhibits tau phosphorylation in mice with Alzheimer disease. Neurochem Res 41:2470–2480
Bihaqi SW (2019) Early life exposure to lead (Pb) and changes in DNA methylation: relevance to Alzheimer's disease. Rev Environ Health 34:187–195
Bihaqi SW, Zawia NH (2012) Alzheimer’s disease biomarkers and epigenetic intermediates following exposure to Pb in vitro. Curr Alzheimer Res 9:555–562
Bihaqi SW, Zawia NH (2013) Enhanced taupathy and AD-like pathology in aged primate brains decades after infantile exposure to lead (Pb). Neurotoxicology 39:95–101
Bird A (2002) DNA methylation patterns and epigenetic memory. Genes Dev 16:6–21
Bjorklund G, Tinkov AA, Dadar M, Rahman MM, Chirumbolo S, Skalny AV, Skalnaya MG, Haley BE, Ajsuvakova OP, Aaseth J (2019) Insights into the potential role of mercury in Alzheimer’s disease. J Mol Neurosci 67:511–533
Block ML, Calderon-Garciduenas L (2009) Air pollution: mechanisms of neuroinflammation and CNS disease. Trends Neurosci 32:506–516
Boal HL, Christensen BK, Goodhew SC (2018) Social anxiety and attentional biases: A top-down contribution? Atten Percept Psychophysiol 80:42–53
Bu G (2009) Apolipoprotein E and its receptors in Alzheimer's disease: pathways, pathogenesis and therapy. Nat Rev Neurosci 10:333–344
Cacciottolo, M., Wang, X., Driscoll, I., Woodward, N., Saffari, A., Reyes, J., Serre, M.L., Vizuete, W., Sioutas, C., Morgan, T.E., et al. (2017). Particulate air pollutants, APOE alleles and their contributions to cognitive impairment in older women and to amyloidogenesis in experimental models, Transl Psychiat 7:e1022.
Calderon-Garciduenas L, Reed W, Maronpot RR, Henriquez-Roldan C, Delgado-Chavez R, Calderon-Garciduenas A, Dragustinovis I, Franco-Lira M, Aragon-Flores M, Solt AC et al (2004) Brain inflammation and Alzheimer’s-like pathology in individuals exposed to severe air pollution. Toxicol Pathol 32:650–658
Calvanese V, Lara E, Kahn A, Fraga MF (2009) The role of epigenetics in aging and age-related diseases. Ageing Res Rev 8:268–276
Castro-Chavira SA, Fernandez T, Nicolini H, Diaz-Cintra S, Prado-Alcala RA (2015) Genetic markers in biological fluids for aging-related major neurocognitive disorder. Curr Alzheimer Res 12:200–209
Champagne FA, Weaver IC, Diorio J, Dymov S, Szyf M, Meaney MJ (2006) Maternal care associated with methylation of the estrogen receptor-alpha1b promoter and estrogen receptor-alpha expression in the medial preoptic area of female offspring. Endocrinology 147:2909–2915
Chang JW, Pai MC, Chen HL, Guo HR, Su HJ, Lee CC (2008) Cognitive function and blood methylmercury in adults living near a deserted chloralkali factory. Environ Res 108:334–339
Chen JC, Schwartz J (2009) Neurobehavioral effects of ambient air pollution on cognitive performance in US adults. Neurotoxicology 30:231–239
Chen NN, Luo DJ, Yao XQ, Yu C, Wang Y, Wang Q, Wang JZ, Liu GP (2012) Pesticides induce spatial memory deficits with synaptic impairments and an imbalanced tau phosphorylation in rats. J Alzheimers Dis 30:585–594
Chin-Chan M, Navarro-Yepes J, Quintanilla-Vega B (2015) Environmental pollutants as risk factors for neurodegenerative disorders: Alzheimer and Parkinson diseases. Front Cell Neurosci 9:124
Cinzia N, Manuel C, Donatella F, Rosita G, Antonio DS, Cerasa LS, Isabel S, Valentina D, Roberto C (2013) Effects of early life permethrin exposure on spatial working memory and on monoamine levels in different brain areas of pre-senescent rats. Toxicology 303:162–168
Coimbra JRM, Marques DFF, Baptista SJ, Pereira CMF, Moreira PI, Dinis TCP, Santos AE, Salvador JAR (2018) Highlights in BACE1 inhibitors for Alzheimer’s disease treatment. Front Chem 6:178
Colomina MT, Peris-Sampedro F (2017) Aluminum and Alzheimer's disease. Adv Neurobiol 18:183–197
Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA (1993) Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261:921–923
Coupland KG, Kim WS, Halliday GM, Hallupp M, Dobson-Stone C, Kwok JB (2016) Role of the long non-coding RNA MAPT-AS1 in regulation of microtubule associated protein tau (MAPT) expression in Parkinson’s disease. PLoS One 11:e0157924
Cropley JE, Suter CM, Beckman KB, Martin DI (2006) Germ-line epigenetic modification of the murine A vy allele by nutritional supplementation. Proc Natl Acad Sci U S A 103:17308–17312
Dai MH, Zheng H, Zeng LD, Zhang Y (2018) The genes associated with early-onset Alzheimer’s disease. Oncotarget 9:15132–15143
Dansokho C, Heneka MT (2018) Neuroinflammatory responses in Alzheimer’s disease. J Neural Transm (Vienna) 125:771–779
Denis PA (2013) Alzheimer’s disease: a gas model. The NADPH oxidase-nitric oxide system as an antibubble biomachinery. Med Hypotheses 81:976–987
Dolinoy DC, Huang D, Jirtle RL (2007) Maternal nutrient supplementation counteracts bisphenol A-induced DNA hypomethylation in early development. Proc Natl Acad Sci U S A 104:13056–13061
Dorsey CD, Lee BK, Bolla KI, Weaver VM, Lee SS, Lee GS, Todd AC, Shi WP, Schwartz BS (2006) Comparison of patella lead with blood lead and tibia lead and their associations with neurobehavioral test scores. J Occup Environ Med 48:489–496
Durga M, Devasena T, Rajasekar A (2015) Determination of LC50 and sub-chronic neurotoxicity of diesel exhaust nanoparticles. Environ Toxicol Pharmacol 40:615–625
Dzamba D, Harantova L, Butenko O, Anderova M (2016) Glial cells - the key elements of Alzheimer’s disease. Curr Alzheimer Res 13:894–911
Faulk C, Dolinoy DC (2011) Timing is everything. The when and how of environmentally induced changes in the epigenome of animals. Epigenetics-Us 6:791–797
Feng Y, Wang X (2012) Antioxidant therapies for Alzheimer’s disease. Oxidative Med Cell Longev 2012:472932
Ferrari C, Nacmias B, Bagnoli S, Piaceri I, Lombardi G, Pradella S, Tedde A, Sorbi S (2014) Imaging and cognitive reserve studies predict dementia in presymptomatic Alzheimer’s disease subjects. Neurodegener Dis 13:157–159
Fratiglioni L, Wang HX (2007) Brain reserve hypothesis in dementia. J Alzheimers Dis 12:11–22
Fratiglioni L, Paillard-Borg S, Winblad B (2004) An active and socially integrated lifestyle in late life might protect against dementia. Lancet Neurol 3:343–353
Fujimura M, Usuki F, Sawada M, Takashima A (2009) Methylmercury induces neuropathological changes with tau hyperphosphorylation mainly through the activation of the c-Jun-N-terminal kinase pathway in the cerebral cortex, but not in the hippocampus of the mouse brain. Neurotoxicology 30:1000–1007
Gomez-Gomez ME, Zapico SC (2019) Frailty, cognitive decline, Neurodegenerative Diseases and Nutrition Interventions. Int J Mol Sci 20:E2842
Gong G, O'Bryant SE (2010) The arsenic exposure hypothesis for Alzheimer disease. Alzheimer Dis Assoc Disord 24:311–316
Gu H, Wei X, Monnot AD, Fontanilla CV, Behl M, Farlow MR, Zheng W, Du Y (2011) Lead exposure increases levels of beta-amyloid in the brain and CSF and inhibits LRP1 expression in APP transgenic mice. Neurosci Lett 490:16–20
Guerrero-Bosagna CM, Sabat P, Valdovinos FS, Valladares LE, Clark SJ (2008) Epigenetic and phenotypic changes result from a continuous pre and post natal dietary exposure to phytoestrogens in an experimental population of mice. BMC Physiol 8:17
Heijmans BT, Tobi EW, Stein AD, Putter H, Blauw GJ, Susser ES, Slagboom PE, Lumey LH (2008) Persistent epigenetic differences associated with prenatal exposure to famine in humans. Proc Natl Acad Sci U S A 105:17046–17049
Heppner FL, Ransohoff RM, Becher B (2015) Immune attack: the role of inflammation in Alzheimer disease. Nat Rev Neurosci 16:358–372
Holliday R (2006) Epigenetics A historical overview. Epigenetics-Us 1:76–80
Hu K, Li Y, Yu H, Hu Y (2019) CTBP1 confers protection for hippocampal and cortical neurons in rat models of Alzheimer’s disease. Neuroimmunomodulation 26(3):139–152 1–14
Huang CL, Hsiao IL, Lin HC, Wang CF, Huang YJ, Chuang CY (2015) Silver nanoparticles affect on gene expression of inflammatory and neurodegenerative responses in mouse brain neural cells. Environ Res 136:253–263
Huang D, Yu M, Yang S, Lou D, Zhou W, Zheng L, Wang Z, Cai F, Zhou W, Li T et al (2018) Ethanol alters APP processing and aggravates Alzheimer-associated phenotypes. Mol Neurobiol 55:5006–5018
Huat TJ, Camats-Perna J, Newcombe EA, Valmas N, Kitazawa M, Medeiros R (2019) Metal toxicity links to Alzheimer’s disease and neuroinflammation. J Mol Biol 431:1843–1868
Huber CM, Yee C, May T, Dhanala A, Mitchell CS (2018) Cognitive decline in preclinical Alzheimer’s disease: amyloid-beta versus tauopathy. J Alzheimers Disease 61:265–281
Ito S, Kuraoka I (2015) Epigenetic modifications in DNA could mimic oxidative DNA damage: a double-edged sword. DNA Repair 32:52–57
Jalilian H, Teshnizi SH, Roosli M, Neghab M (2018) Occupational exposure to extremely low frequency magnetic fields and risk of Alzheimer disease: a systematic review and meta-analysis. Neurotoxicology 69:242–252
Jiang LF, Yao TM, Zhu ZL, Wang C, Ji LN (2007) Impacts of Cd (II) on the conformation and self-aggregation of Alzheimer’s tau fragment corresponding to the third repeat of microtubule-binding domain. Bba-Proteins Proteom 1774:1414–1421
Johansson C, Castoldi AF, Onishchenko N, Manzo L, Vahter M, Ceccatelli S (2007) Neurobehavioural and molecular changes induced by methylmercury exposure during development. Neurotox Res 11:241–260
Jouanne M, Rault S, Voisin-Chiret AS (2017) Tau protein aggregation in Alzheimer’s disease: an attractive target for the development of novel therapeutic agents. Eur J Med Chem 139:153–167
Karri V, Schuhmacher M, Kumar V (2016) Heavy metals (Pb, Cd, As and MeHg) as risk factors for cognitive dysfunction: a general review of metal mixture mechanism in brain. Environ Toxicol Pharmacol 48:203–213
Kilian J, Kitazawa M (2018) The emerging risk of exposure to air pollution on cognitive decline and Alzheimer’s disease - evidence from epidemiological and animal studies. Biom J 41:141–162
Kim DK, Park JD, Choi BS (2014) Mercury-induced amyloid-beta (Abeta) accumulation in the brain is mediated by disruption of Abeta transport. J Toxicol Sci 39:625–635
Kleinman MT, Araujo JA, Nel A, Sioutas C, Campbell A, Cong PQ, Lia H, Bondy SC (2008) Inhaled ultrafine particulate matter affects CNS inflammatory processes and may act via MAP kinase signaling pathways. Toxicol Lett 178:127–130
Larsson SC, Orsini N (2018) Coffee consumption and risk of dementia and Alzheimer’s disease: a dose-response meta-analysis of prospective studies. Nutrients 10:E1501
Li N, Sioutas C, Cho A, Schmitz D, Misra C, Sempf J, Wang MY, Oberley T, Froines J, Nel A (2003) Ultrafine particulate pollutants induce oxidative stress and mitochondrial damage. Environ Health Perspect 111:455–460
Li N, Yu ZL, Wang L, Zheng YT, Jia JX, Wang Q, Zhu MJ, Liu XH, Xia X, Li WJ (2010) Increased tau phosphorylation and beta amyloid in the hippocampus of mouse pups by early life Lead exposure. Acta Biol Hung 61:123–134
Li J, Wang YJ, Zhang M, Xu ZQ, Gao CY, Fang CQ, Yan JC, Zhou HD, Chongqing Ageing Study, G (2011) Vascular risk factors promote conversion from mild cognitive impairment to Alzheimer disease. Neurology 76:1485–1491
Li FJ, Shen L, Ji HF (2012a) Dietary intakes of vitamin E, vitamin C, and beta-carotene and risk of Alzheimer’s disease: a meta-analysis. J Alzheimers Dis 31:253–258
Li X, Lv Y, Yu S, Zhao H, Yao L (2012b) The effect of cadmium on Abeta levels in APP/PS1 transgenic mice. Exp Ther Med 4:125–130
Li G, Kim C, Kim J, Yoon H, Zhou H, Kim J (2015) Common pesticide, dichlorodiphenyltrichloroethane (DDT), increases amyloid-beta levels by impairing the function of ABCA1 and IDE: implication for Alzheimer’s disease. J Alzheimers Dis 46:109–122
Li K, Cheng X, Jiang J, Wang J, Xie J, Hu X, Huang Y, Song L, Liu M, Cai L, Chen L, Zhao S (2017) The toxic influence of paraquat on hippocampal neurogenesis in adult mice. Food Chem Toxicol 106:356–366
Liang RF (2018) Cross talk between aluminum and genetic susceptibility and epigenetic modification in Alzheimer’s disease. Adv Exp Med Biol 1091:173–191
Lindsay J, Laurin D, Verreault R, Hebert R, Helliwell B, Hill GB, McDowell I (2002) Risk factors for Alzheimer’s disease: a prospective analysis from the Canadian Study of Health and Aging. Am J Epidemiol 156:445–453
Liu PD, Huang ZH, Gu N (2013) Exposure to silver nanoparticles does not affect cognitive outcome or hippocampal neurogenesis in adult mice. Ecotox Environ Safe 87:124–130
Lobo V, Patil A, Phatak A, Chandra N (2010) Free radicals, antioxidants and functional foods: impact on human health. Pharmacogn Rev 4:118–126
Luo Y, Niu F, Sun Z, Cao W, Zhang X, Guan D, Lv Z, Zhang B, Xu Y (2009) Altered expression of Abeta metabolism-associated molecules from D-galactose/AlCl(3) induced mouse brain. Mech Ageing Dev 130:248–252
Marques SCF, Oliveira CR, Pereira CMF, Outeiro TF (2011) Epigenetics in neurodegeneration: a new layer of complexity. Prog Neuro-Psychoph 35:348–355
Martinez L, Jimenez V, Garcia-Sepulveda C, Ceballos F, Delgado JM, Nino-Moreno P, Doniz L, Saavedra-Alanis V, Castillo CG, Santoyo ME et al (2011) Impact of early developmental arsenic exposure on promotor CpG-island methylation of genes involved in neuronal plasticity. Neurochem Int 58:574–581
Masoud AM, Bihaqi SW, Machan JT, Zawia NH, Renehan WE (2016) Early-life exposure to lead (Pb) alters the expression of microRNA that target proteins associated with Alzheimer’s disease. Journal of Alzheimers Disease 51:1257–1264
Mawuenyega KG, Sigurdson W, Ovod V, Munsell L, Kasten T, Morris JC, Yarasheski KE, Bateman RJ (2010) Decreased clearance of CNS beta-amyloid in Alzheimer's disease. Science 330:1774
Mendez MF (2017) Early-onset Alzheimer disease. Neurol Clin 35:263–281
Mezzaroba L, Alfieri DF, Colado Simao AN, Vissoci Reiche EM (2019) The role of zinc, copper, manganese and iron in neurodegenerative diseases. Neurotoxicology 74:230–241
Miguel-Alvarez M, Santos-Lozano A, Sanchis-Gomar F, Fiuza-Luces C, Pareja-Galeano H, Garatachea N, Lucia A (2015) Non-steroidal anti-inflammatory drugs as a treatment for Alzheimer’s disease: a systematic review and meta-analysis of treatment effect. Drugs Aging 32:139–147
Monnet-Tschudi F, Zurich MG, Boschat C, Corbaz A, Honegger P (2006) Involvement of environmental mercury and lead in the etiology of neurodegenerative diseases. Rev Environ Health 21:105–117
Mourao RJ, Mansur G, Malloy-Diniz LF, Castro Costa E, Diniz BS (2016) Depressive symptoms increase the risk of progression to dementia in subjects with mild cognitive impairment: systematic review and meta-analysis. Int J Geriatr Psychiatry 31:905–911
Murugadoss S, Lison D, Godderis L, Van den Brule S, Mast J, Brassinne F, Sebaihi N, Hoet PH (2017) Toxicology of silica nanoparticles: an update. Arch Toxicol 91:2967–3010
Mushtaq G, Khan JA, Joseph E, Kamal MA (2015) Nanoparticles, neurotoxicity and neurodegenerative diseases. Curr Drug Metab 16:676–684
Novikova SI, He F, Bai J, Cutrufello NJ, Lidow MS, Undieh AS (2008) Maternal cocaine administration in mice alters DNA methylation and gene expression in hippocampal neurons of neonatal and prepubertal offspring. PLoS One 3:e1919
Oberlander TF, Weinberg J, Papsdorf M, Grunau R, Misri S, Devlin AM (2008) Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics-Us 3:97–106
Okuda M, Hijikuro I, Fujita Y, Wu X, Nakayama S, Sakata Y, Noguchi Y, Ogo M, Akasofu S, Ito Y et al (2015) PE859, a novel tau aggregation inhibitor, reduces aggregated tau and prevents onset and progression of neural dysfunction in vivo. PLoS One 10:e0117511
Olivieri G, Brack C, Muller-Spahn F, Stahelin HB, Herrmann M, Renard P, Brockhaus M, Hock C (2000) Mercury induces cell cytotoxicity and oxidative stress and increases beta-amyloid secretion and tau phosphorylation in SHSY5Y neuroblastoma cells. J Neurochem 74:231–236
Park HS, Park SS, Kim CJ, Shin MS, Kim TW (2019) Exercise alleviates cognitive functions by enhancing hippocampal insulin signaling and neuroplasticity in high-fat diet-induced obesity. Nutrients 11:E1603
Perera F, Tang WY, Herbstman J, Tang D, Levin L, Miller R, Ho SM (2009) Relation of DNA methylation of 5'-CpG island of ACSL3 to transplacental exposure to airborne polycyclic aromatic hydrocarbons and childhood asthma. PLoS One 4:e4488
Peris-Sampedro F, Salazar JG, Cabre M, Reverte I, Domingo JL, Sanchez-Santed F, Colomina MT (2014) Impaired retention in AbetaPP Swedish mice six months after oral exposure to chlorpyrifos. Food Chem Toxicol 72:289–294
Piaceri I, Nacmias B, Sorbi S (2013) Genetics of familial and sporadic Alzheimer's disease. Front Biosci (Elite Ed) 5:167–177
Plassman BL, Havlik RJ, Steffens DC, Helms MJ, Newman TN, Drosdick D, Phillips C, Gau BA, Welsh-Bohmer KA, Burke JR, Guralnik JM, Breitner JC (2000) Documented head injury in early adulthood and risk of Alzheimer's disease and other dementias. Neurology 55:1158–1166
Poirier LA, Vlasova TI (2002) The prospective role of abnormal methyl metabolism in cadmium toxicity. Environ Health Perspect 110:793–795
Pratico D, Uryu K, Sung S, Tang S, Trojanowski JQ, Lee VMY (2002) Aluminum modulates brain amyloidosis through oxidative stress in APP transgenic mice. FASEB J 16:1138
Qi S, Feng Z, Li Q, Qi Z, Zhang Y (2018) Inhibition of ROS-mediated activation Src-MAPK/AKT signaling by orientin alleviates H2O2-induced apoptosis in PC12 cells. Drug Des Devel Ther 12:3973–3984
Rahman MA, Rhim H (2017) Therapeutic implication of autophagy in neurodegenerative diseases. BMB Rep 50:345–354
Ramos-Chavez LA, Rendon-Lopez CR, Zepeda A, Silva-Adaya D, Del Razo LM, Gonsebatt ME (2015) Neurological effects of inorganic arsenic exposure: altered cysteine/glutamate transport, NMDA expression and spatial memory impairment. Front Cell Neurosci 9:21
Reuben A (2018) Childhood lead exposure and adult neurodegenerative disease. J Alzheimers Dis 64:17–42
Richardson JR, Roy A, Shalat SL, von Stein RT, Hossain MM, Buckley B, Gearing M, Levey AI, German DC (2014) Elevated serum pesticide levels and risk for Alzheimer disease. Jama Neurology 71:284–290
Rusanen M, Kivipelto M, Quesenberry CP Jr, Zhou J, Whitmer RA (2011) Heavy smoking in midlife and long-term risk of Alzheimer disease and vascular dementia. Arch Intern Med 171:333–339
Sadakierska-Chudy A, Filip M (2015) A comprehensive view of the epigenetic landscape. Part II: histone post-translational modification, nucleosome level, and chromatin regulation by ncRNAs. Neurotox Res 27:172–197
Sakamoto T, Saito H, Ishii K, Takahashi H, Tanabe S, Ogasawara Y (2006) Aluminum inhibits proteolytic degradation of amyloid beta peptide by cathepsin D: A potential link between aluminum accumulation and neuritic plaque deposition. FEBS Lett 580:6543–6549
Salazar JG, Ribes D, Cabre M, Domingo JL, Sanchez-Santed F, Colomina MT (2011) Amyloid beta peptide levels increase in brain of AbetaPP Swedish mice after exposure to chlorpyrifos. Curr Alzheimer Res 8:732–740
Sananbenesi F, Fischer A (2009) The epigenetic bottleneck of neurodegenerative and psychiatric diseases. Biol Chem 390:1145–1153
Savva GM, Stephan BC, Alzheimer's Society Vascular Dementia Systematic Review, G (2010) Epidemiological studies of the effect of stroke on incident dementia: a systematic review. Stroke 41:e41–e46
Schlosser Covell GE, Hoffman-Snyder CR, Wellik KE, Woodruff BK, Geda YE, Caselli RJ, Demaerschalk BM, Wingerchuk DM (2015) Physical activity level and future risk of mild cognitive impairment or dementia: a critically appraised topic. Neurologist 19:89–91
Sharma HS, Sharma A (2012) Neurotoxicity of engineered nanoparticles from metals. Cns Neurol Disord-Dr 11:65–80
Sharma HS, Ali SF, Hussain SM, Schlager JJ, Sharma A (2009) Influence of engineered nanoparticles from metals on the blood-brain barrier permeability, cerebral blood flow, brain edema and neurotoxicity An Experimental Study in the Rat and Mice Using Biochemical and Morphological Approaches. J Nanosci Nanotechnol 9:5055–5072
Sharp SI, Aarsland D, Day S, Sonnesyn H, Ballard C, Alzheimer's Society Vascular Dementia Systematic Review Group, B (2011) Hypertension is a potential risk factor for vascular dementia: systematic review. Int J Geriatr Psychopharmacol 26:661–669
Shin RW, Kruck TP, Murayama H, Kitamoto T (2003) A novel trivalent cation chelator Feralex dissociates binding of aluminum and iron associated with hyperphosphorylated tau of Alzheimer’s disease. Brain Res 961:139–146
Singh KP, DuMond JW Jr (2007) Genetic and epigenetic changes induced by chronic low dose exposure to arsenic of mouse testicular Leydig cells. Int J Oncol 30:253–260
Smith MA, Harris PLR, Sayre LM, Perry G (1997) Iron accumulation in Alzheimer disease is a source of redox-generated free radicals. P Natl Acad Sci USA 94:9866–9868
Son SM, Kang S, Choi H, Mook-Jung I (2015) Statins induce insulin-degrading enzyme secretion from astrocytes via an autophagy-based unconventional secretory pathway. Mol Neurodegener 10:56
Song JW, Choi BS (2013) Mercury induced the accumulation of amyloid beta (Abeta) in PC12 cells: the role of production and degradation of Abeta. Toxicol Res 29:235–240
Sorgdrager FJH, Vermeiren Y, Van Faassen HJR, Van Der Ley CP, Nollen EAA, Kema IP, De Deyn PP (2019) Age- and disease-specific changes of the kynurenine pathway in Parkinson’s and Alzheimer’s disease. J Neurochem 151(5):656–668
Souza AD, Couto-Lima CA, Catalao CHR, Santos NN, dos Santos JF, da Rocha MJA, Alberici LC (2019) Neuroprotective action of eicosapentaenoic (EPA) and docosahexaenoic (DHA) acids on Paraquat intoxication in Drosophila melanogaster. Neurotoxicology 70:154–160
Suelves M, Carrio E, Nunez-Alvarez Y, Peinado MA (2016) DNA methylation dynamics in cellular commitment and differentiation. Brief Funct Genomics 15:443–453
Takahashi RH, Nagao T, Gouras GK (2017) Plaque formation and the intraneuronal accumulation of beta-amyloid in Alzheimer’s disease. Pathol Int 67:185–193
Tyler CR, Allan AM (2014) The effects of arsenic exposure on neurological and cognitive dysfunction in human and rodent studies: a review. Curr Environ Health Rep 1:132–147
Uddin MS, Stachowiak A, Al Mamun A, Tzvetkov NT, Takeda S, Atanasov AG, Bergantin LB, Abdel-Daim MM, Stankiewicz AM (2018) Autophagy and Alzheimer’s disease: from molecular mechanisms to therapeutic implications. Front Aging Neurosci 10:4
Uddin MS, Mamun AA, Labu ZK, Hidalgo-Lanussa O, Barreto GE, Ashraf GM (2019) Autophagic dysfunction in Alzheimer’s disease: cellular and molecular mechanistic approaches to halt Alzheimer’s pathogenesis. J Cell Physiol 234:8094–8112
Urdinguio RG, Sanchez-Mut JV, Esteller M (2009) Epigenetic mechanisms in neurological diseases: genes, syndromes, and therapies. Lancet Neurol 8:1056–1072
Vahidnia A, Romijn F, van der Voet GB, de Wolff FA (2008) Arsenic-induced neurotoxicity in relation to toxicokinetics: effects on sciatic nerve proteins. Chem Biol Interact 176:188–195
Ventriglia M, Brewer GJ, Simonelli I, Mariani S, Siotto M, Bucossi S, Squitti R (2015) Zinc in Alzheimer’s disease: A meta-analysis of serum, plasma, and cerebrospinal fluid studies. J Alzheimers Dis 46:75–87
Verreault R, Laurin D, Lindsay J, De Serres G (2001) Past exposure to vaccines and subsequent risk of Alzheimer’s disease. CMAJ 165:1495–1498
Vickers MH (2014) Early life nutrition, epigenetics and programming of later life disease. Nutrients 6:2165–2178
Walton JR, Wang MX (2009) APP expression, distribution and accumulation are altered by aluminum in a rodent model for Alzheimer’s disease. J Inorg Biochem 103:1548–1554
Wang WY, Tan MS, Yu JT, Tan L (2015) Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann Transl Med 3:136
Weaver CM, Teegarden D, Welch A, Hwalla N, Lelievre S (2014) International breast Cancer and nutrition: A model for research, training and policy in diet, epigenetics, and chronic disease prevention. Adv Nutr 5:566–567
Wichmann MA, Cruickshanks KJ, Carlsson CM, Chappell R, Fischer ME, Klein BEK, Klein R, Schubert CR (2016) NSAID use and incident cognitive impairment in a population-based cohort. Alzheimer Dis Assoc Disord 30:105–112
Wills J, Credle J, Oaks AW, Duka V, Lee JH, Jones J, Sidhu A (2012) Paraquat, but not Maneb, induces synucleinopathy and tauopathy in striata of mice through inhibition of proteasomal and autophagic pathways. PLoS One 7:e30745
Wingo TS, Cutler DJ, Wingo AP, Le NA, Rabinovici GD, Miller BL, Lah JJ, Levey AI (2019) Association of early-onset Alzheimer disease with elevated low-density lipoprotein cholesterol levels and rare genetic coding variants of APOB. JAMA Neurol 76:809–817
Wirths O, Multhaup G, Czech C, Blanchard V, Moussaoui S, Tremp G, Pradier L, Beyreuther K, Bayer TA (2001) Intraneuronal Abeta accumulation precedes plaque formation in beta-amyloid precursor protein and presenilin-1 double-transgenic mice. Neurosci Lett 306:116–120
Wong P, Appadurai K, Kanagarajah S (2016) Alcohol and the risk of behavioural and psychological symptoms of dementia in men and women. Australas J Ageing 35:29–29
Wu J, Basha MR, Brock B, Cox DP, Cardozo-Pelaez F, McPherson CA, Harry J, Rice DC, Maloney B, Chen D et al (2008) Alzheimer’s disease (AD)-like pathology in aged monkeys after infantile exposure to environmental metal lead (Pb): evidence for a developmental origin and environmental link for AD. J Neurosci 28:3–9
Xu W, Wang HF, Wan Y, Tan CC, Li JQ, Tan L, Yu JT (2017) Alcohol consumption and dementia risk: a dose-response meta-analysis of prospective studies. Eur J Epidemiol 32:31–42
Xu L, Zhang WC, Liu XC, Zhang CL, Wang P, Zhao XL (2018) Circulatory levels of toxic metals (aluminum, cadmium, mercury, lead) in patients with Alzheimer’s disease: A quantitative meta-analysis and systematic review. Journal of Alzheimers Disease 62:361–372
Yamamoto H, Saitoh Y, Yasugawa S, Miyamoto E (1990) Dephosphorylation of tau-factor by protein phosphatase-2a in synaptosomal cytosol fractions, and inhibition by aluminum. J Neurochem 55:683–690
Yan D, Zhang Y, Liu L, Yan H (2016) Pesticide exposure and risk of Alzheimer’s disease: a systematic review and meta-analysis. Sci Rep 6:32222
Yang DJ, Shi SO, Zheng LF, Yao TM, Ji LN (2010) Mercury (II) promotes the in vitro aggregation of tau fragment corresponding to the second repeat of microtubule-binding domain: coordination and conformational transition. Biopolymers 93:1100–1107
Yang XF, He CE, Li J, Chen HB, Ma Q, Sui XJ, Tian SL, Ying M, Zhang Q, Luo YG et al (2014) Uptake of silica nanoparticles: neurotoxicity and Alzheimer-like pathology in human SK-N-SH and mouse neuro2a neuroblastoma cells. Toxicol Lett 229:240–249
Yang MH, Chen SC, Lin YF, Lee YC, Huang MY, Chen KC, Wu HY, Lin PC, Gozes I, Tyan YC (2019) Reduction of aluminum ion neurotoxicity through a small peptide application - NAP treatment of Alzheimer’s disease. J Food Drug Anal 27:551–564
Yao D, Jing T, Niu L, Huang XX, Wang Y, Deng XQ, Wang MG (2018) Amyloidogenesis induced by diet cholesterol and copper in a model mouse for Alzheimer's disease and protection effects of zinc and fluvastatin. Brain Res Bull 143:1–8
Yaoi T, Itoh K, Nakamura K, Ogi H, Fujiwara Y, Fushiki S (2008) Genome-wide analysis of epigenomic alterations in fetal mouse forebrain after exposure to low doses of bisphenol A. Biochem Biophys Res Commun 376:563–567
Zarazua S, Burger S, Delgado JM, Jimenez-Capdeville ME, Schliebs R (2011) Arsenic affects expression and processing of amyloid precursor protein (APP) in primary neuronal cells overexpressing the Swedish mutation of human APP. Int J Dev Neurosci 29:389–396
Zawia NH, Lahiri DK, Cardozo-Pelaez F (2009) Epigenetics, oxidative stress, and Alzheimer disease. Free Radical Bio Med 46:1241–1249
Ze YG, Hu RP, Wang XC, Sang XZ, Ze X, Li B, Su JJ, Wang Y, Guan N, Zhao XY et al (2014) Neurotoxicity and gene-expressed profile in brain-injured mice caused by exposure to titanium dioxide nanoparticles. J Biomed Mater Res A 102:470–478
Zenaro E, Piacentino G, Constantin G (2017) The blood-brain barrier in Alzheimer’s disease. Neurobiol Dis 107:41–56
Zhang QL (2018) Aluminum-induced neural cell death. Adv Exp Med Biol 1091:129–160
Zhang QL, Niu Q, Niu PY, Shi YT, Liu CY, Di Gioacchino M, Zhang L, Zhang C, Braga M (2010) Bax gene silencing: A potential intervention in aluminum-induced neural cell death. J Biol Reg Homeos Agents 24:7–17
Zhang ZY, Miah M, Culbreth M, Aschner M (2016) Autophagy in neurodegenerative diseases and metal neurotoxicity. Neurochem Res 41:409–422
Zong L, Xing J, Liu S, Liu Z, Song F (2018) Cell metabolomics reveals the neurotoxicity mechanism of cadmium in PC12 cells. Ecotoxicol Environ Saf 147:26–33
Funding
This work was supported by the Korea Research Fellowship (KRF) Program (2016H1D3A1908615, 2017H1D3A1A02013844, 2015H1D3A1062189) through the National Research Foundation of Korea and the NRF Research Program (2016M3C7A1913845) funded by the Ministry of Science and ICT, Republic of Korea.
Author information
Authors and Affiliations
Contributions
This collaboration work was carried out among all the authors. MAR designed outlines and wrote the draft of the manuscript. MSR prepared the figures of the manuscript. MJU and ANMMR wrote some part of the manuscript. MGP reviewed the manuscript. HR proposed original idea and reviewed the scientific contents of the manuscript. All authors read and approved the final submitted version of the manuscript.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Responsible Editor: Philippe Garrigues
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Rahman, M.A., Rahman, M.S., Uddin, M.J. et al. Emerging risk of environmental factors: insight mechanisms of Alzheimer’s diseases. Environ Sci Pollut Res 27, 44659–44672 (2020). https://doi.org/10.1007/s11356-020-08243-z
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-020-08243-z