
Abstract
To assess the two most toxicologically relevant species of As, namely arsenite (As(III)) and arsenate (As(V)), chromatographic separations often require two separate chromatographic columns to address the co-elution of arsenobetaine (AsB) with As(III). This issue is typically observed using conventional isocratic methods on anion exchange columns, increasing cost and analysis time. Here, we optimize the extraction of inorganic As from a lichen air biomonitor and develop an isocratic method for the chromatographic separation of five common As species on a PRP X-100 anion exchange column, resulting in the complete baseline separation of all species under study. This method was then applied to lichen biomonitors from an urban and rural site to demonstrate its use. In order of abundance, the various arsenic species in lichens from the urban site in South Africa were As(V) > As(III) > AsB > dimethylarsinic acid (DMA) > monomethylarsonic acid (MMA), and As(V) > AsB > As(III) > DMA > MMA for the rural site, where MMA was present in extremely low, non-quantifiable concentrations in lichens from both sites. Total concentrations of As were higher in samples from the urban site (6.43 ± 0.25 μg/g) than in those from the rural site (1.87 ± 0.05 μg/g), with an overall extraction efficiency of 19% and 40%, respectively. The optimized method utilized relatively inexpensive solvents and is therefore low-cost and eco-friendly in comparison with conventional chromatographic techniques. This is the first study which addresses the optimized extraction and characterization of As species in a South African lichen biomonitor of air pollution.

.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
The speciation of arsenic in biomonitors of air pollution poses an interesting analytical challenge, where samples are often complex, containing several different arsenic species. Such speciation studies are of significance due to the high relative abundance of toxic inorganic arsenic species attached to particulate matter in the air, in comparison with their methylated counterparts (Chung et al. 2014). In some cases, this particulate matter has been reported to be As-enriched by 10–1000 times higher than continental crust concentrations (Johnson and Braman 1975; Cullen and Reimer 1989). Organic arsenic species, although generally of lower abundance in the atmosphere, may arise from a number of different sources. These include the production of volatile organo-As species through microbe and yeast metabolism (Cullen and Reimer 1989; Koch et al. 1999; Bentley and Chasteen 2002; Chung et al. 2014), the spraying of arsenic-containing pesticides such as monosodium methyl arsonate (MSMA), or the use of arsenic-based preservatives such as biocides composed of aryl- and alkyl-arsenicals in wood production (Cullen and Reimer 1989). Organic arsenic species, however, are not as of great toxicological relevance as inorganic forms due to their lower toxicity, where some species are regarded as non-toxic (Machado et al. 2006). Since all arsenic species are not equal in terms of their toxicity, studies evaluating the total arsenic concentration would not be able to accurately reflect upon the bioavailability and toxicity of airborne arsenic (Chakraborti et al. 2013). As such, it is important to have an analytical method which can accurately differentiate between the various chemical forms of arsenic in air.
Lichens are effective biomonitors of air pollution and have been integrated into a number of regional and national air pollution surveys where several arsenic species have been found to be present in the thallus (Machado et al. 2006). There is some uncertainty about the appropriateness of using lichens as biomonitors of As species in the air due to their ability to metabolize and methylate the various chemical forms (Farinha et al. 2004; Mrak et al. 2008). On the other hand, the ability of lichens to methylate As compounds may in fact prove useful in biomonitoring studies in terms of understanding temporal variations in exposure to As; therefore, speciation of As in lichens is worthy of further investigation.
Given the great number of arsenic species present in nature, sufficient resolution of peaks can be difficult to attain in chromatographic applications (Dembitsky and Rezanka 2003; Quaghebeur and Rengel 2005). Moreover, there are a considerable number of factors which can affect the equilibrium between species, including improper sample handling and storage, the introduction of acidic or basic solvents during extraction, and other factors (Kroukamp et al. 2016). It is therefore essential that the methods used in sampling, extraction, and separation ensure that the integrity of the original chemical species remains intact so that meaningful information can be gathered.
There is currently no consensus regarding the best extraction solvent for the various species of arsenic from plant and plant-like materials, although many studies use methanol:deionized water (MeOH:DIW; 1:1) which has proven to be effective in extracting organo-arsenic compounds (Koch et al. 1999; Zhang et al. 2002; Machado et al. 2006; Bergqvist et al. 2014). Other studies, although few in number, use dilute HNO3, such as that used on signalgrass (Brachiaria brizantha) (Amaral et al. 2014), and some recent studies such as those conducted on a hyperaccumulating fern (Pteris vittata), rice seedlings, and tobacco leaf have employed the use of ethanol:DIW (EtOH:DIW; 1:1) (Zhao et al. 2015). Kuehnelt et al. (2000) found that DIW extracted As more efficiently than the more commonly employed MeOH:DIW, where these findings were attributed to the greater extraction of inorganic forms of As and arsenoribose. Despite these findings, summed extraction efficiencies of the various identified As species using these methods are often relatively poor. In the study by Kuehnelt et al. (2000) for example, only 7–25% extraction yield was achieved for fruticose lichens Alectoria ochroleuca and Usnea articulata, respectively, and were based upon the separation and quantification of 12 arseno compounds.
There is also no standardization when it comes to how these species are extracted, where ultrasonic baths (Farinha et al. 2004), mechanical shakers (Machado et al. 2006), and microwave techniques (Quaghebeur et al. 2003) have all been employed. The sample mass-to-volume of extractant matrix has also not been extensively investigated, where masses are often large (0.2–1 g) and the volume of the solvent can vary greatly (15–50 mL/g) (Koch et al. 1999; Kuehnelt et al. 2000; Farinha et al. 2004; Machado et al. 2006; Mrak et al. 2006, 2008; Farinha et al. 2009). The effective separation of arsenic species in plant and plant-like materials is also a challenge, where co-elution of species is commonplace in chromatographic analyses (Kuehnelt et al. 2000; Mrak et al. 2006), the most common of which is the co-elution of arsenobetaine (AsB) and arsenite (As III) when using isocratic anion exchange methods. This issue has up until now been addressed by using different analytical columns and mobile phases, which could introduce additional analytical challenges, or by including cation exchange chromatographic methods, thereby increasing analysis time and cost. Gradient methods have also been used (Watts et al. 2008; Alava et al. 2012), but are often not the method of choice due to difficulties in controlling species interconversions, changes in the chromatographic baseline, hidden peaks due to the solvent gradient, and long re-equilibration times. Since As(III) is often of key analytical interest due to its high toxicity, the baseline separation of As(III) from AsB using a single analytical run and an isocratic method would be highly advantageous.
The critical assessment of these methods in the evaluation of ultra-trace concentrations of arsenic species, such as often found in lichen biomonitors, is crucial to understanding atmospheric exposure levels and the relative toxicity of arseno compounds in the air. Lichens have been extensively used in monitoring arsenic air pollution; however, studies are often limited to total As analysis (Mrak et al. 2007; Pisani et al. 2011). Few speciation studies characterizing As in lichens are available (Koch et al. 1999; Farinha et al. 2004, 2009; Machado et al. 2006; Mrak et al. 2008) and so methods appraising this are still an area requiring further exploration.
In this work, the optimized extraction of inorganic forms of As (As(III) and As(V)) and the subsequent separation and semi-quantification of five common arsenic species in the lichen biomonitor, Parmotrema austrosinense (Zahlbr.) Hale, using high-pressure liquid chromatography–inductively coupled plasma mass spectrometry (HPLC-ICP-MS) was investigated. The extraction parameters were qualitatively optimized for As(III) and As(V) due to their environmental relevance, high toxicity, and species prevalence in atmospheric dusts (Machado et al. 2006). Consequently, less abundant forms of As such as arsenosugars and cationic species were not of interest and were therefore outside the scope of this study. The development of an isocratic anion-exchange chromatographic method which could baseline separate and semi-quantify AsB, As(III), monomethylarsonic acid (MMA), dimethylarsinic acid (DMA), and As(V) was also investigated. Organic As species, MMA, and DMA were evaluated as they are the most common toxic organic forms of arsenic found in lichen, fungi, and algae (Koch et al. 1999; Machado et al. 2006) where MMA is more toxic than DMA (Bissen and Frimmel 2000). Arsenobetaine was evaluated to ensure that co-elution with As(III) was avoided, which would otherwise skew the analytical results for this target inorganic analyte.
The optimized method was thereafter applied to two sampling sites within South Africa to evaluate the species of arsenic present in lichens at an urban and rural site. The intention of this was to demonstrate the appropriateness of the proposed method in the application to lichens as biomonitors of air pollution, where observed differences could be tentatively linked to site impacts. Such a study involving the characterization of arsenic species in the South African lichen biomonitor, P. austrosinense has not been published to date. The method developed in this study is one of the few methods which can demonstrate relatively high extraction yields for the semi-quantitative data gained using simple solvents and laboratory equipment. It is also the only published method, which the authors are aware of, which has been able to baseline resolve As(III) from AsB using an isocratic method on a PRP X-100 anion exchange column. Furthermore, a relatively simple and inexpensive mobile phase, without a large waste footprint, makes the proposed method a cost-effective and eco-friendly option.
Materials and methods
Sample collection
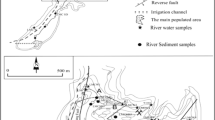
Bulk lichen, P. austrosinense, was collected for initial method development from the Johannesburg Botanical Gardens (JHB), South Africa (GPS coordinates, 26.159875 S, 27.99346 E) which is within 10 km of 6 major hospitals, 3 crematoria, and the city center. It is also between 15 and 40 km of two major gold mines. Moreover, many residential households burn fossil fuels for cooking and heating purposes. This site was chosen as the impacted urban site for this study since As is often associated with the burning of fossil fuels, mining of gold-bearing minerals, and crematoriums, the latter of which can lead to highly localized As contamination (USEPA 1998; Chakraborti et al. 2013). Samples were collected in the same manner as has been done in previous work (Kroukamp et al. 2017), being 100 m away from the main road, between 2 and 4 m above ground level and from all around the tree to prevent bias due to car emissions, soil contamination, and the prevalent wind direction, respectively (Monaci et al. 2012). As done previously (Kroukamp et al. 2017), a large representative sample of lichen material was collected using plastic forceps and care was taken to prevent extraneous (i.e., non-lichen materials such as bark) material from being stored with the sample. Samples were then stored in acid-washed polypropylene bottles.
Upon returning to the laboratory, the samples were further cleaned of any remaining substratum under a magnifying lamp and gently tapped to remove small particles of extraneous material resulting from the cleaning procedure. Since As in the atmosphere is considered to be completely contained within the troposphere and solely present in the particulate form (Farinha et al. 2004), the samples were not washed as this would remove particulate As which had been deposited on the surface of the lichens from the air. This approach supports that of Frati et al. (2005) who found that washing procedures had an effect on the metal content in lichens. Nitrile-gloved hands were used to shred the sample into small pieces to improve the homogeneity of the bulk material, and fractions from 1 to 4.699 mm were collected from an Endecott sieve for further processing. The whole mass of the cleaned sample which was used for further analysis was 10 g. The sample was not freeze-dried, oven-dried, or frozen in liquid nitrogen since a past study found that these procedures may liberate volatile elements, such as As, from the sample matrix (Kroukamp et al. 2017). Instead, samples used for method development were air-dried and stored in acid-washed polypropylene vials in a cool dark place until processing. On the day of the analysis, the lichen samples were ground using a porcelain pestle and mortar and sieved through an Endecott 420-μm sieve to ensure a homogenous particle distribution for extraction. The initial stock samples used for method development were stored for a total of 6 months from the beginning to the end of the optimization process.
Once evaluated, the optimized extraction was applied to freshly collected P. austrosinense lichen samples from the urban site, JHB (as described above), and from a rural site in the Waterberg Mountain area, South Africa (GPS coordinates, 24.4880278 S, 27.8137778 E), where potential sources of pollution are primarily in the form of agriculture and livestock farming. Samples were collected in the same manner as described earlier.
Instrumentation
All analyses took place using a NexION® 300X ICP-MS (PerkinElmer Inc. Shelton, CT). For chromatographic applications, the ICP-MS was coupled to a Flexar™ HPLC with Chromera™ software. In order to ensure that the system was functioning properly prior to use, daily performance checks, including nebulizer gas flow and torch alignment, were done prior to analysis where the instrument was optimized for maximum sensitivity of In with robust plasma conditions such as oxides (CeO/Ce) < 2.5% and doubly charged ions (Ce++/Ce) < 3%. After having met the daily performance requirements, the following parameters were optimized on the ICP-MS to ensure that the As signal was maximized: RF power, torch alignment, nebulizer gas flow, plasma gas flow, auxiliary gas flow, and torch sampling depth. The torch alignment and nebulizer gas flows were subject to daily changes, as is normal for ICP-MS, and will therefore not be mentioned; however, the other optimized parameters for the study are shown in Table S1 (Supplementary Material).
All calibration standards and samples were prepared on the day of analysis. As part of the method development for the analysis of As species in lichens, m/z 75 and 77 were monitored to check for the presence of 75ArCl+ interference on 75As+. Since no ArCl+ could be detected, the lichen samples were analyzed in standard mode. The column was regenerated after every 60 samples using mildly acidified MeOH and a blank was run after every sample to ensure that there were no memory effects or further species eluting from the column.
Optimization of extraction of arsenic from lichens
Preliminary investigations into the elution times of As(III) and As(V)
Standards (100 ppb) of As(III) and As(V) were prepared from stock solutions (1000 mg/L, Inorganic Ventures). Standards were analyzed in triplicate, both independently and as a mixture, using the system described in Tables S1 and S2 (Supplementary Material) to determine the elution times of As(III) and As(V).
Choice of extraction technique and variation of injection volume
To evaluate the most appropriate extraction technique (hot plate/ultrasonic bath) and injection volume for the lichen samples, 0.01 g of homogenous pulverized sample was accurately weighed on an analytical balance (XP205, Mettler Toledo) into centrifuge tubes (Greiner Bio-one), extracted in triplicate using either 5 mL of MeOH:DIW (1:1, AR methanol, MilliQ 18 MΩ/cm) or 5 mL of DIW for 1 h using either an ultrasonic bath (Integral systems) or a magnetic stirrer (FMH instruments, speed 5, room temperature, micromagnetic Teflon-coated stirrer bars), and analyzed with the instrument and parameters described in Tables S1 and S2 (Supplementary Material). In all cases, samples were quantitatively transferred using 4 rinsings of 0.5 mL of water and filtered through pre-wet (using 0.5-mL MeOH) PTFE syringe filters (0.45 μm, Membrane solutions) prior to analysis. Injection volumes of 10, 50, 100, and 120 μL were evaluated to determine which injection volume resulted in the best normalized (according to the individual sample mass) signal-to-noise (S/N) ratios for the As species without resulting in significant peak tailing.
Choice of extraction solvent
The sample to solvent ratio was decreased to improve detection limits, using the optimal injection volume from the previous step. For evaluation purposes, powdered lichen samples (0.05 g) were extracted in triplicate using 1 mL of either deionized water, EtOH:DIW (1:1, AR ethanol), MeOH:DIW (1:1), or 1% HNO3 (65% Suprapur®, Merck in deionized water), respectively. Extractions took place using either an ultrasonic bath or a magnetic stirrer for a period of 1 h (triplicate for each solvent using each of the extraction apparatus). Peak positions for the arsenic species in each solvent were confirmed through spiking of the pure solvent with As(III) and As(V), both individually and as a mixture.
The chromatograms were normalized according to the individual sample mass to ensure that the results were not mass-biased, and the S/N ratios were compared. The method with the best S/N ratio for the inorganic arsenic species (As(III) and As(V)) and which did not result in the interconversion of the arsenic species was selected as the preferred extraction technique.
Mass/volume extraction experiments
The optimized parameters from the previous step were used. Each mass of the pulverized lichen material (0.01 g, 0.025 g, 0.05 g, 0.07 g) was extracted in different volumes of DIW (0.5 mL, 1 mL, 2 mL, 4 mL, 5 mL, 7 mL), using an ultrasonic bath for 1 h.
Chromatograms were normalized according to mass and volume and compared. The results with the best S/N ratio for As(III) and As(V) were chosen for further method development.
Extraction time-dependent study
The optimized parameters from the previous step were used. The sample extraction time using an ultrasonic bath was optimized by evaluating samples in triplicate at extraction times of 5, 10, 20, and 30 min, as well as 1, 2, 3, 5, 10, 15, 20, 24, and 29 h, and the S/N ratio was compared among the various chromatograms. Temperature changes in the ultrasonic bath were monitored to check the dependency of the extraction yield upon the extraction temperature and to determine whether or not there were any observed interconversions of arsenic species, which would indicate temperature dependence.
Chromatograms were compared and the results with the best S/N ratio for As(III) and As(V) chosen for further method development.
Variation of the mobile phase composition to resolve arsenic species
Although As(III) and As(V) are of primary interest due to their toxicity and the fact that they are the most likely forms to be found in airborne dust, this study also included the evaluation of two moderately toxic methylated forms of As, namely MMA and DMA, which have been found in some lichen studies (Machado et al. 2006). Although considered to be non-toxic, AsB was included in the analysis to ensure that the method developed did not allow the co-elution of As(III) with AsB as is commonly found in anion exchange speciation methods.
With the extraction of the most toxicologically relevant arsenic species (As(III) and As(V)) optimized, dilute mixed standards of AsB (Sigma-Aldrich, Fluka ≥ 95%), MMA (prepared from monosodium acid methane arsonate (Sigma-Aldrich, Supelco 99.5%), DMA (prepared from Cacodylic acid, Sigma-Aldrich ≥ 99.0%), As(III), and As(V) were freshly prepared and analyzed with the original mobile phase of 70:30 ammonium nitrate (50 mM, pH 8.6) and DIW (Millipore, MilliQ, 18 MΩ cm-1). Despite being suitable for the initial method development, where the focus was on improving the extraction of As(III) and As(V) from the lichen matrix, it was found that the 70:30 NH4NO3 (50 mM, pH 8.6):DIW solution resulted in the co-elution of AsB and As(III) on the PRP X-100 column and caused salting of the nebulizer and injector when analyzing a large number of samples. To address this issue, other mobile phases such as NH4NO3 (50 mM, pH 8.6):NH4NO3 (80 mM, pH 8.6), NH4HPO4 (8.0 mM, pH 6.2, Fluka TraceSELECT® ≥ 99.999%):NH4NO3 (8.0 mM, pH 6.2), NH4NO3 (50 mM, pH 8.6):(NH4)2CO3 (2 mM, pH 10 with 1% MeOH, Promark Chemicals, AR), and NH4NO3 (50 mM, pH 8.6) in 1% MeOH:1% MeOH (Millipore, MilliQ) were all tested for their suitability for the baseline separation of AsB and As(III) at ratios of A:B 100:0, 90:10, 80:20, 70:30, 60:40, 50:50, 40:60, 30:70, 20:80, 10:90, and 0:100. A gradient method of 2 mM (NH4)2CO3, followed by 40 mM (NH4)2CO3 and followed by 2 mM (NH4)2CO3, as recommended by Alava et al. (2012) was also evaluated for its suitability.
Since lichens contain a number of arsenic species leading to complex chromatograms, the resolution study utilized pure individual and mixed standards of As(III), AsB, DMA, MMA, and As(V). Standards were prepared in deionized water to mimic the extraction solvent used in the extraction of the samples. An argon humidifier was used in all studies to reduce salt buildup at the nebulizer tip, thereby eliminating aspiration issues over a large number of samples.
Analysis of lichen samples
The elution times of the individual As species were identified through the use of freshly prepared standard solutions. The optimized method of 0.07 g of unwashed, air-dried, powdered sample was extracted using 7 mL of H2O (MilliQ) in an ultrasonic bath for 24 h. The ICP-MS conditions shown in Table S1 (Supplementary Material) along with the optimized mobile phase from the previous step were used for further evaluations. To check elution times of the different arsenic species in the lichen matrix, a mixed-species spike of As(III), As(V), MMA, DMA, and AsB was added to the lichen matrix. Ten replicate samples each from the urban and rural sites were prepared as described earlier, and the concentrations of As(III), As(V), MMA, DMA, and AsB were determined. A blank was run after every sample to ensure that there were no further species eluting from the column and to check for memory effects. A method blank was also prepared in the same manner as the samples.
Total As concentration analysis
For total As determinations, powdered lichen samples were prepared as reported previously (Kroukamp et al. 2017) where 0.1 g was weighed in triplicate and digested in a CEM Mars 6 microwave digestion unit using 10 mL of HNO3 (65%, Merck, Suprapur) and 1 mL of H2O2 (30%, Merck, Suprapur) with a ramp time of 20 min to 180 °C and a hold time of 20 min. This digestion method was a modification of the Milestone Application Note for lichen digestions (HPR-FO-55; Milestone 2014) where the volume of HNO3 was increased to 10 mL due to a lower acid volume limit on the Mars 6 microwave digestion system as a result of 100-mL digestion vessels being used. Water was not used to achieve the necessary volume as this would dilute the acid and would likely result in an incomplete sample digestion. Samples were filtered through a quantitative filter paper (Merck, 0.22-μm hardened ashless) and the filtrate was diluted to 50 mL using deionized water.
Quality control (QC) standards for total arsenic analysis included the BCR Reference Material no 482, trace elements in Lichen (Pseudevernia furfuracea), and the Tea Leaf CRM, INCT-TL-1 (Institute of Nuclear Chemistry and Technology), and were prepared in the same manner as the samples.
Samples and the QC standards were analyzed with dilutions of 1:10 and 1:5 and were also analyzed without any dilution where the internal standard, ruthenium, was monitored to determine whether or not there were any significant matrix effects and was also used to compensate for long-term drift. Samples were analyzed on the same day as the dilution and no more than 72 h after the digestion had taken place. Total arsenic concentrations were determined using a PerkinElmer NexION® 300X ICP-MS with a collision gas (He) to manage any polyatomic interferences which may have arisen from acid impurities. Adding a collision gas also had the added benefit of providing collisional focusing of the ion beam thereby improving analyte sensitivity (Tanner et al. 2002).
Data processing
All chromatogram signals from method development were qualitatively evaluated by normalizing according to mass and replotted using Microsoft excel. Data processing of chromatograms for the lichen samples was performed on Chromera Chromatography software. In some cases, the software did not permit the identification of a peak due to software post-processing limitations. In such cases, the results were considered to be outliers and due to this and other factors discussed later, the data is regarded as semi-quantitative.
Results and discussion
Optimization of extraction of arsenic
Preliminary investigations into the elution times of As(III) and As(V) for method development
The purpose of this study was to determine what the elution times were for the toxic, inorganic forms of arsenic (As(III) and As(V)) in a 70:30 50 mM NH4NO3:DIW mobile phase (Table S2, Supplementary Material) using a PRP X-100 column at 0.6 mL/min in the absence of artifacts which could be introduced by the lichen matrix. During the initial method development for this study, As(III) and As(V) were found to elute at approximately 3 min 45 s and 12 min 15 s, respectively.
Choice of extraction technique and variation of injection volume
The extraction method using an ultrasonic bath and an injection volume of 120 μL was found to have the best S/N ratio for As(III) and As(V) when compared with magnetic stirring over the same time period. The increased extraction using ultrasonic bath may be related to the elevated temperature in the ultrasonic bath (42 °C at 1 h) in comparison with the magnetic stirring at ambient temperature. An injection volume of 120 μL was needed in order to achieve good S/N ratios for the As species under study due to the low natural concentrations of these As species in the lichen matrix. Despite the large injection volume, no significant tailing of the As(III) and As(V) peaks was observed. Deionized water showed promise as the extractant of choice as it resulted in the best S/N ratio for the inorganic species (As(III) and As(V)), where MeOH:DIW 1:1 was found to give the best S/N ratio for the other species of arsenic where this is likely due to MeOH facilitating the dissolution of organoarsenicals as further revealed in the next section.
Choice of extraction solvent for further evaluations
Deionized water was found to be the most effective extraction solvent for the inorganic As species since metal arsenites and arsenates are both highly soluble in water (Magalhães 2002), confirming our earlier findings in this study and those of Kuehnelt et al. (2000) who had found that water exhibited the best extraction efficiency in comparison with MeOH:DIW (1:1) in the fruticose lichens, Alectoria ochroleuca and Usnea articulata. The extraction solvent of MeOH:DIW (1:1) provided a good but lower S/N ratio (Table S3, Supplementary Material) of As(III) and As(V) and higher S/N ratio for the unknown As species in the samples, thought to be organic arseno compounds due to the prevalence of organoarsenicals in lichens (Koch et al. 1999; Machado et al. 2006), showing that this solvent is more suited to the extraction of the unidentified organo-arsenic compounds (Fig. 1).

Averaged, mass-normalized HPLC-ICP-MS peak intensities (cps/g) showing the suitability of the various extraction solvents (DIW, MeOH:DIW, EtOH: DIW, 1% HNO3) used in the ultrasonic bath extraction of powdered lichen material over a period of 1 h. Inorganic arsenic species (As(III) and As(V)) and unknown arsenic species (U1–4) are shown. Compounds were separated using a Hamilton® PRP X-100 column (Mobile phase, 70:30 50 mM NH4NO3:DIW, pH 8.6; flow, 0.6 mL/min)
Studies involving the use of EtOH:DIW as an extraction solvent as recommended by Zhao et al. (2015) who analyzed As in the hyperaccumulating fern (Pteris vittata), rice seedlings, and tobacco leaf showed some promise as an appropriate extraction solvent. However, the poor S/N (Table S3, Supplementary Material) and the appearance of an additional peak (U4, Fig. 1) which was not found to be present in any of the other extractions raised caution to the use of this solvent for further method development. It is plausible that these additional peaks resulted from impurities in the ethanol solvent or were due to the improved extraction of an additional arsenic species, although this hypothesis would need to be verified through further experimentation. If an additional As species, or many, are present, this would imply that EtOH is selective to certain arsenicals but poor in its overall recovery of arsenic species as observed by the poor S/N ratios in comparison with the other extraction solvents; however, studies of this type will not be covered in the scope of this study.
Our findings contrasted to those by Amaral et al. (2014) who had recommended the use of 1% nitric acid as an appropriate extraction solvent, as we found that As(V) S/N ratio (Table S3, Supplementary Material) increased dramatically in the presence of 1% HNO3, the As(III) S/N ratio increased slightly, and the organic species which were observed in the other solvents were undetectable (Fig. 1). These results agreed with the finding by Cullen and Reimer (1989), where the use of 1% HNO3 as an extraction solvent compromised the integrity of the various As species in the lichen matrix as it is a strong oxidizer, making it unsuitable as an extraction solvent in such applications. The observed vertical baseline shift between the other extraction solvents and HNO3 (Fig. 1) is likely due to the evaluation taking place after the ICP-MS components (nebulizer, torch, spray chamber) had to be cleaned due to a salt crystallization on the various sample introduction components of the ICP-MS. Conditions between analyses were the same, taking place using the same mobile phase, daily performance parameters, and analyte intensities for the daily tuning solution; therefore, the results are valid. Moreover, decisions regarding the suitability of the solvent were based upon the S/N ratios and the integrity of the various arsenic species and consequently vertical shifts are not of consequence. The observed horizontal shift of As(V) in this matrix (Fig. 1) is likely due to the change in the pH and ionic strength of the solution as peak positions were confirmed through spiking.
Mass/volume extraction experiments
It was found that 0.07 g of lichen sample extracted in 7 mL of DIW in an ultrasonic bath yielded the best S/N ratios for the mass-normalized intensities; therefore, these values were used for further method development.
Effect of extraction time
The triplicate samples were normalized according to mass, averaged for each extraction time, and replotted in Excel. Based upon S/N ratio, an extraction time of 24 h yielded the highest S/N ratio for the inorganic arsenic species of interest. Since the relative abundance of the different peaks remained very much the same over the 24 h extraction period (maximum temperature of ultrasonic bath, 57 °C), the extraction procedure did not appear to affect the integrity of the arsenic species present in the lichen matrix. This confirms the findings of Mrak et al. (2006) who found that at up to 90 °C, the various forms of arsenic remained intact. At temperatures exceeding 90 °C, however, they noted a decrease in the AsB concentration and an increase in trimethylarsine oxide (TMAO).
It can be seen from Figure S1 (Supplementary Material) that an increase in the time of extraction resulted in an increase in temperature in the ultrasonic bath over the first 2 h, thereafter the ultrasonic bath temperature stabilized at 57 °C. As such, the observed improvement in extraction efficiency is in fact most likely dominated by the length of time of extraction and not so much due to the increase in temperature, as suggested in the initial method development in earlier sections of this article. To elaborate, extending the time of extraction would ensure adequate dispersion of the sample slurry through continuous agitation (Blasco et al. 2006), improving sample-to-extraction matrix contact. Moreover, it would expose the sample to a higher number of ultrasonic-generated imploding bubbles which are known to result in high local pressures and temperatures (Kazi et al. 2009). These findings are somewhat different from those of Machado et al. (2006) in Parmelia caperata and Mrak et al. (2006) in Hypogymnia physodes and Cladonia Rei, where an increase in temperature was solely responsible for improved extractability of As from lichens. Although temperature may play a role in improved extraction of the lichen matrix in our study, it is unlikely to be the only causative factor.
Variation of mobile phase composition to resolve arsenic species
None of the tested ratios of mobile phases, NH4NO3 (50 mM, pH 8.6):NH4NO3 (80 mM, pH 8.6) and NH4HPO4 (8.0 mM, pH 6.2):NH4NO3 (8.0 mM, pH 6.2), were able to separate AsB from As(III). The gradient method developed by Alava et al. (2012) showed some degree of separation of these species; however, this mobile phase caused blockages of the nebulizer due to salt buildup after just a few samples. Although a mobile phase of 30:70 NH4NO3 (50 mM, pH 8.6):NH4CO3 (2 mM, pH 10) + 1% MeOH gave a good separation of As species, an elution time of 30 min was required for a complete elution of all of the arsenic species and high throughput was limited by blockages and salt formation in the nebulizer and injector after just a few samples.
It was found that the addition of MeOH (1%) to the DIW in 70:30 NH4NO3 (50 mM, pH 8.6):DIW resulted in a better separation of AsB and As(III). Further optimizations involved the adjustment of this ratio, where an end mobile phase composition of 17.5 mmol NH4NO3 in 1% MeOH at 0.6 mL/min resulted in the complete baseline resolution between all five target arsenic species (Fig. 2). This is the only published method that the authors are aware of which is able to baseline resolved these five arsenic species using an isocratic method on a PRP X-100 anion exchange column. Attempts were made to improve the method elution times by increasing the mobile phase flow rate to 1 mL/min. Although this improved the sharpness of the MMA peak, AsB and As(III) were no longer baseline resolved and so the flow rates were returned to 0.6 mL/min.

The complete separation of five arsenic species (AsB, As(III), DMA, MMA, and As(V)) in deionized water in 25 min, with a mobile phase of 17.5 mmol NH4NO3 in 1% MeOH using a PRP X-100 anion exchange column (4.6 × 150 mm, 5 μm) by HPLC-ICP-MS
Analysis of lichen samples
During initial method validation using the lichen extracts, elution times of the AsB, As(III), and DMA were the same as those observed in pure standards, as confirmed through spiking. Spikes of MMA and As(V) eluted slightly earlier in comparison with spikes in water and are likely due to the change of matrix, although the change could be effectively dealt with by expanding the peak search window. In samples spiked with a mixture of the five target arsenic species, peak positions were similar to those observed in the single spikes of each species into the lichen matrix. The method blank (Fig. S2, Supplementary Material) was not found to contribute to the As baseline when compared to a blank which had not been through the sample preparation procedure.
Initial studies involving the bulk lichen material from the urban site showed a change in species over an extended storage time. At the start of method development, the S/N ratios were highest for the inorganic arsenic species and by the end of method development, S/N ratios were highest for the organic forms of arsenic. The storage of shredded (but not pulverized) lichens for a period of 6 months in a cool dark place could have provided the lichens with the time and conditions needed to metabolize and methylate the inorganic forms of As initially found to be present in the sample. This finding confirms the hypotheses of Farinha et al. (2004) and Mrak et al. (2008) that lichens are actively involved in the metabolism and biotransformation of arsenic species. Although some may consider the biotransformation of arsenic by a biomonitor as non-beneficial, such a process can be useful in providing insight into how recent the impacts are, allowing changes in arsenic contamination within an environment as a result of air pollution to be determined.
Due to these effects, a new bulk lichen material sample was collected from each site for the final method assessments. Here, the elution times for MMA and As(V) were found to have increased to 21 and 43 min, respectively, as a consequence of column degradation over time. Despite this change in elution times, the separation was proven to remain unaffected and the results allowed for an understanding of arsenic speciation in lichens. As such, elution times for all species were re-evaluated in water and the lichen matrix, and the optimized method run accordingly. Correlation coefficients for AsB, As(III), DMA, MMA, and As(V) (n = 5 + blank) were 0.99999, 0.99990, 0.99998, 0.99999, and 0.99995, respectively. As can be seen in Fig. 3, the repeatability of the developed method was excellent.

Mass-normalized intensities (cps/g) of As species found in freshly collected lichen, P. austrosinense, from the urban site, JHB, as determined by HPLC-ICP-MS. Results show 10 replicate analyses of lichen samples from this site which took place using a DIW extraction carried out in an ultrasonic bath over 24 h, separated on a Hamilton PRP X-100 column (mobile phase, 17.5 mmol NH4NO3 in 1% MeOH, pH 8.6; flow, 0.6 mL/min; injection volume, 120 μL). Five arsenic species (AsB, As(III), DMA, MMA, and As(V)), as well as unknown arsenic species (U1–3), are shown
It can be seen from Figs. 3 and 4 that AsB and As(III) were no longer baseline resolved, likely due to the elution of an additional As species from the lichen, P austrosinense (Fig. 4, U1), just after AsB. Despite this, the most toxicologically relevant form of As, As(III), was still sufficiently resolved for semi-quantitation. Figure 3 shows that there were two additional unidentified As species in the urban site (U2 and U3) in comparison with the rural site (Fig. 5). These are likely to be other organo-arsenic species or arsenosugars as has been found in studies by Koch et al. (1999) and may have resulted from different sources of pollution such as the application of pesticides in this park or nearby industrial activities, the volatile metabolic products of different microbes in surrounding soils (Huang et al. 2011), meteorological factors, or lichen metabolic processes. Since the same lichen species was used throughout the study, the latter scenario is less likely. Concentrations of As(III) and As(V) were lower in the rural site in comparison with those in the urban site, indicating differences in ambient air quality (Table 1). Arsenobetaine +U1 were found to be in high abundance in lichens from both sites, although concentrations for the urban site were higher than in the rural site. Since lichens are the product of a symbiotic relationship between algae and fungi (Forbes et al. 2015), it is highly likely that AsB in these samples had arisen from the fungal component, where many fungi are known to possess high concentrations of this arseno compound (Nearing et al. 2014a, b). Based upon the semi-quantitative results, the summative As concentration of freshly collected lichens from the urban and rural sites were 1.22 ± 0.50 μg/g and 0.74 ± 0.06 μg/g, respectively. In order of abundance, the various arsenic species in lichens from the urban site were As(V) > As(III) > AsB+U1 > DMA > MMA and As(V) > AsB+U1 > As(III) > DMA > MMA for the rural site, where MMA was present in extremely low, non-quantifiable concentrations in lichens from both sites.

Magnification of chromatogram (Fig. 3, 2–5 min) showing extracted AsB and As(III) in lichens from an urban impacted site as determined by HPLC-ICP-MS. The dotted line shows the hypothetical elution time and peak shape for the unknown arsenic species eluting just after AsB which prevents the complete baseline separation of AsB from As(III)

Mass-normalized intensities (cps/g) of As species found in freshly collected lichen, P. austrosinense, from the rural site, Waterberg, as determined by HPLC-ICP-MS. Results show 10 replicate analyses of lichen samples from this site which took place using a DIW extraction carried out in an ultrasonic bath over 24 h, separated on a Hamilton PRP X-100 column (mobile phase, 17.5 mmol NH4NO3 in 1% MeOH, pH 8.6; flow, 0.6 mL/min; injection volume, 120 μL). Five arsenic species (AsB, As(III), DMA, MMA, and As(V)), as well as an unknown arsenic species (U1), are shown
Total arsenic analysis and recoveries from speciation analysis
A dilution factor of five was found to be most appropriate for the lichen samples. Total recoveries for the lichen and tea leaf samples were 104 and 121% for the lichen and tea leaf CRMs respectively and were within the 95% confidence interval stipulated on the CRM certificate. The internal standard recoveries for all samples were well within the limits of 70—130% set by the USEPA (Keith 1996).
Since arsenic may enter the environment from gold mining (Villaescusa and Bollinger 2008; Chakraborti et al. 2013), crematoriums, medical waste incineration (USEPA 1998), burning of fossil fuels (Garelick et al. 2005), and smelting activities (Crecelius et al. 1974), it can be expected that urban sites, such as JHB, would exhibit higher concentrations of arsenic than rural site which does not have these activities. This hypothesis agrees with the findings from our study where the total concentrations of As were higher in samples from the urban site (total, 6.43 ± 0.25 μg/g) than in those from the rural site (1.87 ± 0.05 μg/g). The burning of fossil fuels, such as coal, and the burning of pressure-treated wood for the cooking of food and heat generation are believed to be the major contributors to arsenic concentrations at the urban site. Cremation services and gold mining are also likely sources of As air pollution at this site, although probably minor contributors. Sources of As at the rural site are likely to be predominantly natural in origin, although neighboring farming activities may also be a contributing factor.
The sum of the water-extractable concentrations of the monitored As species was 19% and 40% of the total arsenic concentration in lichens from the urban and rural sites, respectively (Table 1). The difference in the extraction yields between sites can partly be contributed to the higher percentage of AsV (88% of the summed species concentrations) in lichens from the rural site in comparison with that in the urban site (81% of the summed species concentrations) which will cause higher yields since As(V) is water-soluble. The rest of the difference could be due to the high RSDs for AsB+U1 from the rural site resulting from the apparent higher concentration of the unknown interfering As species eluting on the tail end of AsB in lichen samples from this site. Since only five As species were under investigation in the present study, the extraction yields are an improvement on those reported by Kuehnelt et al. (2000) who evaluated 12 As species in the fruticose lichens, Alectoria ochroleuca and Usnea articulata, with yields of 7% and 25%, respectively. Koch et al. (1999) reported high extraction yields of As from the lichens, Bryoria sp. and Alectoria sp.; however, most reported values were below the LOD and LOQ, and the actual measurable recoveries accounted for only 0.625–10% of the total arsenic species.
In agreement with studies by Koch et al. (1999), Machado et al. (2006), and Farinha et al. (2009), we found that the As(III) and As(V) were the dominant As species in the freshly collected lichen, P. austrosinense, from the urban impacted site of our study. For the rural site, As(V) was the dominant arsenic species; however, As(III) was found in low concentrations in lichens from this site pointing to a different source of As contamination. In agreement with the findings by Farinha et al. (2009) and Koch et al. (1999), we too found arsenate in all of our samples and this was considered to be the main arsenic species. Our results contrasted to those of Machado et al. (2006) who found that arsenite was the most abundant As species at their sites, where the order of abundance of As species in lichens from a background site of their study was As (III) > As(V) > DMA > MMA and As(III) = As(V) > DMA > MMA in discontinuously exposed transplanted lichens which had been exposed to pollution for a period of 2 months. This difference could be due to a different lichen metabolic processes or rates, or different pollutant sources, as the pollution sources for the study by Machado et al. (2006) were a coal-fired power plant and industrial area, respectively. The higher abundance of As(III) observed in their study is counter to what one would expect, given that the main emission by-product from coal combustion has been found to be As(V) (Goodarzi and Huggins 2005; Shah et al. 2007). As such, the differences between two lichen species metabolisms could be a major factor affecting the As species observed, where the study by Machado et al. (2006) used the foliose lichen Parmelia caperata. Moreover, the length of storage time before processing could also be a factor since As(V) usually converts to As(III) before it is methylated (Challenger 1945; Cullen 2014), although it is hard to draw conclusions regarding this, since the storage period was not mentioned in their study. It is also plausible that this lichen species may be able to convert As(V) to As(III) more readily than P. austrosinense following the mechanism proposed by the Challenger pathway (Challenger 1945; Cullen 2014).
Conclusions
This study has shown that the total concentrations of As were higher in lichen samples from the urban site (total, 6.43 ± 0.25 μg/g) than those from the rural site (1.87 ± 0.05 μg/g). In the method development related to the extraction of inorganic As species, the ultrasonic extraction of pulverized lichen material over a period of 24 h using deionized water resulted in the highest S/N ratio for the As(III) and As(V) species present. Optimized chromatographic parameters included an injection volume of 120 μL and a mobile phase of 17.5 mmol NH4NO3 in 1% MeOH, pH 8.6 at 0.6 mL/min which resulted in the complete baseline resolution of five target As species, namely AsB, As(III), DMA, MMA, and As(V), using a PRP X-100 anion exchange column. This study confirmed that lichens methylate inorganic arsenic species over an extended storage time. As such, if a direct comparison of the lichen with the environment is needed, fresh samples should be collected, where metabolites can help to elucidate information regarding how recent the impact is. Since lichens have a complex array of arsenic species present in their thallus, it is possible that additional As species may elute shortly after AsB which could affect the baseline separation of AsB and As(III). Nevertheless, meaningful semi-quantitative toxicological information can still be gathered in a relatively short time. Based upon the semi-quantitative results, the summative As concentrations of freshly collected lichens from the urban and rural sites in this preliminary study were 1.22 ± 0.50 μg/g and 0.74 ± 0.06 μg/g, respectively. Consequently, the extraction efficiency of As species from the lichen using the proposed method was 19% and 40% of the total arsenic concentration in lichens from the urban and rural sites, respectively, demonstrating an improved extraction efficiency in comparison to other published methods. It should be noted that more samples would need to be collected in future studies and other factors affecting speciation, such as meteorological data, should be included to further substantiate these results. In order of abundance, the various arsenic species in lichens from the urban site were As(V) > As(III) > AsB > DMA > MMA and As(V) > AsB > As(III) > DMA > MMA for the rural site, where MMA was present in extremely low, non-quantifiable concentrations in lichens from both sites. These differences in the speciation patterns between an urban and rural site were likely reflective of the different sources. Future studies could involve the adjustment of pH to determine whether or not a lower pH could help resolve AsB and As(III); however, care should be taken to ensure that this does not promote the interconversion of As species.
References
Alava P, Tack F, Du Laing G, Van de Wiele T (2012) HPLC-ICP-MS method development to monitor arsenic speciation changes by human gut microbiota. Biomed Chromatogr 26:524–533. https://doi.org/10.1002/bmc.1700
Amaral CDB, Nóbrega JA, Nogueira ARA (2014) Investigation of arsenic species stability by HPLC-ICP-MS in plants stored under different conditions for 12 months. Microchem J 117:122–126. https://doi.org/10.1016/j.microc.2014.06.018
Bentley R, Chasteen TG (2002) Microbial methylation of metalloids: arsenic, antimony, and bismuth. Microbiol Mol Biol Rev 66:250–271. https://doi.org/10.1128/MMBR.66.2.250-271.2002
Bergqvist C, Herbert R, Persson I, Greger M (2014) Plants influence on arsenic availability and speciation in the rhizosphere, roots and shoots of three different vegetables. Environ Pollut 184:540–546. https://doi.org/10.1016/j.envpol.2013.10.003
Bissen M, Frimmel FH (2000) Speciation of As (III), As (V), MMA and DMA in contaminated soil extracts by HPLC-ICP-MS. Fresenius J Anal Chem 367:51–55. https://doi.org/10.1007/s002160051597
Blasco M, Domeño C, Nerin C (2006) Use of lichens as pollution biomonitors in remote areas: comparison of PAHs extracted from lichens and atmospheric particles sampled in and around the Somport Tunnel ( Pyrenees ). Environ Sci Technol 40:6384–6391. https://doi.org/10.1021/es0601484
Chakraborti D, Rahman MM, Murrill M, Das R, Siddayya S, Patil S et al (2013) Environmental arsenic contamination and its health effects in a historic gold mining area of the Mangalur greenstone belt of northeastern Karnataka, India. J Hazard Mater 262:1048–1055. https://doi.org/10.1016/j.jhazmat.2012.10.002
Challenger F (1945) Biological methylation. Chem Rev 36:315–361. https://doi.org/10.1021/cr60115a003
Chung J-Y, Yu S-D, Hong Y-S (2014) Environmental source of arsenic exposure. J Prev Med Public Health 47:253–257. https://doi.org/10.3961/jpmph.14.036
Crecelius EA, Johnson CJ, Hofer GC (1974) Contamination of soils near a copper smelter by arsenic, antimony and lead. Water Air Soil Pollut 3:337–342. https://doi.org/10.1007/BF00226464
Cullen WR (2014) Chemical mechanism of arsenic biomethylation. Chem Res Toxicol 27:457–461. https://doi.org/10.1021/tx400441h
Cullen WR, Reimer KJ (1989) Arsenic speciation in the environment. Chem Rev 89:713–764. https://doi.org/10.1021/cr00094a002
Dembitsky VM, Rezanka T (2003) Natural occurance of arseno compounds in plants, lichens, fungi, algal species, and microorganisms. Plant Sci 165:1177–1192. https://doi.org/10.1016/j.plantsci.2003.08.007
Farinha MM, Šlejkovec Z, van Elteren JT, Wolterbeek HT, Freitas MC (2004) Arsenic speciation in lichens and in coarse and fine airborne particulate matter by HPLC – UV – HG – AFS. J Atmos Chem 49:343–353. https://doi.org/10.1007/s10874-004-1248-1
Farinha MM, Freitas MC, Šlejkovec Z, Wolterbeek HT (2009) Arsenic speciation in Portuguese in situ lichen samples. Appl Radiat Isot 67:2123–2127. https://doi.org/10.1016/j.apradiso.2009.04.018
Forbes PBC, van der Wat L, Kroukamp EM (2015) Chapter 3: Biomonitors, in Comprehensive Analytical Chemistry vol. 70: Monitoring of Air Pollutants: Sampling, Sample Preparation and Analytical Techniques, Patricia Forbes (ed). 53-108, Elsevier, Netherlands. https://doi.org/10.1016/bs.coac.2015.09.003
Frati L, Brunialti G, Loppi S (2005) Problems related to lichen transplants to monitor trace element deposition in repeated surveys: a case study from central Italy. J Atmos Chem 52:221–230. https://doi.org/10.1007/s10874-005-3483-5
Garelick H, Dybowska A, Valsami-Jones E, Priest ND (2005) Remediation technologies for arsenic contaminated drinking waters. J Soils Sediments 5:182–190. https://doi.org/10.1065/jss2005.06.140
Goodarzi F, Huggins FE (2005) Speciation of arsenic in feed coals and their ash byproducts from canadian power plants burning sub-bituminous and bituminous coals. Energy Fuel 19:905–915. https://doi.org/10.1021/ef049738i
Huang JH, Hu KN, Decker B (2011) Organic arsenic in the soil environment: speciation, occurrence, transformation, and adsorption behaviour. Water Air Soil Pollut 219:401–415. https://doi.org/10.1007/s11270-010-0716-2
Johnson DL, Braman RS (1975) Alkyl- and inorganic arsenic in air samples. Chemosphere 6:333–338. https://doi.org/10.1016/0045-6535(75)90027-2
Kazi TG, Jalbani N, Arain MB, Jamali MK, Afridi HI, Shah AQ (2009) Determination of toxic elements in different brands of cigarette by atomic absorption spectrometry using ultrasonic assisted acid digestion. Environ Monit Assess 154:155–167. https://doi.org/10.1007/s10661-008-0386-3
Keith LH (1996) Compilations of EPA’s sampling and analysis methods, 2nd edn. CRC Press, Florida
Koch I, Feldmann J, Wang L, Andrewes P, Reimer KJ, Cullen WR (1999) Arsenic in the Meager Creek hot springs environment, British Columbia, Canada. Sci Total Environ 236:101–117. https://doi.org/10.1016/S0048-9697(99)00273-9
Kroukamp EM, Wondimu TW, Forbes PBC (2016) Metal and metalloid speciation in plants: overview, instrumentation, approaches and commonly assessed elements. Trends Anal Chem 77:87–99. https://doi.org/10.1016/j.trac.2015.10.007
Kroukamp EM, Godeto TW, Forbes PBC (2017) Comparison of sample preparation procedures on metal(loid) fractionation patterns in lichens. Environ Monit Assess 189:451. https://doi.org/10.1007/s10661-017-6155-4
Kuehnelt D, Lintschinger J, Goessler W (2000) Arsenic compounds in terrestrial organisms. IV. Green plants and lichens from an old arsenic smelter site in Austria. Appl Organomet Chem 14:411–420. https://doi.org/10.1002/1099-0739(200008)14:8%3C411::AID-AOC24%3E3.0.CO;2-M
Machado A, Šlejkovec Z, van Elteren JT, Freitas MC, Baptista MS (2006) Arsenic speciation in transplanted lichens and tree bark in the framework of a biomonitoring scenario. J Atmos Chem 53:237–249. https://doi.org/10.1007/s10874-006-9013-2
Magalhães MCF (2002) Arsenic. An environmental problem limited by solubility. Pure Appl Chem 74:1843–1850. https://doi.org/10.1351/pac200274101843
Milestone (2014) Milestone application note HPR-FO-55, Lichen, Rev 05_11. www.milestonesrl.com. Accessed 30 February 2017
Monaci F, Fantozzi F, Figueroa R, Parra O, Bargagli R (2012) Baseline element composition of foliose and fruticose lichens along the steep climatic gradient of SW Patagonia (Aisén Region, Chile). J Environ Monit 14:2309–2316. https://doi.org/10.1039/c2em30246b
Mrak T, Šlejkovec Z, Jeran Z (2006) Extraction of arsenic compounds from lichens. Talanta 69:251–258. https://doi.org/10.1016/j.talanta.2005.10.011
Mrak T, Simčič J, Pelicon P, Jeran Z, Reis MA, Pinheiro T (2007) Use of micro-PIXE in the study of arsenate uptake in lichens and its influence on element distribution and concentrations. Nucl Instrum Methods Phys Res Sect B 260:245–253. https://doi.org/10.1016/j.nimb.2007.02.029
Mrak T, Šlejkovec Z, Jeran Z, Jaćimović R, Kastelec D (2008) Uptake and biotransformation of arsenate in the lichen Hypogymnia physodes (L.) Nyl. Environ Pollut 151:300–307. https://doi.org/10.1016/j.envpol.2007.06.011
Nearing MM, Koch I, Reimer KJ (2014a) Arsenic speciation in edible mushrooms. Environ Sci Technol 48:14203–14210. https://doi.org/10.1021/es5038468
Nearing MM, Koch I, Reimer KJ (2014b) Complementary arsenic speciation methods: a review. Spectrochim Acta B At Spectrosc 99:150–162. https://doi.org/10.1016/j.sab.2014.07.001
Pisani T, Munzi S, Paoli L, Bačkor M, Loppi S (2011) Physiological effects of arsenic in the lichen Xanthoria parietina (L.) Th. Fr. Chemosphere 82:963–969. https://doi.org/10.1016/j.chemosphere.2010.10.079
Quaghebeur M, Rengel Z (2005) Arsenic speciation governs arsenic uptake and transport in terrestrial plants. Microchim Acta 151:141–152. https://doi.org/10.1007/s00604-005-0394-8
Quaghebeur M, Rengel Z, Smirk M (2003) Arsenic speciation in terrestrial plant material using microwave-assisted extraction, ion chromatography and inductively coupled plasma mass spectrometry. J Anal At Spectrom 18:128–134. https://doi.org/10.1039/b210744a
Shah P, Strezov V, Stevanov C, Nelson PF (2007) Speciation of arsenic and selenium in coal combustion products. Energy Fuel 21:506–512. https://doi.org/10.1021/ef0604083
Tanner SD, Baranov VI, Bandura DR (2002) Reaction cells and collision cells for ICP-MS: a tutorial review. Spectrochim Acta B At Spectrosc 57:1361–1452. https://doi.org/10.1016/S0584-8547(02)00069-1
USEPA (1998) Locating and estimating air emissions from sources of arsenic and arsenic compounds, EPA-454/R-98-013. North Carolina. Available from: https://www3.epa.gov/ttnchie1/le/arsenic.pdf
Villaescusa I, Bollinger J-C (2008) Arsenic in drinking water: sources, occurrence and health effects (a review). Rev Environ Sci Biotechnol 7:307–323. https://doi.org/10.1007/s11157-008-9138-7
Watts MJ, Button M, Brewer TS, Jenkin GRT, Harrington CF (2008) Quantitative arsenic speciation in two species of earthworms from a former mine site. J Environ Monit 10:753–759. https://doi.org/10.1039/b800567b
Zhang W, Cai Y, Tu C, Ma LQ (2002) Arsenic speciation and distribution in an arsenic hyperaccumulating plant. Sci Total Environ 300:167–177. https://doi.org/10.1016/S0048-9697(02)00165-1
Zhao D, Li HB, Xu JY, Luo J, Ma LQ (2015) Arsenic extraction and speciation in plants: method comparison and development. Sci Total Environ 523:138–145. https://doi.org/10.1016/j.scitotenv.2015.03.051
Acknowledgments
The authors would like to thank Johannesburg City Parks and Zoo and the Burri family for allowing sampling to take place on their premises. Special thanks are also given to the following people from PerkinElmer Inc. for their support of this study: Dr. Kenneth Neubauer for his guidance on HPLC-ICP-MS methods; Dr. Mark Upton for training; and Lisa Koorts, Chris de Jager, and Dr.Fadi Abou-Shakra for arranging a loan instrument. Further thanks are given to Rufaro Bhero for his assistance in the preparation of the organic arsenic stock solutions.
Funding
The financial assistance of the University of Pretoria, University of Johannesburg, and the Department of Higher Education and Training towards this research is hereby acknowledged.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Disclaimer
The opinions expressed and conclusions arrived at are those of the authors and are not necessarily to be attributed to these Universities.
Additional information
Responsible editor: Severine Le Faucheur
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Electronic supplementary material
ESM 1
(DOCX 5797 kb)
Rights and permissions
About this article

Cite this article
Kroukamp, E.M., Godeto, T.W. & Forbes, P.B.C. Optimized extraction of inorganic arsenic species from a foliose lichen biomonitor. Environ Sci Pollut Res 26, 29896–29907 (2019). https://doi.org/10.1007/s11356-019-06073-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-019-06073-2