
Abstract
The soil microbial fuel cell (MFC) is a promising biotechnology for the bioelectricity recovery as well as the remediation soil. Moreover, there were no studies on the heavy metal pollution in a soil MFC yet. A soil MFC was constructed to remediate the contaminated soil, and the electric field was generated from the oxidation of the acetate at the anode. We demonstrated the copper migration, the power generation, and the pH variation in the soil and the electrodes. The maximal voltage and the power density of 539 mV and 65.77 m W/m2 were obtained in the soil MFC. The chemical fractionation of copper (Cu) was analyzed with a modified BCR sequential extraction method. The soluble Cu form and the total Cu contents from the anode to the cathode increased, and the difference between them kept growing over time. The Cu fractions in the soil and the electrodes were converted with the change of the dramatic pH from the anode to the cathode. There was a focusing effect leading to the change of the Cu forms, and the extractable acid form content increased in the three fifths where the acid and the alkali fronts met.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Soil is fundamental for agricultural production (Kong et al. 2014). However, agricultural soil contamination by copper happens after long-term use of pig manure and fungicides (de Boer et al. 2012). The heavy metals in soil gradually release into pore water results in the pollution of surface and ground water, contaminating agricultural land and crops. Thus, the heavily contaminated soils in industrial areas are a great concern due to their direct and indirect harmful effects on human health (Wada 2002). Therefore, soil remediation to minimize downstream damage is essential (Powlson et al. 2011). A range of remediation techniques have been developed for soil contamination, including biological, chemical, and physical treatments (Barbosa et al. 2015; Ma and Jiao 2012). However, the application of these traditional methods in situ is usually expensive and may generate new problems, such as fertility loss and soil erosion (Khodadad et al. 2011; Kumpiene et al. 2008). Therefore, microbial fuel cell (MFC) technique becomes an alternative worthy of attention.
MFCs have advantages of low cost, long sustaining periods, and minimal impacts on environment (Logan and Regan 2006; Rinaldi et al. 2008). Abundant microbes exist in organic matter-rich soils and sediments, which can be alternative energy source in MFCs. The soil MFC device filled with the soil collected from the forest and agriculture and the MFCs used organic and mineral soil saturated with deionized water. The results showed that the performance of agricultural soil MFCs was about 17-fold higher than the forest soil MFCs (Dunaj et al. 2012). Therefore, it is feasible to construct MFCs in soils and get energy from soil MFCs. An H-style or two-chamber MFC was constructed by Tandukar (Tandukar et al. 2009), and Wang (Wang et al. 2008), respectively. Their MFCs were applied to remove hexavalent chromium (Cr(VI)) which was reduced at the cathode. Heijne et al. (2010) proposed a metallurgical MFC to achieve a combination of copper removal and recovery at the cathode for detoxification. In these MFCs, the organic materials are oxidized at the anode and the heavy metals are reduced at the anode. For example, Cr6+ and Cu2+ can be reduced to Cr3+ and Cu, respectively (Heijne et al. 2010; Tandukar et al. 2009; Wang et al. 2008). However, to the best of our knowledge, currently, there are no studies on metal removal by soil MFCs. The outstanding challenge is that there are three phases in the soil MFC: solid, liquid, and gas, but only the liquid phase is in the metallurgical MFC.
Electricity is produced by applying biodegradable organic compounds in a soil MFC. In the restoration of electric field, the contaminants are transported to the electrode in the electric field from where they can be brought to the surface. The electric field drives the contaminants toward the electrodes by two mechanisms, electroosmosis and electromigration (Hicks and Tondorf 1994). In order to make the metal transport by electromigration, the metal must be in the solution and carry a charge. In this study, rhamnolipid is used to realize metal ion removal from aqueous solutions (Dahrazma and Mulligan 2007; Hong et al. 2009; Mulligan and Wang 2006; Mulligan et al. 1999). A new soil MFC device was constructed in this paper; the performance of soil MFCs was studied for the generation of voltage and polarization curves and the migration and fractionations of copper during soil MFC treatment.
Materials and methods
Experimental soil
The soil sample was collected from the agricultural plots near Yangtze River in Nanjing, China. It was dried sieved (<2 mm) and stored at room temperature. The pH was 7.91, the organic carbon was 3.71 ± 0.14 g/kg, the total nitrogen was 0.20 ± 0.08 g/kg, and the total phosphorus was 0.42 ± 0.18 g/kg. The chosen soil was contaminated artificially by adding copper to the soil. The detailed procedures were as below: 5 kg soil was weighted and added with copper solution containing 8.792 g Cu(NO3)2 (purity 99.0 %), stirred, air dried, and aged for 1 month for homogeneity. The total copper content in the contaminated soil should be 600 mg/kg according to the third grade in “Environmental quality standard for soils of China” (GB15618 1995).
Soil MFC construction
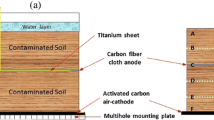
The soil MFCs was constructed in the top soil, and its schematic diagram was illustrated in Fig. 1. The constructions were fabricated from a glass cylinder with an internal diameter of 35 mm and a length of 150 mm. About 1 cm of soil was placed at the base of the MFC container before anode installation which was made of granular activated carbon (GAC, 3–5 mm in diameter with a specific area of 500–900 m2/g), and the contaminated soil was deposited on the top of the anode with a depth of 10 cm. The GAC cathode was placed above the contaminated soil, allowing oxygen interaction with the cathode. The total mass of soil and GAC in soil MFC systems were 130 g (dry weight) and 20 g (dry weight), respectively. All the assembled MFCs were then incubated at 30 ± 1 °C in the dark until final destructive sampling and analysis (total 56 days).

Configuration of the soil microbial fuel cell
Each MFC contained a mixture of contaminated soil and 12 ml concentrated anaerobic sludge from a wastewater treatment plant that were homogenized and saturated with the nutrient solution. The nutrient medium was consisted of (per L) 0.78 g KCl, 0.58 g NaCl, 0.68 g KH2PO4, 0.80 g K2HPO4, 0.10 g MgCl2, 0.28 g NH4Cl, 0.10 g CaCl2 · 2H2O (Tandukar et al. 2009), and 1 mL of trace elements (Rabaey et al. 2005b). MgCl2 was used instead of MgSO4 to avoid sulfate reduction and abiotic Cu2+ reduction by sulfide (Tandukar et al. 2009). In order to get more water-soluble copper and make microorganism growth better, 2.0 % rhamnolipids (Huzhou Zijin Biological Technology Co., Ltd., China) and 6.00 g/L CH3COONa were added into the distilled water with pH = 7.00.
There were two groups named test group and CK group in this experiment. The anode and the cathode embedded with carbon cloth (30 × 10 mm) were connected by titanium wires (1 mm in diameter). The epoxy was used to prevent the titanium wire from making a direct electrical contact with the cathode electrode. There was a closed circuit with an external resistance of 1000 Ω in the test group and an open circuit in the CK group. All the experiments were conducted in triplicate and each sample was measured three times.
Soil sampling and chemical speciation of copper
The MFCs were put in the ultra-low temperature freezer (−79 ± 1 °C, Haier BioMedical, DW-86l388A, China) for 1 h before sampling in order to prevent soil disturbance. The MFC was divided into six sections and marked as S1–S6 from the cathode to the anode, respectively (see Fig. 1), for further analysis. The activated carbon in the cathode and the anode was marked as S1 and S6, respectively. The contaminated soil part was evenly divided into four sections that were marked as S2–S5. The six subsamples were freeze dried, grinded, and sieved (100 mesh) before analysis. All the data in this paper were reported on a dry weight basis.
The soil pH was measured by mixing soil with water at a ratio of 1:2.5 using pH meter (Sartorius, PB-10, Germany). Soil samples were sequentially extracted according to an improved BCR (European Communities Bureau of Reference, now Standards, Measurements and Testing Programme) sequential extraction method. The five fractions could be categorized as step 1—water soluble fraction (F1), step 2—acid-extractable fraction (F2), step 3—reducible fraction (F3, bound to Fe-Mn oxides), step 4—oxidizable fraction (F4, bound to organic matter and sulfides), and step 5—residue fraction (F5) (Rauret et al. 1999; Ure et al. 1993). To determine the total concentration of Cu in the soil samples, 0.5 g of sample was digested with a mixture of acids (9 ml of concentrated HNO3 + 3 ml of concentrated HCl) in a microwave unit (MDS-6G, Sineo, China), according to EPA method 3051A. The concentration of every fraction was analyzed by atomic absorption spectrometer (Perkin-Elmer AAS400). The results of the original contaminated soil and activated carbon obtained in the BCR extraction procedure were shown in Table 1.
Statistical analysis
The voltages (V) generated from soil MFCs were recorded every 8 h by a data acquisition module (DAM-3057 and DAM-3210, Art Technology Co. Ltd., China). The power density (W/m2) was calculated according to P = IU/V. Where I is the current, U is the voltage, and V is the working volume of the anode. The internal resistance was calculated by the linear region of the polarization curve (Puig et al. 2012). The results were analyzed using an analysis of variance (ANOVA) procedure, and the multiple comparisons was performed by Duncan’s new multiple range method by SAS 8.0, and all the figures were plotted by Sigmaplot 11.0.
Results
The electrical characteristics of soil MFC
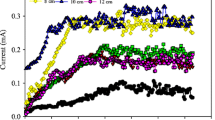
The experiment lasted for 56 days. Figure 2 showed soil MFCs’ voltage and the polarization curve. In the first 7 days, the voltage was stable around 30 mV and rose quickly to 108 mV when the soil MFC was injected with sodium acetate nutrient solution. When sodium acetate solution was added into the system on the 14th day, 21st day, 28th day, and 42nd day, the voltage peak appeared on the 26th day, 35th day, and 46th day, and the voltages were 336, 539, 488, and 499 mV, respectively. The polarization curve of MFC was obtained by transforming resistance from 100 to 100,000 Ω. The internal resistance of soil MFC was 984 Ω, and the maximum power density was 65.77 m W/m2.

Voltage variation and polarization curve
pH variation
The pH from the cathode to the anode in the soil MFC changed significantly, while the pH in the CK group was generally stable at about 8.00 as shown in Fig. 3. The pH in the test group reduced significantly from S1 to S6. The pHs in sections S1 and S2 were dramatically higher than that in CK, while the pHs in sections S3–S6 were lower (P < 0.05). Hence, there was a pH jump zone in section S3.

pH variation in different sections. Different capital letters indicated significant difference among different sections in the same group, and different lowercase letters indicated significant difference of the same sections under different groups at 0.05 level
Spatial and temporal distribution of soluble copper
Figure 4 showed the spatial and temporal distribution profiles of water-soluble Cu (F1) in the CK and the test groups. There was no significant difference in every period of S2–S5 for the CK group, and the F1 concentration ranged from 157.32 kg to 173.04 mg/kg (Fig. 3b). The F1 concentration in the cathode and the anode was significantly higher than in the original activated carbon, with the contents ranging from 47.14 to 53.53 mg/kg, and no dramatic difference with time which was varied with time in the different locations for the test group (Fig. 3a). The F1 fraction in the soil part began to migrate gradually by electricity although the voltage was below 200 mV at the beginning of the experiment. The concentrations of F1 fraction were 193.99, 203.91, and 183.15 mg/kg from S2 to S4, respectively, which were higher than that in the CK with an average value of 63.46 mg/kg. It showed that F1 formed in the soil migrated from the anode to the cathode and was mainly concentrated in sections S2 and S3 at the end of the 14th day. The F1 concentration escalated from sections S5 to S2 in sequence from 222.35 to 140.27 mg/kg when the voltage continued to rise to 539 mV. Meanwhile, the contents of F1 in sections S2 and S3 were higher than that in the CK (164.39 mg/kg), and the content of F1 in section S5 was lower than that in the CK. This suggested that Cu in the soil moved from the anode to the cathode and accumulated in section S2 gradually, and the migration of F1 fraction did not cease in the soil MFC but continue to migrate until reaching the cathode.

The time and space distribution characteristics of water-soluble copper. Different capital letters indicated significant difference among different sections in the same time, and different lowercase letters indicated significant difference of the same sections under different time at 0.05 level
When the voltage increased to 488 mV, there was an extremely significant change between the F1 concentration in the anode and that in the cathode. The F1 concentration in the cathode (80.91 mg/kg) was higher than that in the anode (70.25 mg/kg), while the F1 concentration in the anode began to decline. The F1 concentration increased in the soil part from S5 to S2, and the F1 concentration of section S2 was higher than that in S5 and CK by 95.06 and 68.85 mg/kg, respectively. At the end of the experiment, the F1 concentraton in S6 increased from 60.88 to 83.34 mg/kg and declined with the value of 40.19 mg/kg. The reason was that H+ released in the anode could neutralize carbonate and OH− and cause copper to be released from the insoluble form and migrated from the anode to the cathode in the electric field. The change of concentration of the F1 fraction in S1 was different from that in S6 and continuously increased to be higher than that in S6 by 55.28 mg/kg at the end of the experiment. Whereas, the S1 content was lower than section S2 by 141.89 mg/kg. This may be due to the transformation of copper, and the accumulated F1 was precipitation in S1 with high pH, for example, acid-extractable and oxidizable contents increased by 37.77 and 28.18 mg/kg, respectively, compared to that in the CK (Fig. 5). Simultaneously, there was high mass transfer resistance between the soil and the activated carbon. In the soil part, the F1 concentration increased significantly from S5 of 138.20 to S2 of 237.36 mg/kg (P < 0.05). The results suggested that Cu in the contaminated soil moved from the anode to the cathode and accumulated in sections S1 and S2. Furthermore, the difference between them became larger with the increase of the MFC operation time until the end of the experiment. These results showed that the soil MFC performance improved with electric field and the time which strengthened the migration of F1 fraction in the soil from the anode to the cathode and was enriched constantly in the S1 and S2 (Fig. 4). However, the similar phenomena were not observed in the CK group.

Spatial distribution and fractionation of Cu by BCR sequential extraction. Different capital letters indicated significant difference among different sections in the same group, and different lowercase letters indicated significant difference of the same sections under different groups at 0.05 level. The sample was collected at the 56th day
Spatial distribution and chemical speciation of total copper
Figure 5 indicated the copper distribution and its fractions. The initial total copper contents were 635.27 and 8.10 mg/kg in the contaminated soil and activated carbon. In the test groups, total copper content increased from sections S5 to S2 with their contents ranging from 468.33 to 668.01 mg/kg. The migration showed that copper in the soil transferred from the anode to the cathode, and the migration quantity was 199.69 mg/kg. The total copper content in the cathode was 278.25 mg/kg, higher than 125.14 mg/kg in the anode, and was lower in S3–S5 than that in the CK group which meant the total copper contents reduced in these sections. Water-soluble (F1) and acid-extractable (F2) fractions were the two main forms of copper in the soil and the electrodes, whose concentrations were 124.55 and 405.07 mg/kg in the soil, and 35.92 and 63.13 mg/kg in the electrodes, respectively. Compared with the CK group, the F1 fraction increased by 7.46 % and rose from S5 to S2 in turn (see Fig. 4). The F2 fraction decreased significantly by 16.18 % in the test group. In the soil part, the highest concentration of F2 fraction was 313.81 mg/kg in S3, and the lowest was 250.21 mg/kg in S5. The F2 fraction concentration in the cathode was 97.70 mg/kg, higher than 34.57 mg/kg in the anode. Reducible (F3) and oxidizable (F4) contents in the cathode were higher than that in the anode and in the CK group (Fig. 5a).
Discussion
In a soil MFC, the process of anoxic organic matter oxidation would be stimulated and possibly accelerated. The basis hereof lies within the spatial separation of the oxidative, electron-generating half-reaction at the anode and the electron-consuming half-reaction at the cathode (De Schamphelaire et al. 2008a; Lakaniemi et al. 2012). The soil organic matter or external carbon source (acetate) in the anode could be oxidized to electrons, protons, and CO2. At the cathode, oxygen, the final electron acceptor, is reduced to H2O. The electrons transfer from anode to cathode through electrical circuit, containing a power user (Fig. 6). The peak power output of soil MFCs was fairly similar to or higher than other MFC studies that were constructed using organic-rich soils or sediments. In order to make a better comparison, the voltages and the powers obtained from the soil and sediment MFCs were presented in Table 2.

Working concept of Cu migration and electricity generation in the soil MFC
The soil MFCs constructed in this study realized electricity production which played an important role in the Cu migration process. Moreover, the rhamnolipid could enhance the removal rate in alkaline condition and improve the removal of copper significantly (Dahrazma and Mulligan 2007). The F1 form changed dramatically with electricity produced in the soil MFC. Furthermore, the difference among them became larger with the increase of MFC operation time until the end of the experiment. These results showed that the soil MFC performance improved with time which strengthened the migration of F1 form in the soil from the anode to the cathode and enriched constantly in the S2. In the typical SMFC, the efflux of the degradation products in the anode, such as the electron and the proton, was influenced by the diffusion, the charge of the molecules, and the internal resistance which caused a potential loss between the anode and the cathode (De Schamphelaire et al. 2008a; Logan et al. 2006; Rabaey et al. 2005a, 2007). However, there was a significant concentration gradient of F1 from sections S2 to S5. The gradient slope increased over time and was opposite to the diffusion that spread from the high concentration place to low (Figs. 4 and 5). It manifested that the migration and enrichment of F1 were driven by the force rather than the diffusion. The driver was the electric field produced by the soil MFC that made the positively charged water-soluble copper migrate from the anode to the cathode. The distribution trends of H+ and Cu2+ were completely different though they both carried positive charges. Under the effect of electric field, Cu2+ migrated to the cathode area constantly. The Cu2+ content in the cathode was higher than in the anode and increased from sections S5 to S2. The release of H+ in the anode and the electricity production in the soil MFC would cause H+ migration to the cathode and could lead to the lower pH in the cathode than that in the anode under the same electric field theoretically. However, the fact was that pH in the cathode was higher than that in the anode. That was because oxygen in the air accepted electrons and was reduced to hydroxide which could neutralize the abundant H+. Moreover, oxygen is a more preferred electron acceptor than Cu2+ (O2 + 2H2O + 4e− = 4OH−, 0.40 V vs NHE, and Cu2++2e− = Cu, 0.34 V vs NHE). Cu2+ cannot accept electrons under the alkaline condition and may precipitate as CuO, Cu2O, or other forms and may not be available for reduction (Heijne et al. 2010). Therefore, a Cu2+ enrichment area appeared near the cathode area.
A mixed potential of both copper and oxygen reduction could result in a higher cathode potential. There was a competitive relationship between Cu2+ and oxygen to accept electrons (Heijne et al. 2010). At the same time, these dissociative copper were converted into Cu and its oxide form by reduction reactions. In this study, the contents of F3 and residual fraction (F5) in the test group were higher than those in the CK by 2.81 and 41.17 mg/kg, respectively. A metallurgical microbial fuel cell (MFC) was constructed by Heijne et al., which was used for copper recovery. Pure copper crystals were formed on the cathode, and no CuO or Cu2O was detected. The high performance of the MFC with Cu2+-reducing cathode (Cu2+ removal efficiencies >99.88 %) could be caused by the following reasons: the oxygen reduction reaction has a high overpotential, while the overpotential for Cu2+ reduction is much lower, and copper might function as a catalyst for the oxygen reduction reaction (Heijne et al. 2010). The similar result also appeared in another literature that Cu2+ was reduced to cuprous oxide and metal copper in the cathodes of the MFC (Tao et al. 2011). As a consequence, the reaction could be more advantageous to reduce Cu2+ and enhance the efficiency of enriched copper in the soil if the oxygen content was reduced in the cathode; the electrode potential was appropriate, and the competition of accepting electron between oxygen and Cu2+ was moderate. Under the effects of the soil MFC, not only the F1 content but also the total copper content changed in the soil MFC. These results illustrated that Cu migrated to the cathode by electricity and accumulated in the S1 and S2.
As shown in Fig. 3, the pH gap between the cathode and the anode was 2.25 units. Gil et al. (2003) also observed the similar pH decrease in the anode chamber and an increase of pH in the cathode chamber. It might be because that the proton transported through the Nafion seemed to be slower than the proton production rate in the anode chamber and the proton consumption rate in the cathode chamber. Furthermore, the oxygen in the air accepted electrons from the anode and was reduced to hydroxide (O2 + 2H2O + 4e− = 4OH−, 0.40 V vs NHE). The hydrogen and hydroxyl ion production rates were fixed by the current, which were dependent on the applied voltage and the conductivity of the medium (Hicks and Tondorf 1994). Anaerobic environment in the soil MFC could help fermentation; the proton transfer rate might be limited by the poor permeability of the water-logged soil, which resulted in a delay in the proton transfer to the cathode and the accumulation of protons in the anode (Huang et al. 2011). It was worth notice that the pH in section S3 was 7.86, which significantly increased by 1.88 in the cathode and decreased by 0.37 in the anode. Meanwhile, the F2 mainly existed in the form of carbonate, which could be dissolved when the pH decreased, and the F1, water soluble, could be migrated in soil MFCs by electricity. During the electrokinetic remediation operation, the acid and the alkali fronts migrated toward the opposing electrodes and interacted at approximately one third of the distance from the cathode to form a zone of abrupt pH change (Abou-Shady and Peng 2012). In other words, there was a pH jump and focusing effect in section S3 where H+ met with OH−. This is because the effective ionic hydrogen mobility is about 1.8 times that of the hydroxyl ion (Acar and Alshawabkeh 1993). In other words, there was a distinct pH change zone and focusing effect leading to changing copper forms and the F2 form increasing in the section S3 where acid and alkali fronts met.
Conclusions
The migration of heavy metals and pH variation were closely related to the electricity production in a soil MFC. In the CK group, the F1 form and total copper did not change in the whole soil and electrode area over time. In the test group, they migrated due to the effect of internal electric field in the soil MFC. The concrete manifestations were (1) pH increased from the anode to the cathode, (2) the content of copper increased from S5 to S2 and the content in S1 was significantly higher than S6, and (3) the concentration difference among these sections were much larger. Hence, the electric field produced by soil MFC played an important role in cation (H+ and Cu2+) migration, and the effect could overcome the diffusion to migrate to an opposite direction compared to the ion charge.
This new soil MFC combines electricity production with copper enrichment in the cathode and on the top of soil. As the soil MFC is still under development, it cannot be directly compared to the electrokinetic remediation. However, significant attention has been paid here for copper recovery in the soil MFC within the generated electricity field. In addition, chemical speciation of Cu changed from water-soluble form to other reduced and oxidized forms, or pure copper in the cathode. Thus, the different reactions in the cathode are worth further study for the improvement of MFC performance, as we have shown that it can compete with oxygen reduction as an efficient cathode option. The copper removal in the soil enhanced by the soil MFC expands the application range of MFC and illustrates once more the flexibility of MFC applications.
References
Abou-Shady A, Peng C (2012) New process for ex situ electrokinetic pollutant removal. I: process evaluation. J Ind Eng Chem 18:2162–2176
Acar YB, Alshawabkeh AN (1993) Principles of electrokinetic remediation. Environ Sci Technol 27:2638–2647
Barbosa B, Boleo S, Sidella S, Costa J, Duarte MP, Mendes B, Cosentino SL, Fernando AL (2015) Phytoremediation of heavy metal-contaminated soils using the perennial energy crops Miscanthus spp. and Arundo donax L. Bioenerg Res 8:1500–1511
Cao X, Song HL, Yu CY, Li XN (2015) Simultaneous degradation of toxic refractory organic pesticide and bioelectricity generation using a soil microbial fuel cell. Bioresour Technol 189:87–93
Dahrazma B, Mulligan CN (2007) Investigation of the removal of heavy metals from sediments using rhamnolipid in a continuous flow configuration. Chemosphere 69:705–711
de Boer TE, Tas N, Braster M, Temminghoff EJ, Roling WF, Roelofs D (2012) The influence of long-term copper contaminated agricultural soil at different pH levels on microbial communities and springtail transcriptional regulation. Environ Sci Technol 46:60–68
De Schamphelaire L, Rabaey K, Boeckx P, Boon N, Verstraete W (2008a) Outlook for benefits of sediment microbial fuel cells with two bio-electrodes. Microb Biotechnol 1:446–462
De Schamphelaire L, Van den Bossche L, Dang HS, Hofte M, Boon N, Rabaey K, Verstraete W (2008b) Microbial fuel cells generating electricity from rhizodeposits of rice plants. Environ Sci Technol 42:3053–3058
Deng H, Wu YC, Zhang F, Huang ZC, Chen Z, Xu HJ, Zhao F (2014) Factors affecting the performance of single-chamber soil microbial fuel cells for power generation. Pedosphere 24:330–338
Dunaj SJ, Vallino JJ, Hines ME, Gay M, Kobyljanec C, Rooney-Varga JN (2012) Relationships between soil organic matter, nutrients, bacterial community structure, and the performance of microbial fuel cells. Environ Sci Technol 46:1914–1922
Fang Z, Song HL, Cang N, Li XN (2013) Performance of microbial fuel cell coupled constructed wetland system for decolorization of azo dye and bioelectricity generation. Bioresour Technol 144:165–171
GB15618 (1995) Environmental quality standard for soils of China
Gil GC, Chang IS, Kim BH, Kim M, Jang JK, Park HS, Kim HJ (2003) Operational parameters affecting the performance of a mediator-less microbial fuel cell. Biosens Bioelectron 18:327–334
Heijne AT, Liu F, Weijden R, Weijma J, Buisman CJ, Hamelers HV (2010) Copper recovery combined with electricity production in a microbial fuel cell. Environ Sci Technol 44:4376–4381
Hicks RE, Tondorf S (1994) Electrorestoration of metal contaminated soils. Environ Sci Technol 28:2203–2210
Hong SW, Chang IS, Choi YS, Chung TH (2009) Experimental evaluation of influential factors for electricity harvesting from sediment using microbial fuel cell. Bioresour Technol 100:3029–3035
Huang DY, Zhou SG, Chen Q, Zhao B, Yuan Y, Zhuang L (2011) Enhanced anaerobic degradation of organic pollutants in a soil microbial fuel cell. Chem Eng J 172:647–653
Khodadad CLM, Zimmerman AR, Green SJ, Uthandi S, Foster JS (2011) Taxa-specific changes in soil microbial community composition induced by pyrogenic carbon amendments. Soil Biol Biochem 43:385–392
Kong LL, Liu WT, Zhou QX (2014) Biochar: an effective amendment for remediating contaminated soil. In: Whitacre DM (ed) Reviews of environmental contamination and toxicology, vol 228. Springer, New York, pp 83–99
Kumpiene J, Lagerkvist A, Maurice C (2008) Stabilization of As, Cr, Cu, Pb and Zn in soil using amendments—a review. Waste Manag 28:215–225
Lakaniemi AM, Tuovinen OH, Puhakka JA (2012) Production of electricity and butanol from microalgal biomass in microbial fuel cells. Bioenerg Res 5:481–491
Logan BE, Regan JM (2006) Microbial fuel cells—challenges and applications. Environ Sci Technol 40:5172–5180
Logan BE, Hamelers B, Rozendal R, Schröder U, Keller J, Freguia S, Aelterman P, Verstraete W, Rabaey K (2006) Microbial fuel cells: methodology and technology. Environ Sci Technol 40:5181–5192
Ma WJ, Jiao BQ (2012) Review of contaminated sites remediation technology. Res J Chem Environ 16:137–139
Mulligan CN, Wang SL (2006) Remediation of a heavy metal-contaminated soil by a rhamnolipid foam. Eng Geol 85:75–81
Mulligan CN, Yong RN, Gibbs BF (1999) Removal of heavy metals from contaminated soil and sediments using the biosurfactant surfactin. J Soil Contam 8:231–254
Powlson DS, Gregory PJ, Whalley WR, Quinton JN, Hopkins DW, Whitmore AP, Hirsch PR, Goulding KWT (2011) Soil management in relation to sustainable agriculture and ecosystem services. Food Policy 36:S72–S87
Puig S, Coma M, Desloover J, Boon N, Colprim J, Balaguer MD (2012) Autotrophic denitrification in microbial fuel cells treating low ionic strength waters. Environ Sci Technol 46:2309–2315
Rabaey K, Boon N, Hofte M, Verstraete W (2005a) Microbial phenazine production enhances electron transfer in biofuel cells. Environ Sci Technol 39:3401–3408
Rabaey K, Ossieur W, Verhaege M, Verstraete W (2005b) Continuous microbial fuel cells convert carbohydrates to electricity. Water Sci Technol 52:515–523
Rabaey K, Rodriguez J, Blackall LL, Keller J, Gross P, Batstone D, Verstraete W, Nealson KH (2007) Microbial ecology meets electrochemistry: electricity-driven and driving communities. ISME J 1:9–18
Rauret G, Lopez-Sanchez JF, Sahuquillo A, Rubio R, Davidson C, Ure A, Quevauviller P (1999) Improvement of the BCR three step sequential extraction procedure prior to the certification of new sediment and soil reference materials. J Environ Monit 1:57–61
Reimers CE, Tender LM, Fertig S, Wang W (2001) Harvesting energy from the marine sediment-water interface. Environ Sci Technol 35:192–195
Rinaldi A, Mecheri B, Garavaglia V, Licoccia S, Di Nardo P, Traversa E (2008) Engineering materials and biology to boost performance of microbial fuel cells: a critical review. Energy Environ Sci 1:417–429
Tandukar M, Huber SJ, Onodera T, Pavlostathis SG (2009) Biological chromium(VI) reduction in the cathode of a microbial fuel cell. Environ Sci Technol 43:8159–8165
Tao HC, Liang M, Li W, Zhang LJ, Ni JR, Wu WM (2011) Removal of copper from aqueous solution by electrodeposition in cathode chamber of microbial fuel cell. J Hazard Mater 189:186–192
Ure AM, Quevauviller P, Muntau H, Griepink B (1993) Speciation of heavy metals in soils and sediments. An account of the improvement and harmonization of extraction techniques undertaken under the auspices of the BCR of the Commission of the European Communities. Int J Environ Anal Chem 51:135–151
Wada S-I (2002) Effect of clay mineralogy on the feasibility of electrokinetic soil decontamination technology. Appl Clay Sci 20:283–293
Wang G, Huang LP, Zhang YF (2008) Cathodic reduction of hexavalent chromium [Cr(VI)] coupled with electricity generation in microbial fuel cells. Biotechnol Lett 30:1959–1966
Acknowledgments
We thank the Provincial Natural Science Foundation of Jiangsu, China (BK20141330), the National Natural Science Foundation of China (21277024) and the Fundamental Research Funds for the Central Universities for financial support.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Bingcai Pan
Rights and permissions
About this article

Cite this article
Wang, H., Song, H., Yu, R. et al. New process for copper migration by bioelectricity generation in soil microbial fuel cells. Environ Sci Pollut Res 23, 13147–13154 (2016). https://doi.org/10.1007/s11356-016-6477-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-016-6477-8