
Abstract
The aim of this study was to determine the impacts of anthropogenic disturbances on bacterial community composition in an urban river (Mooi River). Physico-chemical analysis, bacterial enumeration and 454-pyrosequencing were conducted on the Mooi River system upstream and downstream of an urban settlement in the North West Province, South Africa. Pyrosequencing and multivariate analysis showed that nutrient inputs and faecal pollution strongly impacted the physico-chemical and microbiological quality at the downstream sites. Also, bacterial communities showed higher richness and evenness at the downstream sites. Multivariate analysis suggested that the abundances of Betaproteobacteria, Epsilonproteobacteria, Acidobacteria, Bacteroidetes and Verrucomicrobia are related to temperature, pH, dissolved oxygen (DO), sulphate and chlorophyll-a levels. These results suggest that urbanisation caused the overall water quality of this river to deteriorate, which in turn affected the bacterial community composition. In addition, our work identified potential indicator groups that may be used to track faecal and organic pollution in freshwater systems.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Increase in human population growth as well as economic and industrial development have caused natural freshwater systems to markedly deteriorate in terms of water quality, biodiversity, in-stream processes, watershed hydrological regimes and landscape (O’Driscoll et al. 2010; Martinuzzi et al. 2014). Discharge from anthropogenic activities (e.g., municipal, industrial, mining, wastewater treatment plants, and agricultural activities) expose freshwater systems to a variety of organic and inorganic pollutants, nutrient stress, heavy metals and biological material (Ford 2000). For urban rivers, domestic sewage and industrial effluent are the main pollution sources in which nutrients and heavy metals are the general contaminants (Iwegbue et al. 2012; Li et al. 2012). In addition, dry land agriculture further contributes to nutrient loadings (e.g. nitrates and phosphates) and toxic compounds in the form of fertilisers, herbicides and pesticides (Bricheux et al. 2013; Kamjunke et al. 2013).
Anthropogenic disturbances on freshwater systems hold major repercussions on the overall bacterial structure and function of these habitats (Paerl et al. 2003). In addition, the water quality is compromised to such an extent that it may no longer be fit for recreational and several other human purposes (De Figueiredo et al. 2007). Chemicals may alter the bacterial community composition (BCC) as bacteria are highly sensitive to nutrient availability, concentrations of pollutants and altered environmental conditions (Paerl et al. 2003; Yergeau et al. 2012). Changes in the BCC include selection for more resistant or contaminant-specific species with an associated change in overall diversity (Ford 2000). Such changes have been predominantly observed in urban/suburban rivers and streams impacted by wastewater treatment plants (Drury et al. 2013), industrial effluent (Lu and Lu 2014), sewage discharge (Lin et al. 2014), landfill leachate (Markúsdóttir et al. 2013), and pesticides (Pesce et al. 2008). As such, changes in BCC affect the functional dynamics of whole ecosystems by altering the ecosystem processes (physical, chemical and biological) through metabolic feedback. Changes in the abundance of minor species can thus affect the vitality and success of larger organisms (Zarraonaindia et al. 2013). It is conceivable that the short generation times of bacteria, their high diversity, and quick reaction and recovery from environmental changes give them the advantage to be used as indicators of both physical and chemical stresses in freshwater systems (Stabili and Cavallo 2011).
Important objectives of using bacterial communities as biological indicators are to understand their structure, dynamics and causes of variability (Paerl et al. 2003). Recent metagenomic approaches, such as 454-pyrosequencing, have simplified and accelerated this process by allowing scientists to study bacterial diversity in more detail (De Figueiredo et al. 2007). Metagenomic efforts further the understanding of BCC changes at group level over spatial and temporal scales (Zarraonaindia et al. 2013). Also, metagenomics data assist in determining how environmental conditions such as pollution shape BCC and how these conditions affect diversity of genes associated with biogeochemical cycles (Singh et al. 2009). This information, together with future development of metagenomic techniques and statistical models, will permit possible prediction of changes in microbial communities on the basis of present knowledge (Larsen et al. 2012).
The Mooi River catchment (1800 km2) is located in the western Gauteng and North West Provinces of South Africa. It has been the sole water supply of Potchefstroom, which currently has a population of approximately 124,000 residents (Stats SA 2011). The water quality of the Mooi River and its tributaries (Wonderfontein and Loop Spruit) has been affected in various ways by human activities including agriculture (>20 % of land coverage) and urban and informal settlements (Van der Walt et al. 2002). An informal settlement is defined as “an unplanned settlement on land which has not been surveyed or proclaimed as residential, consisting mainly of informal dwellings (shacks)” (HDA 2012). The Wonderfontein Spruit receives sewage wastewater from informal settlements, runoff from agriculture and large amounts of gold mining effluent. In addition, high salt levels and various trace elements are frequently detected in the Wonderfontein Spruit. The Mooi River and Loop Spruit tributaries are predominantly impacted by dry land agricultural activities. However, several gold mines discharge effluent in the Loop Spruit, whilst small-scale diamond mining in the Mooi River sub-catchment area destroyed the floodplain and riparian habitats (Van der Walt et al. 2002).
The goals of the present study were to (i) determine the impacts of anthropogenic inputs on BCC along the Mooi River system (South Africa) and (ii) statistically analyse the effects of pollution on the spatial distribution of bacterial communities.
Materials and methods
Study area
The Mooi River catchment receives its water supply mainly from dolomitic eyes and springs (Van der Walt et al. 2002). The catchment has three main tributaries including (i) the Wonderfontein Spruit, (ii) the northern stretch of the Mooi River, and (iii) the Loop Spruit. Four major dams are situated in the Mooi River catchment including the Klerkskraal Dam, Boskop Dam, Klipdrift Dam and Potchefstroom Dam (Van der Walt et al. 2002). The Loop Spruit merges with the Mooi River at the Prozetsky Bird sanctuary downstream of Potchefstroom City. The catchment bypasses the Potchefstroom sewage treatment plant and correctional services before it flows into the Vaal River (Kromdraai confluence).
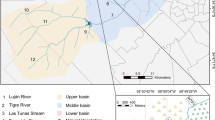
The study sites were specifically selected to represent a range of water quality data and the impact of human settlements on the Mooi River (Fig. 1). Study sites included Muiskraal (site 1; S 26° 26′ 42.2; E 27° 07′ 06.1), sites below the Boskop Dam (site 2; S 26° 34′ 19.3; E 27° 06′ 12.5), below the Potchefstroom Dam (site 3; S 26° 40′ 43.3; E 27° 05′ 56.2), opposite the sewage treatment plant on the outer reaches of Potchefstroom City (site 4; S 26° 45′ 10.9; E 27° 06′ 01) and above the Kromdraai confluence (site 5; S 26° 52′ 49.5; E 26° 57′ 51.4). Muiskraal served as the reference site due to lowest anthropogenic activities in the vicinity.

Geographical map of the Mooi River system. Illustrated is the general location of the study site in the North West Province, with a detailed view of the sampling sites examined for bacterial community composition
Sample collection
Freshwater samples were collected in sterile 2-L containers from the five sites between June and July 2012 (pre-rainfall winter season). Samples were taken in triplicate and placed at 4 °C in the dark until further treatment within 8 h after collection. Physical parameters that were measured in situ included temperature, pH, electrical conductivity (EC) and dissolved oxygen (DO). Selected chemical and microbiological parameters were determined by Midvaal Water Company, South Africa. These included total chlorine (Clˉ), nitrate (NO3 −) and nitrite (NO2 −), phosphate (PO4 3−), sulphate (SO4 2−), chemical oxygen demand (COD), chlorophyll-a, total coliforms (TC) and Escherichia coli. Total and free chlorine were determined using a contour comparator consistent with the Palinstest method (Palintest Ltd, England, UK). Nitrate and nitrite levels were measured according to the South African Bureau of Standards (SABS) method SANS 5210 (SABS, 2010). Phosphate analyses were performed as described by Rice et al. (2012), section 4500-P F, using a Skalar SANplus autoanalyser (Skalar Analytical B.V., Breda, The Netherlands). Sulphates were determined according to Rice et al. (2012), section 4500-SO4 2− E. Chemical oxygen demand analyses were based on standard methods described by Rice et al. (2012), section 5220 D and the HACH COD method 8000 (HACH Company, CO, USA). Chlorophyll-a concentrations were measured by spectrophotometric analysis according to methods described by Rice et al. (2012) and Sartory and Grobbelaar (1984). The TC and E. coli counts were determined by the most probable number (MPN) method using the Colilert® test (INDEXX Laboratories Inc., Westbrook, ME, USA).
Microbiological analysis of water samples
Water samples were collected in sterile 100-mL containers from the five sites (June and July 2012) and stored as described above. Heterotrophic plate count (HPC) bacteria for each site were enumerated by serially diluting water samples in sterile 8.5 % (w/v) sodium chloride (NaCl) solution. Aliquots from serial dilutions were spread-plated on R2A agar (Difco Laboratories Inc., Franklin Lakes, NJ, USA) and incubated aerobically at 26 °C for 5–6 days. The number of colony-forming units (CFU) was recorded and bacterial colonies that were morphologically distinct were further sub-cultured on R2A. Gram-staining was used to assess the purity of the isolates. HPC measurements were performed in triplicate for each site.
DNA isolation and PCR amplification
DNA isolation of pure bacterial isolates was achieved using the colony PCR method as described by Zampini et al. (2015) with a few modifications. Briefly, a single colony was picked from agar plates with a sterile pipette tip and re-suspended in 10 μL of dH2O in sterile PCR tubes. The samples were briefly mixed and heated in a microwave at 1000 W for 2–3 min to lyse the cells. Samples were then centrifuged for 30 s at 13,400 rpm and placed on ice. One microliter of the eluate was used as DNA template for PCR amplification. Amplification was performed in a 25 μL reaction mix containing single strength PCR master mix [(5 U/μL Taq DNA polymerase (recombinant) in reaction buffer, 2 mM MgCl2, 0.2 mM of each dNTP, Thermo Fisher Scientific, Waltham, MA, USA)], 50 pmol of the universal 16S ribosomal RNA (rRNA) primer pair 27F and 1492R (Lane 1991) and PCR-grade water (Thermo Fisher Scientific, Waltham, MA, USA). Amplification was performed in a preheated Bio-Rad iCycler Thermal Cycler (Bio-Rad Laboratories, Hercules, CA, USA) with an initial denaturation at 95 °C for 5 min followed by 30 cycles of denaturation at 95 °C for 30 s, annealing at 51 °C for 1 min and extension at 72 °C for 1 min. Final extension was performed at 72 °C for 5 min. PCR-amplified DNA fragments were observed by standard electrophoresis on 1 % (w/v) agarose gels and visualised by ethidium bromide staining and UV illumination. PCR products were purified and sequenced using the BigDye® Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems Life Technologies, Carlsbad, CA, USA) and Genetic Analyzer 3130 (Applied Biosystems Life Technologies, Carlsbad, CA, USA) according to manufacturer’s instructions. Sequences were examined using the BLASTN algorithm (http://www.ncbi.nlm.nih.gov/BLAST) to detect the closest bacterial match within the GenBank database. Nucleotide sequences obtained from pure bacterial isolates were deposited in the GenBank database under the accession numbers KC515572–KC515642.
Total DNA was isolated from water samples by filtering 250 to 2,000 mL water through sterile 0.2-μm nitrocellulose membrane filters (Whatman GE Healthcare Life Sciences, Buckinghamshire, UK). The filters were subsequently placed in a 1-mg/mL lysozyme solution that contained 0.25–0.50-mm glass beads (Sigma-Aldrich Corporation, St. Louis, MO, USA) for bacterial cell disruption. The lysis solution was incubated at 37 °C for 10 min whilst agitated in a vortex. Proteinase K (1 mg/mL) was then added and the lysis solution was incubated at 56 °C for an additional 30 min. DNA was isolated from the crude lysate using the PeqGold Bacterial DNA Kit (PEQLAB Biotechnologie GmbH, Erlangen, Germany). The quality and quantity of the isolated nucleic acids were determined using the Nanodrop ND1000 (NanoDrop Technologies, Wilmington, DE, USA) and agarose electrophoresis. The isolated DNA was stored at −20 °C until further analysis.
454-pyrosequencing
454-pyrosequencing was performed by Inqaba Biotech, South Africa using the Roche 454 GS-FLX chemistry. The V1 and V3 regions of the 16S rRNA gene were targeted using universal bacterial primer pair 27F and PRUN518R (Lane 1991), containing the 454 FLX adaptors and sample-specific identifiers. Due to budgetary constraints, only one sample per site was sequenced. Raw sequence data was quality trimmed and checked for chimaeras following the MOTHUR v.1.28 pipeline (Schloss et al. 2009; 2011). Sequences were assigned to operational taxonomic units (OTU’s) at a 97 % similarity. Rarefaction curves were constructed from 454-pyrosequencing data using MOTHUR v.1.28. Taxonomic classification of phylotypes was determined using the Ribosomal Database Project (RDP) classifier (Wang et al. 2007) at a 97 % bootstrap confidence threshold. Alpha and beta diversity calculations were performed using reduced data sets in which the number of sequences per samples was made equal with random resampling (516 sequences per samples). Alpha diversity (richness and evenness) was calculated in MOTHUR v.1.28 using rarefaction and the Simpson diversity index. Beta diversity was determined in XLSTAT version 2013.5.04 (Addinsoft SARL, New York, NY, USA) through the Bray-Curtis dissimilarity coefficient to obtain a beta diversity matrix. The resulting distance matrix was mapped on a 2D-multidimensional scaling (MDS) plot with 999 repetitions. Dissimilarity between samples (ANOSIM) was tested in MOTHUR v.1.28.
All DNA sequences were deposited in the GenBank database under the accession numbers KC515643–KC516708.
Statistical analysis
Multivariate analysis was used to assess the effects of physico-chemical water characteristics on BCC. Environmental and microbiological data was log transformed [log(x + 1)] before analysis. The distribution of BCC according to physico-chemical parameters was tested through canonical correspondence analysis (CCA) with Monte Carlo permutation tests based on 1000 unrestricted permutations, 80 % confidence level, and 5 % significance level. All statistical analyses were performed using XLSTAT (v. 2013.5.04).
Results
Physico-chemical and microbiological analysis
Physico-chemical and microbiological water characteristics are summarised in Table 1. River temperature ranged from 8.3 to 11.8 °C, pH varied between 8.03 and 8.57, and DO concentrations ranged from 7.40 to 10.00 mg/L. Conductivity gradually increased from the upstream to downstream sites with the highest value recorded at site 4 (~73 mS/m). Average concentrations for chlorine, nitrate/nitrite, and phosphate were within the recommended water quality objectives (RWQO’s) prescribed by the Department of Water Affairs (DWA), South Africa, for the Mooi River catchment (Table 2) (DWAF 2009). However, sulphate concentrations were consistently higher than the RWQO with highest levels measured at sites 4 and 5 (>90 mg/L).
E. coli levels for the June samples decreased steadily from sites 1 to 3 (99 to 61 MPN/100 mL) but was considerably higher at site 4 (548 MPN/100 mL). In contrast, E. coli levels for the July samples gradually increased from site 1 to site 4 and ranged from 139 to 488 MPN/100 mL. The results indicate that E. coli levels for sites 1, 2, 3 and 5 complied with the target water quality range (TWQR) for recreational use (0–130 cfu/mL) and livestock watering (0–1000 cfu/mL) (DWAF 1996). Total coliforms, however, exceeded the TWQR for recreation (0–150 cfu/100 mL) and agriculture (livestock watering [0–200 cfu/100 mL] and irrigation [<1 cfu/100 mL]) at all sites and sampling periods (DWAF 1996).
Heterotrophic bacteria
Heterotrophic bacterial levels ranged between 1.3 × 105 and 9 × 105 CFU/mL (Table 1). A total of 94 bacterial isolates that represented different morphotypes were recovered from water samples. Of these, 14 % were Gram-positive and 86 % Gram-negative. Alphaproteobacteria was the predominant class detected at site 1 (June and July), representing up to 40 % of the isolates. Species identified include Novosphingobium, Rhizobium, Xanthobacter and Paracoccus spp. Dominant groups detected at sites 2 and 3 during the June sampling period consisted of Firmicutes and Betaproteobacteria (26 %). In contrast, during July, these two sites were dominated by Gammaproteobacteria (37 %), Bacteroidetes (21 %) and Betaproteobacteria (16 %). The latter three groups also occurred in highest numbers (> 69 %) at sites 4 and 5 during June and July.
The phylum Firmicutes predominantly consisted of Gram-positive rod-shaped bacteria in the class Bacilli. Within the Bacteroidetes phylum, Flavobacterium was most frequently detected. A variety of Betaproteobacteria species was identified and grouped into two families (Comamonadaceae and Oxalobacteraceae) and six genera (Massilia sp., Limnohabitans parvus, Dunganella sp., Rhodoferax sp., Curvibacter sp. and Herbaspirillum sp.). Pseudomonas fluorescens, Pseudomonas koreensis and Pseudomonas putida were the main species detected in the Gammaproteobacteria group. These three species accounted up to 40 % of the isolates. Some isolates were also identified as Pseudomonas but could not be classified further by the National Center for Biotechnology Information (NCBI) database to species level.
Bacterial community structure and diversity
To characterise the bacterial community structure along the Mooi River, DNA samples were subjected to 454-pyrosequencing and subsequent analysis. A total of 24,374 pyrosequencing reads were obtained with an average read length of 500 ± 20 bp. Following quality trimming of sequences shorter than 150 bp, 13,984 sequences were used for further analysis. Overall, a total of 900 unique OTU’s were assigned to a class at a confidence threshold of 97 % (Fig. 2a). Of the 900 OTU’s, 60 % were identified up to genus level. The greatest number of OTU’s was associated with site 3 (291 OTU’s in June and 231 OTU’s in July). The lowest estimates were obtained for site 4 with a library size between 200 and 213 OTU’s (June and July, respectively). Sequence libraries for sites 1, 2 and 5 ranged in size between 203 and 251 OTU’s. Rarefaction curves suggested that the bacterial diversity did not reach saturation/plateau at a 97 % similarity level (Fig. 2b).

Bacterial alpha and beta diversity estimates at all sampling sites (June and July) based on 454-pyrosequencing reads. Data sets were normalised to the same number of reads (516 reads) before calculations. a Estimated number of bacterial OTU’s at the 97 % similarity level for each sampling site. b Rarefaction curves for the ten samples estimating the number of bacterial OTU’s at the 97 % similarity level. c Alpha diversity estimates calculated with Simpson diversity index. d MDS diagram showing beta diversity among the five sampling sites
Alpha diversity was calculated using rarefaction and Simpson diversity index. Results indicate that bacterial richness and evenness was in general higher for the June than July samples (Fig. 2b, c). Sites 2 and 3 June were found to be the most diverse, whereas site 4 June was the least diverse. In contrast, sites 4 and 5 July displayed the greatest bacterial richness and evenness, whilst site 1 July had the lowest number of species. These results indicate that species richness positively associated with evenness. Sites with a large number of species showed a degree of equitability among species abundance. On the other hand, sites that displayed low species richness had many individuals belonging to the same species. Variation of beta diversity is visualised with a MDS graph (Fig. 2d) and Bray-Curtis dendrogram (Fig. 3a, b). Results highlighted marked differences in BCC between the June and July sampling periods. However, analysis of similarity (ANOSIM) did not indicate a statistical significance (p = 0.072) between the two sampling periods. Bacterial communities for sites 1, 2 and 3 June were similar as indicated by their clustering on the MDS graph and dendrogram (Fig. 3a). Likewise, sites 2–5 July grouped together, indicating their relatedness in BCC. The July cluster showed to be distantly related to site 5 June, suggesting a slight similarity in bacterial diversity. MDS analysis of site 4 June and site 1 July showed no resemblance in community composition to any of the other sites.

Bray-Curtis dissimilarity dendrogram showing the relatedness of the bacterial communities in terms of phyla (a) and class (b) among the five sampling sites in June and July. The relative abundance of taxonomic groups is expressed as the percentage of the total community. The dendrogram and bacterial community profiles were calculated from 454-pyrosequencing data sets
The BCC in the Mooi River consisted of ten phyla (Fig. 3a), 16 classes (Fig. 3b) and 75 genera. For all samples, Proteobacteria and Bacteroidetes were encountered most frequently, representing 22–46 % and 18–60 % of each sequence library, respectively (Fig. 3a). Actinobacteria was also dominant, but with large sample-to-sample variation. The Proteobacteria were distributed in descending order as Betaproteobacteria, Alphaproteobacteria, Gammaproteobacteria, Epsiolonproteobacteria and Deltaproteobacteria (Fig. 3b). The majority of Betaproteobacteria sequences were affiliated within the family Comamonadaceae in Burkholderiales. Within the family Comamonadaceae, Hydrogenophaga, Limnohabitans and Polaromonas predominated at most sites during both sampling periods. Among the Bacteroidetes members detected, Sphingobacteria and Flavobacteria dominated bacterial communities (Fig. 3B). Genera that occurred most frequently and in high abundance throughout the river included Arcicella, Solitalea and Flavobacterium. The relative abundance of Planctomycetes, Verrucomicrobia and Cyanobacteria were noticeably higher in June compared to the July samples, even though their abundance varied across sites.
Associations between physico-chemical water characteristics and bacterial community structures
CCA was performed to gain an overview on the relatedness between the bacterial community structures and physico-chemical water characteristics (Fig. 4a, b). The CCA biplot showed in total a 100 % species-environment correlation (Fig. 4a). Monte Carlo permutation tests indicated that the overall species-environment relationships were statistically significant (p = 0.007). The first two axes explained 93.06 % of the total variance in the abundance and distribution of taxa tested. Results suggest that four of the environmental variables tested (pH, DO, sulphate and chlorophyll-a) accounted for variability in the spatial succession of bacterial communities. A significant positive correlation existed between Bacteroidetes and DO (p = 0.007). In contrast, Verrucomicrobia (Cerasicoccus) and Betaproteobacteria (Malikia and Leadbetterella) were negatively associated with DO levels (0.001 ≤ p ≤ 0.04). In accordance with the CCA data, the relative abundance of Verrucomicrobia declined substantially from June to July at site 4, whilst the DO levels at this site increased from June to July.

Canonical correspondence analysis (CCA) biplots of bacterial communities at a phylum/class level and b genera in correlation with environmental variables. Significant correlations (p < 0.05) between bacterial groups and environmental parameters are indicated by circles. 454-pyrosequencing data were normalised to the same number of reads (516 reads) before analysis
The abundance of Betaproteobacteria was significantly affected by the slight pH variability (p = 0.046) and changes in sulphate concentrations (p = 0.022). Although the pH remained relatively constant throughout the river, the sulphate levels increased from sites 1 to 5 (June and July).
From Fig. 4a, b, it is evident that Acidobacteria and Epsilonproteobacteria had a close relationship with chlorophyll-a levels (0.023 ≤ p ≤ 0.031). Results suggest that Acidobacteria favoured low chlorophyll-a concentrations whilst elevated levels of this variable stimulated an increase in the abundance of Epsilonproteobacteria. Furthermore, various Betaproteobacterial genera demonstrated positive and negative associations with chlorophyll-a levels. Limnohabitans (p = 0.024) was negatively associated with chlorophyll-a, where Pigmentiphaga, Duganella and Pseudorhodoferax (0.003 ≤ p ≤ 0.029) showed a positive correlation. Ordination and Spearman correlation also established a negative association between chlorophyll-a and Acidobacteria_Gp6 (Acidobacteria) (p = 0.031), whilst Singulispaera (Planctomycetes) (p = 0.034) was positively associated.
Discussion
The main aim of this study was to assess the impacts of anthropogenic inputs on bacterial communities in the Mooi River using physico-chemical analysis, culture-dependent techniques and 454-pyrosequencing. Due to the known limitation of phenotypic methods, cultivation techniques and pyrosequencing were combined to obtain a better overview of the BCC in the Mooi River. Physico-chemical and microbiological data indicated signs of disturbances from the reference (site 1) to downstream sites (sites 2 to 5). The environmental variables DO, EC, sulphate and phosphate levels were higher at the downstream sites and similar to what has been observed for previous freshwater systems that were impacted by urbanisation (Liu et al. 2012; Drury et al. 2013). As indicated by Van der Walt et al. (2002), deterioration in the water quality below the Boskop Dam is the result of urban and industrial storm water runoff, as well as the Potchefstroom sewage works (site 4) which mainly causes an increase in phosphate levels at Kromdraai (confluence of the Mooi and Vaal River). In addition, higher counts of indicator and heterotrophic bacteria were enumerated at this site (site 4). This might be explained by inputs from the sewage works, but also urban runoff and livestock farming along the river. Furthermore, dry land farming practices might add substantial quantities of nitrogen and phosphorus to the river in the form of fertilisers and animal manure. It is likely that the high number of property developments in the northern region of Potchefstroom (downstream from site 3) may result into several sewage leaks. In such an event, sewage may wash into storm water systems and descend toward the river that will negatively affect the water quality.
The taxonomic composition of bacterial communities significantly altered from the upstream to downstream sites due to changes in major taxonomic groups. For instance, in both June and July Betaproteobacteria (25~60 %) was the major taxonomic group detected at the reference site, followed by Bacteroidetes (15~20 %) and Actinobacteria. The contribution of the various phyla differed at the downstream sites, particular at sites 2 and 4 (July) and site 5. Bacterial communities were mainly dominated by Bacteroidetes, Betaproteobacteria, Actinobacteria, Alphaproteobacteria and Verrucomicrobia. The temporal shift in BCC across sites might be attributed to environmental conditions. The climate in Potchefstroom region during the winter season (June and July) is typically dry (<20 mm rainfall) and cold with an average midday temperature of 22 ± 0.45 °C. However, the northern part of South Africa experienced a cold front in mid-July (12 days before sampling) and daytime temperatures dropped below 16 °C. The effects of the cold front lasted for 3 days. The drop in temperature likely created a stress-induced habitat that reduced bacterial growth rates and even survival ability of some species. Although temperature is the best predictor of the temporal variability in BCC, other parameters such as biotic and chemical factors also might have influenced community composition.
A striking result to emerge from the data is the low bacterial diversity (richness and evenness) calculated for the reference site (Muiskraal), whereas the downstream sites were more diverse. Also, BCC at the downstream sites was highly similar, particularly for the July sites. The higher bacterial diversity at the downstream sites is not surprising since increased concentrations of nutrients has been shown to stimulate planktonic bacterial growth (Goñi-Urriza et al. 1999) and benthic bacterial numbers (Wakelin et al. 2008). Anthropogenic inputs, such as septic tanks, storm water runoff, sewage treatment plant overflow and agricultural runoff may create conditions of greater habitat heterogeneity to allow the development of higher community diversity. Moreover, similarity in bacterial communities might be attributed to biotic homogenisation, which suggests that anthropological modifications of the environment are decreasing the biological differences between natural ecosystems (McKinney 2006; Drury et al. 2013). Consequently, these ecosystems consistently support a subset of naturally occurring species that can tolerate human activities (McKinney 2006). Our results imply that environmental inputs may be a key factor in biotic homogenisation of riverine bacterial communities.
CCA suggested that the altered environmental conditions significantly affected the spatial succession of bacterial communities in the Mooi River. Although the results should be interpreted with caution considering the low number of sampling events, some trends appeared. Multivariate analysis showed that pH, temperature, DO, sulphate and chlorophyll-a levels were the major factors to determine variation in BCC. Spatial variance in the abundances of Betaproteobacteria, Epsilonproteobacteria, Acidobacteria, Bacteroidetes and Verrucomicrobia were linked to physico-chemical variables measured in this study. These results are consistent with previous findings that associated the impact of environmental factors with BCC in freshwater systems (Crump and Hobbie 2005; Lindström et al. 2005; De Figueiredo et al. 2012).
Betaproteobacteria is often the dominant group in freshwater systems (Van Der Gucht et al. 2005; Newton et al. 2011) and its abundance has been associated with pH, conductivity, temperature, total suspended solids (TSS), chlorophyll-a, soluble reactive phosphorus and ammonium levels (Brümmer et al. 2000; Altmann et al. 2003; De Figueiredo et al. 2012). In this study, Betaproteobacteria abundance correlated positively with pH and negatively with sulphate levels. Our results are supported by previous studies that show that the abundance of Betaproteobacteria favours environments with a higher pH (De Figueiredo et al. 2012). The negative correlation between Betaproteobacteria and sulphate levels is not entirely clear as a previous study has shown that members of this phylotype can utilise various forms of organic and inorganic sulphate as energy source (Bhowal and Chakraborty 2011).
Limnohabitans and three other minor Betaproteobacterial genera (Duganella, Pigmentiphaga and Pseudorhodoferax) indicated strong associations with chlorophyll-a levels. Limnohabitans showed a negative correlation to chlorophyll-a values, whilst Duganella, Pigmentiphaga and Pseudorhodoferax positively related to this variable. Notably, the latter three genera also showed positive correlations to each other. Previous studies reported significant associations between certain Betaproteobacterial groups, phytoplankton populations and/or phytoplankton-derived organic material (Šimek et al. 2008; Paver and Kent 2010). Šimek et al. (2008) and Paver and Kent (2010) demonstrated the ability of Limnohabitans (R-BT lineage) and Polynucleobacter necessarius to utilise specific algal exudates as a key substrate for growth. Conversely, other studies reported inverse relationships between Limnohabitans (R-BT, Lhab-A2 and Lhab-A4 lineages) and phytoplankton species (Horňák et al. 2008; Eiler et al. 2012). Our findings suggest that the negative relationship between Limnohabitans and phytoplankton may be the result of direct competition for nutrients and/or antagonistic activities such as the production of algal antimicrobial substances (Sigee 2005; Eiler et al. 2012). In contrast, Duganella, Pigmentiphaga and Pseudorhodoferax likely developed a symbiotic relationship with phytoplankton producers with close metabolic coupling. Bacteria have high efficiencies of nitrogen and phosphorus uptake when inorganic nutrients are limited and may provide an important pathway for algae to absorb nitrogen and phosphorus under these conditions (Sigee 2005). This phenomenon may be responsible for the interdependent relationship between Duganella, Pigmentiphaga, Pseudorhodoferax and phytoplankton producers. Duganella and Pigmentiphaga are capable of reducing nitrate to nitrite (Madhaiyan et al. 2013) and/or dephosphorylate inorganic phosphorus compounds (Chen et al. 2009), thereby providing inorganic nutrients for algal growth and development. In return, phytoplankton provided autochthonous dissolved organic carbon (DOC) that favoured the establishment of these genera (Eiler et al. 2003; Laque et al. 2010). The co-occurrences of Duganella, Pigmentiphaga and Pseudorhodoferax would seem to reflect similar or complementary functions (Eiler et al. 2012).
Besides the above-mentioned associations, Malikia was the only Betaproteobacteria genus that statistically correlated with environmental variables (DO and phosphates) other than chlorophyll-a. This genus was detected mainly at the downstream sites (particularly sites 4 and 5) which showed elevated phosphate levels. There is evidence to suggest that Malikia is capable of degrading aromatic hydrocarbons such as polyhydroxyalkanoates (PHA’s) and polyphosphates (Spring et al. 2005; Táncsics et al. 2010). This genus accumulates high quantities of polyphosphates as intracellular granules (Gavigan et al. 1999) and is believed to play a major role in the enhanced biological phosphorus removal (EBPR) process of wastewater treatment plants (Spring et al. 2005). This theory is validated by previous studies that isolated Malikia from activated sludge of a municipal wastewater treatment plant (Spring et al. 2005), polluted rivers in urban and suburban areas (Yi et al. 2011; Drury et al. 2013) and groundwater contaminated by aromatic hydrocarbons (Táncsics et al. 2010). Our results agree with Yi et al. (2011) and Drury et al. (2013) that detected high abundance of Malikia in freshwaters impacted by anthropogenic activities such as industrial discharge, effluent from wastewater treatment plants and sewage runoff from urban and rural communities. This further supports the idea that the downstream sites were polluted by sewage originating from urban infrastructures and/or agricultural activities.
Bacteroidetes sequences were more abundant at the downstream sites with Arcicella and Flavobacterium as the two dominant genera. The Bacteroidetes group, in particular Flavobacterium, is often found in high abundance in mesotrophic, eutrophic and hypertrophic water bodies (Haller et al. 2011; Drury et al. 2013) and usually correlates with high nutrient levels (De Figueiredo et al. 2012). Members are well known to be proficient in degrading dissolved organic material (DOM), especially in nutrient-rich waters where biomacromolecules accumulate (Eiler and Bertilsson 2007; Kirchman 2012). Despite the high concentrations of inorganic nutrients at the downstream sites, no direct link could be established between Bacteroidetes and nutrient levels. Instead, its abundance appeared to correlate with higher DO concentrations. These findings are in contrast with previous studies that reported close associations between Bacteroidetes and nitrogen sources (De Figueiredo et al. 2007; 2010). The relationship between Bacteroidetes and DO needs to be interpreted with caution, particularly in the absence of data on the organic matter in the water column. Flavobacterium species thrive in the presence of complex macromolecules (Kirchman 2012) as they are important metabolisers of various high-molecular-weight (HMW) DOM (Kisand et al. 2002). High-molecular-weight DOM is degraded via photochemical processes (Kisand et al. 2005) that require dissolved oxygen as an electron acceptor (Zafiriou et al. 1984). Photochemical consumption of DOM may explain the positive association between Bacteroidetes and DO levels. In a previous study, we have provided evidence that the abundance of Flavobacterium could potentially be associated with algal blooms (Jordaan and Bezuidenhout 2013) Future studies are required to determine the levels and effects of anthropogenic inputs on riverine DOM and the response of Bacteroidetes to these compounds.
Verrucomicrobia, especially the genus Cerasicoccus, showed a significant inverse relationship to DO levels. The ecological roles of Verrucomicrobia remained largely unexplored, but its presence has been associated with eutrophic or nutrient-rich waters where phosphorus levels are high (Lindström et al. 2004; Jordaan and Bezuidenhout 2013) and environments contaminated with hydrocarbons, heavy metals and pesticides (Pereira et al. 2006; Paissé et al. 2008; Vishnivetskaya et al. 2011).
Finally, potential obligate and/or opportunistic pathogenic genera within the Actinobacteria (Leifsonia and Mycobacterium), Alphaproteobacteria (Brevundimonas, Roseomonas, Rhodobacter and Sphingomonas), Epsilonproteobacteria (Arcobacter), and Gammaproteobacteria (Aeromonas and Pseudomonas) groups were detected mainly at the downstream sites (Decker et al. 1992; Struthers et al. 1996; Evtushenko et al. 2000; Ho et al. 2006; Parker and Shaw 2011; Magee and Ward 2012; Djordjevic et al. 2013). Their presence in the Mooi River may be regarded as a potential risk for human and animal health, considering that the river is used for recreational activities and agricultural purposes. Members of these genera are found in various natural environments (Lee et al. 2001; Rickard et al. 2003), but are also associated with polluted waters (Edwards et al. 2001; Marcel et al. 2002; Kalwasińska et al. 2008). Arcobacter species in environmental waters often correlate with high faecal indicator counts (Fong et al. 2007; Collado et al. 2008). These bacteria are found in high numbers in sewage water inflow to wastewater treatment plants (McLellan et al. 2010) and livestock farming effluents (Chinivasagam et al. 2007). Their high abundance in surface waters further indicates contamination by the above-mentioned sources. In addition, Aeromonas and Sphingomonas thrive in polluted environments because they are able to degrade various recalcitrant compounds (Pinyakong et al. 2003; Ghosh et al. 2004). Although they are not considered to be of faecal origin, their presence is of interest since they hold potential to be used as indicators of aromatic hydrocarbon pollution. Future studies on the abundance and distribution of Acrobacter, Aeromonas and Sphingomonas in freshwater systems and their relationship to environmental variables are required to establish if they can be used as indicators of anthropogenic stress.
Conclusions
The overall results demonstrated that spatial variation in BCC in the Mooi River was influenced by human activities related to urbanisation and agricultural land use impacts. Physico-chemical and microbiological data indicated that water quality deteriorated from the upstream rural sector until the confluence with the Vaal River. Multivariate statistical analysis indicated that temperature, pH, DO, sulphate and chlorophyll-a levels had the greatest impact on BCC. Our work also identified potential alternative pollution indicator groups (Acrobacter, Aeromonas and Sphingomonas) that may be used to track faecal and organic pollution in freshwater systems. A number of potential limitations need to be considered. First, we are aware that part of the spatial variance in BCC could be related to other variables not measured here. Secondly, it is plausible that the small sample size and frequency could have influenced the BCC results obtained. Nevertheless, these results contribute to aspects of the understanding of urbanisation on riverine BCC, particularly on major taxonomic groups and genera. As metagenomics sequencing and freshwater metabolism techniques continue to advance, we believe that this approach has the potential to (i) measure BCC responses to anthropogenic perturbations, (ii) measure the overall ecosystem functioning, (iii) quantify primary production and respiration rates to evaluate the trophic status of the river and (iv) estimate organic matter transfer between and within aquatic systems. Future research on the catchment should sample at least once a month for two/three consecutive years. Not only will this improve the understanding of how urbanisation affects BCC and its activities, but it will also provide priori knowledge of diversity before and after perturbations at a given site by comparing a perturbed site with pristine controls (Ager et al. 2010). Dominant taxa detected at polluted sites deserve to be studied in more detail since information about their roles in polluted freshwater systems is scarce. Lastly, the use of biofilms (on natural rocks and stones) and sediment bacteria as indicators of anthropogenic disturbances should be assessed. Several studies have demonstrated the usefulness of both biofilms and sediment bacteria to determine the impact of urbanisation on freshwater systems (Drury et al. 2013; Lin et al. 2014). The datasets can then be compared with the current dataset to ultimately conclude which type of community (planktonic, biofilm or sediment) accurately represents the ecological nature of the freshwater system.
References
Ager D, Evans S, Li H, Lilley AK, Van der Gast CJ (2010) Anthropogenic disturbance affects the structure of bacterial communities. Environ Microbiol 12(3):670–678
Altmann D, Stief P, Amann R, De Beer D, Schramm A (2003) In situ distribution and activity of nitrifying bacteria in freshwater sediment. Environ Microbiol 5(9):798–803
Bhowal S, Chakraborty R (2011) Five novel acid-tolerant oligotrophic thiosulfate-metabolizing chemolithotrophic acid mine drainage strains affiliated with the genus Burkholderia of Betaproteobacteria and identification of two novel soxB gene homologues. Res Microbiol 162(4):436–445
Bricheux G, Morin L, Le Moal G, Coffe G, Balestrino D, Charbonnel N, Bohatier J, Forestier C (2013) Pyrosequencing assessment of prokaryotic and eukaryotic diversity in biofilm communities from a French river. Microbiol Open 2(3):402–414
Brümmer IHM, Fehr W, Wagner-Döbler I (2000) Biofilm community in polluted rivers: abundance of dominant phylogenetic groups over a complete annual cycle. Appl Environ Microb 66(7):3078–3082
Chen Y-G, Zhang Y-Q, Huang K, Tang S-K, Cao Y, Shi J-X, Xiao H-D, Cui X-L, Li W-J (2009) Pigmentiphaga litoralis sp. nov., a facultatively anaerobic bacterium isolated from a tidal flat sediment. Int J Syst Evol Micr 59(3):521–525
Chinivasagam HN, Corney BG, Wright LL, Diallo IS, Blackall PJ (2007) Detection of Arcobacter spp. in piggery effluent and effluent irrigated soils in southeast Queensland. J Appl Microbiol 103(2):418–426
Collado L, Inza I, Guarro J, Figueras MJ (2008) Presence of Arcobacter spp. in environmental waters correlates with high levels of fecal pollution. Environ Microbiol 10(6):1635–1640
Crump BC, Hobbie JE (2005) Synchrony and seasonality in bacterioplankton communities of two temperate rivers. Limnol Oceanogr 50(6):1718–1729
De Figueiredo DR, Pereira MJ, Moura A, Silva L, Bárrios S, Fonseca F, Henríques I, Correia A (2007) BCC over a dry winter in meso- and eutrophic Portuguese water bodies. FEMS Microbiol Ecol 59(3):638–650
De Figueiredo DR, Pereira MJ, Correia A (2010) Seasonal modulation of bacterioplankton community at a temperate eutrophic shallow lake. World J Microb Biot 26(6):1067–1077
De Figueiredo DR, Ferreira RV, Cerqueira M, Condesso De Melo T, Pereira MJ, Castro BB, Correia A (2012) Impact of water quality on bacterioplankton assemblage along Cértima River Basin (central western Portugal) assessed by PCR-DGGE and multivariate analysis. Environ Monit Assess 184(1):471–485
Decker CF, Hawkins RE, Simon GL (1992) Infections with Pseudomonas paucimobilis. Clin Infect Dis 14(3):783–784
Djordjevic Z, Folic MM, Zivic Z, Markovic V, Jankovic SM (2013) Nosocomial urinary tract infections caused by Pseudomonas aeruginosa and Acinetobacter species: sensitivity to antibiotics and risk factors. Am J Infect Control 41(12):1182–1187
Drury B, Rosi-Marshall E, Kelly JJ (2013) Wastewater treatment effluent reduces the abundance and diversity of benthic bacterial communities in urban and suburban rivers. Appl Environ Microb 79(6):1897–1905
DWAF (1996) Department of Water Affairs and Forestry. South African Water Quality Guidelines, 1st ed. Volume 8: Field Guide.
DWAF (2009) Directorate national water resource planning. Department of Water Affairs and Forestry, South Africa, September 2009. Integrated water quality management plan for the Vaal River System: task 2: water quality status assessment of the Vaal River system. Report No. P RSA C000/00/2305/1.
Edwards ML, Lilley AK, Timms-Wilson TH, Thompson IP, Cooper I (2001) Characterisation of the culturable heterotrophic bacterial community in a small eutrophic lake (Priest Pot). FEMS Microbiol Ecol 35(3):295–304
Eiler A, Langenheder S, Bertilsson S, Tranvik LJ (2003) Heterotrophic bacterial growth efficiency and community structure at different natural organic carbon concentrations. Appl Environ Microb 69(7):3701–3709
Eiler A, Bertilsson S (2007) Flavobacteria blooms in four eutrophic lakes: linking population dynamics of freshwater bacterioplankton to resource availability. Appl Environ Microb 73(11):3511–3518
Eiler A, Heinrich F, Bertilsson S (2012) Coherent dynamics and association networks among lake bacterioplankton taxa. ISME J 6(2):330–342
Evtushenko LI, Dorofeeva LV, Subbotin SA, Cole JR, Tiedje JM (2000) Leifsonia poae gen. nov., sp. nov., isolated from nematode galls on Poa annua, and reclassification of ‘Corynebacterium aquaticum’ Leifson 1962 as Leifsonia aquatica (ex Leifson 1962) gen. nov., nom. rev., comb. nov. and Clavibacter xyli Davis et al. 1984 with two subspecies as Leifsonia xyli (Davis et al. 1984) gen. nov., comb. nov. Int J Syst Evol Micr 50(1):371–380
Fong TT, Mansfield LS, Wilson DL, Schwab DJ, Molloy SL, Rose JB (2007) Massive microbiological groundwater contamination associated with a waterborne outbreak in Lake Erie, South Bass Island, Ohio. Environ Health Persp 115(6):856–864
Ford TE (2000) Response of marine microbial communities to anthropogenic stress. J Aquat Ecosyst Stress Recovery 7(1):75–89
Gavigan J-A, Leonard MM, Dobson ADW (1999) Regulation of polyphosphate kinase gene expression in Acinetobacter baumannii 252. Microbiology 145(10):2931–2937
Ghosh M, Verma SC, Mengoni A, Tripathi AK (2004) Enrichment and identification of bacteria capable of reducing chemical oxygen demand of anaerobically treated molasses spent wash. J Appl Microbiol 96(6):1278–1286
Goñi-Urriza M, Capdepuy M, RAymond N, Quentin C, Caumette P (1999) Impact of an urban effluent on the bacterial community structure in the Arga River, Spain, with special reference to culturable Gram-negative rods. Can J Microbiol 45(10):826–832
Haller L, Tonolla M, Zopfi J, Peduzzi R, Wildi W, Poté J (2011) Composition of bacterial and archaeal communities in freshwater sediments with different contamination levels (Lake Geneva, Switzerland). Water Res 45(3):1213–1228
HDA (2012) The Housing Development Agency. Informal settlement status, South Africa
Ho HT, Lipman LJ, Gaastra W (2006) Arcobacter, what is known and unknown about a potential foodborne zoonotic agent! Vet Microbiol 115(1–3):1–13
Horňák K, Jezbera J, Šimek K (2008) Effects of a Microcystis aeruginosa bloom and bacterivory on bacterial abundance and activity in a eutrophic reservoir. Aquat Microb Ecol 52(2):107–117
Iwegbue CMA, Arimoro FO, Nwajei GE, Eguavoen OI (2012) Concentrations and distribution of trace metals in water and streambed sediments of Orogodo River, Southern Nigeria. Soil Sediment Contam 21(3):382–406
Jordaan K, Bezuidenhout CC (2013) The impact of physico-chemical water quality parameters on bacterial diversity in the Vaal River, South Africa. Water SA 39(3):365–376
Kalwasińska A, Kęsy J, Donderski W, Lalke–POrczyk E (2008) Biodegradation of carbendazim by planktonic and benthic bacteria of eutrophic lake Chełmżyńskie. Pol J Environ Stud 17(4):515–523
Kamjunke N, Buttner O, Jager CG, Marcus H, Von Tumpling W, Halbedel S, Norf H, Brauns M, Baborowski M, Wild R, Borchardt D, Weitere M (2013) Biogeochmical patterns in a river network along a land use gradient. Environ Monit Assess 185(11):9221–36
Kirchman DL (2012) Processes in microbial ecology. Oxford University Press, New York
Kisand V, Cuadros R, Wikner J (2002) Phylogeny of culturable estuarine bacteria catabolizing riverine organic matter in the northern Baltic Sea. Appl Environ Microb 68(1):379–388
Kisand V, Andersson N, Wikner J (2005) Bacterial freshwater species successfully immigrate to the brackish water environment in the northern Baltic. Limnol Oceanogr 50:945–956
Lane DJ (1991) 16S/23S rRNA sequencing. In: Stackebrandt E, Goodfellow M (eds) Nucleic acid techniques in bacterial systematics. John Wiley & Sons, New York, pp 115–175
Laque T, Farjalla VF, Rosado AS, Esteves FA (2010) Spatiotemporal variation of bacterial community composition and possible controlling factors in tropical shallow lagoons. Microbial Ecol 59(4):819–829
Larsen PE, Field D, Gilbert JA (2012) Predicting bacterial community assemblages using an artificial neural network approach. Nat Methods 9:621–625
Lee J-S, Shin YK, Yoon J-H, Takeuchi M, Pyun Y-R, Park Y-H (2001) Sphingomonas aquatilis sp. nov., Sphingomonas koreensis sp. nov. and Sphingomonas taejonensis sp. nov., yellow-pigmented bacteria isolated from natural mineral water. Int J Syst Evol Micr 51(4):1491–1498
Li W, Shen Z, Tian T, Liu R, Qiu J (2012) Temporal variation of heavy metal pollution in urban stormwater runoff. Front Environ Sci En 6(5):692–700
Lin S, Wang Y, Lin J, Quan C (2014) Response of planktonic and benthic microbial community to urban pollution from sewage discharge in Jilin reach of the second Songhua River, China. Clean – Soil Air Water 42(10):1376–1383
Lindström ES, Vrede K, Leskinen E (2004) Response of a member of the Verrucomicrobia, among the dominating bacteria in a hypolimnion, to increased phosphorus availability. J Plankton Res 26(2):241–246
Lindström ES, Kamst-van Agterveld MP, Zwart G (2005) Distribution of typical freshwater bacterial groups is associated with pH, temperature, and lake water retention time. Appl Environ Microb 71(12):8201–8206
Liu Z, Huang S, Sun G, Xu Z, Xu M (2012) Phylogenetic diversity, composition and distribution of bacterioplankton community in the Dongjiang River, China. FEMS Microbiol Ecol 80(1):30–44
Lu X-M, Lu P-Z (2014) Characterization of bacterial communities in sediments receiving various wastewater effluents with high-throughput sequencing analysis. Microbial Ecol 67(3):612–623
Madhaiyan M, Poonguzhali S, Saravanan VS, Hari K, Lee K-C, Lee J-S (2013) Duganella sacchari sp. nov. and Duganella radices sp. nov., two novel species isolated from rhizosphere of field-grown sugar cane. Int J Syst Evol Micr 63:1126–1131
Magee JG, Ward AC (2012) Genus I. Mycobacterium. In: Goodfellow M, Kämpfer P, Busse H-J, Trujillo ME, Suzuki K, Ludwig W, Whitman B (eds) Bergey’s manual of systematic bacteriology, vol 5, 2nd edn. The Actinobacteria. Springer, New York, pp 312–375
Marcel KA, Antoinette AA, Mireille D (2002) Isolation and characterization of Aeromonas species from an eutrophic tropical estuary. Mar Pollut Bull 44(12):1341–1344
Markúsdóttir M, Heiðmarsson S, Eyþórsdótti A, Magnússon KP, Vilhelmsson O (2013) The natural and anthropogenic microbiota of Glerá, a sub-arctic river in northeastern Iceland. Int Biodeter Biodegr 84:192–203
Martinuzzi S, Januchowski-Hartley SR, Pracheil BM, McIntyre PB, Plantinga AJ, Lewis DJ, Lewis DJ, Radeloff VC (2014) Threats and opportunities for freshwater conservation under future land use change scenarios in the United States. Glob Change Biol 20(1):113–24
Mckinney ML (2006) Urbanization as a major cause of biotic homogenization. Biol Conserv 127(3):247–260
Mclellan SL, Huse SM, Mueller-Spitz SR, Andreishcheva EN, Sogin ML (2010) Diversity and population structure of sewage-derived microorganisms in wastewater treatment plant influent. Environ Microbiol 12(2):378–392
Newton RJ, Jones SE, Eiler A, Mcmahon K, Bertilsson S (2011) A guide to the natural history of freshwater lake bacteria. Microbiol Mol Biol Rev 75(1):14–49
O’Driscoll M, Clinton S, Jefferson A, Manda A, McMillan S (2010) Urbanization effects on watershed hydrology and in-stream processes in the southern United States. Water 2(3):605–648
Paerl HW, Dyble J, Moisander PH, Noble RT, Piehler MF, Pinckney JL, Steppe TF, Twomey L, Valdes LM (2003) Microbial indicators of aquatic ecosystems change: current applications to eutrophication studies. FEMS Microbiol Ecol 46(3):233–246
Paissé S, Coulon F, Goñi-Urriza M, Peperzak L, McGenity TJ, Duran R (2008) Structure of bacterial communities along a hydrocarbon contamination gradient in a coastal sediment. FEMS Microbiol Ecol 66(2):295–305
Parker JL, Shaw JG (2011) Aeromonas spp. clinical microbiology and disease. J Infect 62(2):109–118
Paver SF, Kent AD (2010) Temporal patterns in glycolate-utilizing bacterial community composition correlate with phytoplankton population dynamics in humic lakes. Microbial Ecol 60(2):406–418
Pereira RM, DA Silveira EL, Scaquitto DC, Pedrinho EAN, Val-Moraes SP, Wickert E, Carareto-Alves LM (2006) Molecular characterization of bacterial populations of different soils. Braz J Microbiol 37(4):439–447
Pesce S, Fajon C, Bardot C, Bonnemoy F, Portelli C, Bohatier J (2008) Longitudinal changes in microbial planktonic communities of a French river in relation to pesticide and nutrient inputs. Aquat Toxicol 86(3):352–360
Pinyakong O, Habe H, Omori T (2003) The unique aromatic catabolic genes in sphingomonads degrading polycyclic aromatic hydrocarbons (PAHs). J Gen Appl Microbiol 49(1):1–19
Rice EW, Baird RB, Eaton AD, Clesceri LS (2012). Standard methods for the examination of water and wastewater, 22nd ed. American Public Health Association, American Water Works Association, Water Environment Federation
Rickard AH, McBain AJ, Ledder RG, Handley PS, Gilbert P (2003) Coaggregation between freshwater bacteria within biofilm and planktonic communities. FEMS Microbiol Lett 220(1):133–140
SABS (2010) South African Bureau of Standards, Water–Nitrate and nitrite content. 2nd ed. Standards South Africa
Sartory DP, Grobbelaar JU (1984) Extraction of chlorophyll a from freshwater phytoplankton for spectrophotometric analysis. Hydrobologia 114(3):177–187
Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF (2009) Introducing MOTHUR: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microb 75(23):7537–7541
Schloss PD, Gevers D, Westcott SL (2011) Reducing the effects of PCR amplification and sequencing artifacts on 16S rRNA-based studies. PloS ONE 6:e27310
Sigee DC (2005) Freshwater microbiology: biodiversity and dynamic interactions of microorganisms in the aquatic environment. John Wiley & Sons, England
Šimek K, Horňák K, Jezbera J, Nedoma J, Znachor P, Hejzlar J, Sed’a J (2008) Spatio-temporal patterns of bacterioplankton production and community composition related to phytoplankton composition and protistan bacterivory in a dam reservoir. Aquat Microb Ecol 51(3):249–262
Singh J, Behal A, Singla N, Joshi A, Birbian N, Singh S, Bali V, Batra N (2009) Metagenomics: concept, methodology, ecological inference and recent advances. Biotechnol J 4(4):480–494
Spring S, Wagner M, Schumann P, Kämpfer P (2005) Malikia granosa gen. nov., sp. nov., a novel polyhydroxyalkanoate- and polyphosphate accumulating bacterium isolated from activated sludge, and reclassification of Pseudomonas spinosa as Malikia spinosa comb. nov. Int J Syst Evol Micr 55(2):621–629
Stabili L, Cavallo RA (2011) Microbial pollution indicators and culturable heterotrophic bacteria in a Mediterranean area (Southern Adriatic Sea Italian coasts). J Sea Res 65(4):461–469
Stats SA (2011) Census 2011. http://www.statssa.gov.za/census2011/default.asp. Accessed 10 February 2013.
Struthers M, Wong J, Janda JM (1996) An initial appraisal of the clinical significance of Roseomonas species associated with human infections. Clin Infect Dis 23(4):729–733
Táncsics A, Szabó I, Baka E, Szoboszlay S, Kukolya J, Kriszt B, Márialigeti K (2010) Investigation of catechol 2,3-dioxygenase and 16S rRNA gene diversity in hypoxic, petroleum hydrocarbon contaminated groundwater. Syst Appl Microbiol 33(7):398–406
Van der Gucht K, Vandekerckhove T, Vloemans N, Cousin S, Muylaert K, Sabbe K, Gillis M, Declerk S, De Meester L, Vyverman W (2005) Characterization of bacterial communities in four freshwater lakes differing in nutrient load and food web structure. FEMS Microbiol Ecol 53(2):205–220
Van der Walt IJ, Winde F, Nell B (2002) Integrated catchment management: the Mooi River (North West Province, South Africa) as a case study. Cuadernos de Investigación Geográfica 28:109–126
Vishnivetskaya TA, Mosher JJ, Palumbo AV, Yang ZK, Podar M, Brown SD, Brooks SC, Gu B, Southworth GR, Drake MM, Brandt CC, Elias DA (2011) Mercury and other heavy metals influence bacterial community structure in contaminated Tennessee streams. Appl Environ Microb 77(1):302–311
Wakelin SA, Colloff MJ, Kookana RS (2008) Effect of wastewater treatment plant effluent on microbial function and community structure in the sediment of a freshwater stream with variable seasonal flow. Appl Environ Microb 74(9):2659–2668
Wang Q, Garrity GM, Tiedje JM, Cole JR (2007) Naïve Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl Environ Microb 73(16):5261–5267
Yergeau E, Lawrence JR, Sanschagrin S, Waiser MJ, Korber DR, Greera CW (2012) Next-generation sequencing of microbial communities in the Athabasca River and its tributaries in relation to oil sands mining activities. Appl Environ Microb 78(21):7626–7637
Yi H, Li Z, Shuying Z, Shuguang X (2011) Comparison of bacterioplankton communities in three heavily polluted streams in China. Biomed Environ Sci 24(2):140–145
Zafiriou OC, Joussot-Dubien J, Zepp RG, Zika RG (1984) Photochemistry of natural waters. Environ Sci Technol 18(12):358A–371A
Zampini M, Stevens PR, Pachebat JA, Kingston-Smith A, Mur LAJ, Hayes F (2015) RapGene: a fast and accurate strategy for synthetic gene assembly in Escherichia coli. Scientific Reports 5:11302. doi:10.1038/srep11302
Zarraonaindia I, Smith DP, Gilbert JA (2013) Beyond the genome: community-level analysis of the microbial world. Biol Philos 28(2):261–282
Acknowledgments
This work was supported by the Water Research Commission (WRC), South Africa, under project K5/1966. The authors kindly thank the cartographer, Liesl De Swardt (North-West University, Potchefstroom campus), for generating the geographical map.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Robert Duran
Rights and permissions
About this article

Cite this article
Jordaan, K., Bezuidenhout, C.C. Bacterial community composition of an urban river in the North West Province, South Africa, in relation to physico-chemical water quality. Environ Sci Pollut Res 23, 5868–5880 (2016). https://doi.org/10.1007/s11356-015-5786-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-015-5786-7