
Abstract
For in situ groundwater remediation, polyelectrolyte-modified nanoscale zerovalent iron particles (NZVIs) have to be delivered into the subsurface, where they degrade pollutants such as trichloroethylene (TCE). The effect of groundwater organic and ionic solutes on TCE dechlorination using polyelectrolyte-modified NZVIs is unexplored, but is required for an effective remediation design. This study evaluates the TCE dechlorination rate and reaction by-products using poly(aspartate) (PAP)-modified and bare NZVIs in groundwater samples from actual TCE-contaminated sites in Florida, South Carolina, and Michigan. The effects of groundwater solutes on short- and intermediate-term dechlorination rates were evaluated. An adsorbed PAP layer on the NZVIs appeared to limit the adverse effect of groundwater solutes on the TCE dechlorination rate in the first TCE dechlorination cycle (short-term effect). Presumably, the pre-adsorption of PAP “trains” and the Donnan potential in the adsorbed PAP layer prevented groundwater solutes from further blocking NZVI reactive sites, which appeared to substantially decrease the TCE dechlorination rate of bare NZVIs. In the second and third TCE dechlorination cycles (intermediate-term effect), TCE dechlorination rates using PAP-modified NZVIs increased substantially (~100 and 200%, respectively, from the rate of the first spike). The desorption of PAP from the surface of NZVIs over time due to salt-induced desorption is hypothesized to restore NZVI reactivity with TCE. This study suggests that NZVI surface modification with small, charged macromolecules, such as PAP, helps to restore NZVI reactivity due to gradual PAP desorption in groundwater.
Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Nanoscale zerovalent iron particles (NZVIs) have rapidly received much attention as a novel, in situ subsurface remediation agent to treat various kinds of vexing environmental contaminants, including chlorinated organics such as trichloroethylene (TCE) (Lowry 2007; Tratnyek and Johnson 2006; Zhang 2003). Various organic macromolecule surface modifiers, such as xanthane, guar gum, poly(aspartate) (PAP), poly(styrene sulfonate), carboxymethyl cellulose (CMC), poly(methyl methacrylate), poly(acrylic acid), and tri-block copolymers, are used to engineer NZVIs to inhibit their aggregation (Golas et al. 2010; Phenrat et al. 2008; Sakulchaicharoen et al. 2010; Saleh et al. 2005; Wang et al. 2010), increase their mobility in the subsurface (Kim et al. 2009; Phenrat et al. 2009a, 2010; Saleh et al. 2008; Vecchia et al. 2009), and provide pollutant selectivity (Bishop et al. 2010; Phenrat et al. 2011; Saleh et al. 2005; Wang and Zhou 2010), all of which are necessary for effective in situ remediation. Several promising pilot- and field-scale tests using surface-modified NZVIs have been reported (Bennett et al. 2010; He et al. 2010; Henn and Waddill 2006; Kocur et al. 2014).
From the materials science point of view, significant research progress has been made in understanding the important properties of bare and polymeric-modified NZVIs that affect NZVI reactivity with chlorinated organics (Liu et al. 2005a, 2007; Liu and Lowry 2006; Nurmi et al. 2005; Phenrat et al. 2009a; Sarathy et al. 2008). Different dechlorination rates (Liu et al. 2005a, b; Nurmi et al. 2005; Sakulchaicharoen et al. 2010; Song and Carraway 2008; Zhang et al. 1998), dechlorination pathways (Liu et al. 2005a, b; Nurmi et al. 2005; Song and Carraway 2008; Zhang et al. 1998), electron utilization efficiencies (Liu et al. 2005a, b; Song and Carraway 2008), and longevities (Liu et al. 2005a; Sarathy et al. 2008) have been attributed to the crystallinity and chemical composition of bare NZVIs (i.e., the presence of noble metals, such as Pd and Ni, on the NZVI surfaces). As for polymer-modified NZVIs, Phenrat et al. (2009b) revealed that when the surfaces of pre-synthesized NZVIs were modified by the physisorption of polyelectrolytes, the TCE dechlorination rate constant decreased nonlinearly with increasing adsorbed mass of the polyelectrolytes, with a maximum 24-fold decrease in reactivity, due to reactive site blocking and a decrease in the aqueous TCE concentration at the surfaces of the NZVIs due to the partitioning of TCE to the adsorbed polyelectrolytes. A similar finding was also reported by Wang and Zhou (2010) using solvent-responsive, polymer-coated NZVIs to degrade TCE. Nevertheless, increases in TCE reactivity with polymer-modified Fe-Pd bimetallic nanoparticles at low polyelectrolyte concentrations compared with bare Fe-Pd bimetallic nanoparticles were also observed (He and Zhao 2008; Sakulchaicharoen et al. 2010). This difference is probably because the Fe-Pd bimetallic nanoparticles were synthesized in the presence of polymers such as CMC, guar gum, and polyvinylpyrrolidone (PVP), thereby yielding smaller particles that were resistant to aggregation and, thus, more reactive than larger, non-stabilized Fe-Pd particles (He and Zhao 2008; Sakulchaicharoen et al. 2010).
However, in addition to the properties of the particles themselves, the interaction between NZVIs and other (non-target) inorganic ions and dissolved natural organic matter (NOM) in groundwater can affect the reactivity, longevity, and, thus, the performance of NZVIs. Because of geochemical cycles (dissolution and precipitation) of minerals in the subsurface, groundwater normally consists of various cationic and anionic species, such as Na+, Ca2+, Mg2+, NO3 −, Cl−, SO4 2−, HCO3 −, and HPO4 −2. Groundwater chemistry is known to affect the performance of the ZVI permeable reactive barrier (PRB) by controlling the Fe0 corrosion rate (El-Naggar 2006; Scherer et al. 2000), dechlorination rate (Klausen et al. 2003; Kohn et al. 2005; Su and Puls 2004), H2 production (Scherer et al. 2000), microbial activity (Scherer et al. 2000; Van Nooten et al. 2008), the formation of mineral precipitates on the surfaces of iron filings (Agrawal et al. 2002; Kohn et al. 2005), and the dissolution of the iron oxide layer on Fe0 (Agrawal et al. 2002). Some similar effects of anionic species on the performances of bare and bimetallic NZVIs were experimentally observed (Lim and Zhu 2008; Liu et al. 2007). At low concentrations (0.2–1 mM), reducible solutes, such as NO3 −, did not significantly affect the NZVI-mediated TCE dechlorination rate. However, at high concentrations (~5 mM), NO3 − reduced the reactivity of NZVIs with TCE after 3 days, even though Fe0 remained in the NZVIs. Presumably, at high NO3 − concentrations, the surface reaction was shifted from cathodic control (i.e., the reduction of TCE) to anodic control (i.e., the release of Fe2+ and electrons) and, thus, facilitated the formation of a passivating FeOOH layer (Liu et al. 2007). In the presence of high NO3 − and NO2 − concentrations, a similar decrease in reactivity and particle passivation was also observed for trichlorobenzene (TCB) dechlorination using bimetallic Fe-Pd nanoparticles (Lim and Zhu 2008).
In contrast, anions such as Cl−, SO4 2−, HCO3 −, and HPO4 −2 are not reducible by Fe0. Their effects on dechlorination using iron filings varied with their concentration. Several studies (Agrawal et al. 2002; Devlin and Allin 2005; Johnson et al. 1998) reported that high concentrations of non-reducible ionic species decreased ZVI reactivity through the formation of a passivating oxide layer. In contrast, other studies (Agrawal et al. 2002; Johnson et al. 1998) reported the opposite, i.e., that these ions promoted the dissolution of the iron oxide layer, leading to increased reactivity at low concentrations. As for NZVIs, a recent study (Liu et al. 2007) revealed that non-reducible ionic species decreased the TCE dechlorination rate by up to a factor of seven compared with deionized (DI) water, and the order of their effect followed their affinity for hydrous ferric oxide, i.e., Cl− < SO4 2− < HCO3 − < HPO4 2− at pH 8.9 (Liu et al. 2007). This implies that the inhibitory effect of these solutes on TCE degradation may be caused by reactive site blocking due to the formation of Fe-anion complexes on the NZVI surface.
The effect of non-catalytic cations, such as Na+, Ca2+, and Mg2+, on TCE dechlorination using NZVIs has not been systematically studied, although these cations, which normally coexist with anions in groundwater, are likely to accumulate more closely to the surfaces of NZVIs than anions according to the Boltzmann distribution of ions in a solution that contains negatively charged surfaces (Israelachvili 1992). At a neutral pH, the electrostatic interaction between the negatively charged NZVI surface and cations might exhibit reactive site blocking, which might affect the TCE dechlorination rate.
Field applications of NZVIs for in situ remediation cannot avoid interactions with a mixture of non-target, dissolved ionic and organic species that are commonly present in the subsurface, as mentioned previously. Understanding the effects of these dissolved species on the short- and intermediate-term TCE dechlorination rates of NZVIs is essential to determine the amount of NZVIs that should be injected into the subsurface to achieve a particular clean-up goal. While such effects on bare NZVIs are known, the effects on polymer-modified NZVIs, which are more practical for field applications, are not (He and Zhao 2008; Karn et al. 2009; Sakulchaicharoen et al. 2010). Dechlorination using NZVIs is an interfacial phenomenon that is substantially affected by the presence of adsorbed polymer layers (Phenrat et al. 2009b). Therefore, it is not appropriate to assume that the effects of groundwater solutes on TCE dechlorination using polymer-modified NZVIs will be similar to those using bare NZVIs.
The objective of this study was to experimentally examine the short- and intermediate-term effects of a mixture of dissolved ionic and organic species in natural groundwater on TCE dechlorination using polymer-modified NZVIs. Poly(aspartate) (PAP), a bio-polyelectrolyte, was used as a representative polymeric modifier in this study (Phenrat et al. 2009b, c). The three cycles of TCE dechlorination using PAP-modified and bare NZVIs were conducted in three different natural groundwater samples from Florida (FL), South Carolina (SC), and Michigan (MI). The difference between TCE dechlorination rates using bare and PAP-modified NZVIs in the same groundwater sample was discussed and attributed to the presence of adsorbed PAP layers. An initial spike of TCE was used to evaluate short-term TCE dechlorination, while the second and third spikes of TCE were used to evaluate intermediate-term TCE dechlorination. The presence of a Donnan potential inside the adsorbed polyelectrolyte layers was used to mechanistically explain the altered distributions of ionic species surrounding the NZVI surfaces that might hypothetically contribute to decreasing the adverse effects of dissolved ionic species on TCE dechlorination rates using polymer-modified NZVIs in comparison with bare NZVIs. A conceptual model that explains the decrease in solute concentrations at the NZVI surface, as well as the decrease in site blocking by (ionic and organic) solutes, due to the presence of adsorbed polyelectrolyte layers was proposed to illustrate how a polymeric surface coating limits the adverse effects of groundwater solutes on NZVI reactivity. In addition, the desorption of PAP from the surface of NZVIs in the presence of groundwater solutes was indirectly observed using electrophoretic mobility measurements and was attributed to the experimentally observed improvement in the intermediate-term TCE dechlorination rate for PAP-modified NZVIs.
Materials and methods
Chemicals
TCE (99.5+ %) was from Sigma-Aldrich (St. Louis, MO, USA). Acetylene (1000 ppm and 1 %), ethylene (1 %), ethane (1 %), vinyl chloride (VC) (10 ppm), and hydrogen (1.08 %) standards were from Alltech Chemicals (Subiaco, Australia). The balance gas standard was N2. Ultra high-purity argon, hydrogen (5.18 %), and N2 were from Butler Gas products (Pittsburgh, PA, USA).
Bare NZVIs
Reactive nanoscale iron particles (RNIPs), consisting of reactive Fe0/Fe3O4 core-shelled NZVI particles, were obtained from Toda Kogyo (Onada, Japan). The physical and chemical properties of the RNIPs have been previously reported (Liu et al. 2005b; Nurmi et al. 2005; Phenrat et al. 2007). Prior to use, RNIPs were stored as an aqueous slurry (pH 10.6) at approximately 300 g/L in an anaerobic chamber. From this slurry, a stock dispersion (10 mL at ~120 g/L) was prepared in 1 mM NaHCO3, followed by ultrasonication for 30 min to break up any aggregates that formed during storage. The N2-BET specific surface area of the RNIPs was ~15 m2/g. The Fe0 content of the particles determined from acid digestion and monitoring hydrogen evolution as previously described (Liu et al. 2005b) was ~15 % by mass.
Poly(aspartate)-modified NZVIs
Sodium PAP (MW 2000–3000 g/mol) stabilized NZVIs (MRNIP) were obtained from Toda Kogyo. The physical and chemical properties of the PAP-modified NZVIs have been previously reported (Phenrat et al. 2008; Saleh et al. 2007; 2008). The PAP monomer unit is aspartate, one of the 20 natural amino acid building blocks of proteins, making PAP of potential interest as an environmentally benign modifier. Prior to use, MRNIPs were stored as an aqueous slurry (pH 10.6) at approximately 180 g/L in an anaerobic chamber. From this slurry, a stock dispersion (10 mL at ~30 g/L) was prepared in DI water followed by ultrasonication for 30 min to break up any aggregates that formed during storage. The excess PAP in the MRNIP dispersion was removed by ultracentrifugation prior to redispersion by ultrasonication for 3 min before the TCE dechlorination study. The N2-BET specific surface area of MRNIP was found to be similar to that of bare RNIPs, i.e., ~15 m2/g. The Fe0 content of the particles determined as described above was similar to the bare particles, i.e., ~15 % by mass.
Zeta potential of bare NZVIs
The electrophoretic mobility of bare RNIPs was measured for dilute dispersions (~30 mg/L) in 1 mM NaCl (pH 8) with a Malvern Zetasizer (Southborough, MA, USA). The measured electrophoretic mobilities were converted to apparent ζ-potentials using the Helmholtz-Smoluchowski relationship (Israelachvili 1992).
Adsorbed PAP layer characterization
The adsorbed polymer layer properties of polymer-modified NZVIs govern the TCE dechlorination rate (Phenrat et al. 2009b). The adsorbed PAP layer on the MRNIPs was characterized using electrophoretic mobility (EPM) measurements and Ohshima’s soft particle theory (Ohshima 1995) as previously described (Phenrat et al. 2008). Details of Ohshima’s method can be found in Ohshima (1995) and Phenrat et al. (2008)), as well as in the Additional file 1. Briefly, EPM was measured for 10 mg/L solutions of the washed, PAP-modified NZVIs at NaCl concentrations ranging from 1 to 61 mM (pH 8.0 ± 0.1). EPM was measured in triplicate (25 °C) using a Malvern Zetasizer. The mean and standard deviation (σ) of the measured EPM (u e) were calculated. The procedure for extracting the adsorbed polyelectrolyte layer properties from the EPM data involves fitting Ohshima’s model to obtain the best fit layer properties, including the charge density in the adsorbed polyelectrolyte layer (N), the softness parameter (λ), the adsorbed layer thickness (d) for the mean u e, and the mean u e ± σ as a function of ionic strength using a MATLAB (the Mathworks, Novi, MI, USA) code employing an iterative least-squares minimization.
Groundwater
Three different groundwater samples from three actual TCE-contaminated sites in FL, SC, and MI were used in this study. Groundwater samples were stored at 4 °C and deoxygenated by N2 sparging prior to use. All VOCs were also purged from the groundwater samples by N2 sparging prior to the addition of known concentration of TCE for dechlorination study described next. The pH of the solution was measured after N2 sparging. Concentrations of dissolved anions and cations were determined by a commercial laboratory (Severn Trent Laboratories, Pittsburgh, PA, USA) for the FL and SC groundwater samples, and they were provided by the Michigan Department of Environmental Quality Environmental Laboratory (Lansing, MI) for the MI groundwater samples. Total organic carbon (TOC) was measured using UV/persulfate wet oxidation (OI Analytical Model 1100). Table 1 summarizes the solute concentrations in the three groundwater samples.
TCE dechlorination
TCE dechlorination rates and by-products were measured in 70-mL serum bottles containing 40 mL of headspace, 30 mL of liquid, and a Mininert™ closure. All reactors were prepared in an anaerobic glove box (argon-filled) and contained 3 g/L of either bare or PAP-modified NZVIs in one of the three groundwater samples. An aliquot of 0.15 mL of saturated TCE solution (8.4 mM) was added to provide an initial TCE concentration of 40 μM. Experiments were performed in duplicate. The reactors were rotated on an end-over-end rotator at 30 rpm at 23 ± 2 °C. In control experiments without NZVIs, it was demonstrated that the TCE loss by mechanisms (e.g., photodegradation, adsorption, leakage) other than degradation by Fe0 was negligible. Mass transfer resistance at the vapor/liquid interface was not considered, as these phases are assumed to be in equilibrium (Burris et al. 1996). Kinetically, a series of 100-μL headspace samples were withdrawn from the reactors and analyzed for TCE and its products using a 30-m GSQ PLOT capillary column on a HP 6890 GC/FID. Following the complete degradation of the first spike of TCE, the reactors were sparged with nitrogen, purged in a glove box, and then an additional spike of TCE was added right away. This process was repeated to achieve three cycles of TCE dechlorination or until the NZVIs were no longer reactive.
A previously reported model of the TCE degradation pathway by bare (Liu et al. 2007) (Additional file 1: Fig. S1a) and PAP-modified NZVIs (Phenrat et al. 2009a) was used to interpret the dechlorination kinetics in this study. It was assumed that all TCE reduction was via β-elimination to form acetylene, and that both ethane and ethene resulted from the reduction of acetylene. The reaction rate constants, k TCE (TCE dechlorination to acetylene), k 2 (ethene formation from acetylene), and k 3 (ethane formation from acetylene), were determined using a kinetic modeling software package, Scientist, v.2.01 (Micromath, St. Louis, MO, USA), in which the loss of TCE and the formation of products (acetylene, ethane, and ethane) were fit concurrently to determine the rate constants and 95 % confidence intervals for the fits. The observed reaction rate constants determined from headspace measurements, k obs-h, were converted to the observed rate constants without headspace, k obs, to compare TCE and acetylene, which have different Henry’s law constants (Additional file 1: Fig. S1b) (Liu and Lowry 2006).
Quantifying the change of adsorbed PAP layers on NZVIs after TCE dechlorination
To determine the change in the adsorbed PAP layers on NZVIs due to a possible interaction with organic and inorganic solutes in groundwater samples during the dechlorination process, PAP-modified and bare NZVIs were recovered from the TCE dechlorination reactors using magnetic separation and washed with N2-purged DI water several times to remove solutes that were attached to the NZVI surfaces. Then, EPM measurements were conducted on the recovered particles. To compare the influence of groundwater solutes on the adsorbed PAP layer on the NZVIs, the EPM of the recovered PAP-modified NZVIs was compared to that of PAP-modified NZVIs aged in DI water for the same period of time.
Acridine orange counting of microorganisms
To evaluate if the change of adsorbed biomacromolecules, such as PAP, can be driven by microbial activity, microorganisms were enumerated by acridine orange counting according to the methods of Kepner and Pratt (1994). Briefly, sample aliquots were dispersed in 0.01 M tetrasodium pyrophosphate (Na4PP-i) (Sigma-Aldrich). Ten milliliters of dispersed sample was stained in the dark for 5 min in a 100 μg/mL acridine orange solution (Sigma-Aldrich). The suspension was filtered onto a 0.22-μm black polycarbonate track-etched membrane (Millipore, Billerica, MA, USA) and visualized by epi-fluorescent microscopy using a Zeiss Observer Z1 microscope. Twenty random view fields were counted for each measurement to obtain total cell counts.
Results and discussion
Charge density of bare NZVIs and adsorbed layer properties of PAP-modified NZVIs
The EPM of bare RNIPs in 1 mM NaCl at pH 8 was −3.3 μm V−1 s−1 cm, which corresponds to a zeta potential (ζ) of −42.5 mV using the Helmholtz-Smoluchowski relationship. The apparent bare NZVI charge density (σ) of −3 × 10−4 C/m2 was obtained from the apparent ζ-potentials using Eq. 1 (Evans and Wennerstrom 1999).
where e is the electron charge, z is the valence of the ionic species of interest, k B is Boltzmann’s constant, T is absolute temperature, ε 0 is the permittivity of a vacuum, and ε r is the relative permittivity. κ is the Debye-Hückel parameter of the solution (Eq. 2).
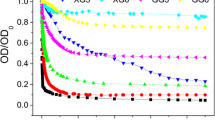
The adsorbed PAP layer properties for PAP-modified NZVIs, based on Ohshima’s soft particle analysis and EPM (Fig. 1), are as follows: N = −2.1 × 1023/m3, d = ~40 nm, and 1/λ = ~24 nm. These adsorbed layer properties are in good agreement with the properties of PAP-modified NZVIs that were reported in recent papers (Phenrat et al. 2008, 2009b).

Electrophoretic mobility of fresh PAP-modified NZVIs (filled triangles), PAP-modified NZVIs recovered from MI groundwater after TCE dechlorination (open triangles), bare NZVIs recovered from MI groundwater after TCE dechlorination (open circles), and PAP-modified NZVI aged in DI water for the same period of time (open squares) as a function of the NaCl concentration (mM) at pH 8.0 ± 0.1. The lines represent the best-fit theoretical curves obtained using Ohshima’s soft particle analysis.
Effect of groundwater solutes on TCE dechlorination using bare NZVIs
Figure 2 illustrates the TCE degradation kinetics and by-product formation kinetics using bare and PAP-modified NZVIs in MI groundwater as a representative example of the groundwater samples evaluated in this study. Acetylene was the intermediate, while ethane and ethene were the main by-products. Mass balance of TCE and dechlorination by-products was from 96 to 127 % for all the cases (Additional file 1: Fig. S2). The experimental rate constants are also reported in Table 2 for bare NZVIs and in Table 3 for PAP-modified NZVIs. The presence of ionic and organic solutes in natural groundwater decreased the TCE dechlorination rates using bare NZVIs to a similar extent as that previously reported by Liu et al. (2007). We found that the TCE dechlorination rates in the SC and MI groundwater samples for the first TCE spike were around ~22 % of the TCE dechlorination rate using bare NZVIs in DI water (Fig. 3a). Similarly, the TCE dechlorination rate using bare NZVIs in FL groundwater, in which the solute concentrations were in the range of those for the SC and MI groundwater, was around 13 % of the TCE dechlorination rate in DI water. Presumably, the decline in the TCE dechlorination rate in groundwater is attributed to the surface complexation of the NZVIs by cationic and anionic solutes (Liu et al. 2007) and reactive site blocking via the adsorption of natural organic matter onto the surface of the NZVIs.

TCE degradation kinetics and by-product formation kinetics using bare NZVIs for the a first, c second, and e third TCE degradation cycles, and using PAP-modified NZVIs for the b first, d second, and f third TCE degradation cycles in MI groundwater

a TCE dechlorination of bare and PAP-modified NZVIs in FL, SC, and MI groundwater standardized by TCE dechlorination in DI water. b Change in TCE dechlorination rates of bare and PAP-modified NZVIs in FL, SC, and MI groundwater from the first to the third TCE spike
The TCE dechlorination rates in the second TCE spike for MI and FL groundwater were similar to the dechlorination rates of the first spike, which is in good agreement with Liu and Lowry’s observation that TCE dechlorination rates remained constant over the life time of NZVIs under a particular solution chemistry (i.e., pH) (Liu and Lowry 2006). However, the TCE dechlorination rate of bare NZVIs in SC groundwater declined in the second spike. In the third TCE spike (around 30 days after the first TCE spike), bare NZVIs stopped reacting with TCE in all three groundwater samples, presumably due to the depletion of Fe0 at the end of the particles’ life time (Fe0 content <3 % by mass for all the cases).
Effect of groundwater solutes on TCE dechlorination using PAP-modified NZVIs
Groundwater solutes did not alter TCE dechlorination pathways using PAP-modified NZVI. Similar to PAP-modified NZVIs in DI water (Phenrat et al. 2009b), for TCE dechlorination using PAP-modified NZVIs in the groundwater samples, acetylene was the intermediate and ethane and ethene were the main by-products (Fig. 2). For the first TCE spike, the TCE dechlorination rate using PAP-modified NZVIs was less than that of bare NZVIs in all the groundwater samples. This is because the adsorbed PAP blocks the NZVI reactive sites and decreases the aqueous TCE concentration at the NZVI surface due to the partitioning of TCE to the adsorbed PAP layer. This observation is in good agreement with our recent study (Phenrat et al. 2009b).
However, the adsorbed PAP layer on the NZVI surface limited the adverse effect of groundwater solutes on TCE dechlorination rates. As shown in Fig. 3a, for the first TCE spike, TCE dechlorination rates using PAP-modified NZVIs in the MI and SC groundwater samples were 70 and 85 % of the TCE dechlorination rate using PAP-modified NZVIs in DI water (Phenrat et al. 2009b). Moreover, the TCE dechlorination rate using MRNIPs in FL groundwater was even greater than that of PAP-modified NZVIs in DI water; a possible explanation for this observation will be discussed in the last section regarding the desorption of PAP from the surfaces of the NZVIs. Overall, the TCE dechlorination rate using PAP-modified NZVIs in the groundwater samples was much less affected by the presence of groundwater solutes than that using bare NZVIs. A possible explanation, the effect of the Donnan potential in the PAP layer on solute distributions, for this finding is discussed in the next section.
Interestingly, unlike bare NZVIs, for which the TCE dechlorination rates in the second spike either decreased or remained the same, the TCE dechlorination rates of PAP-modified NZVIs in all groundwater samples increased substantially (~100 % greater than that in the first spike). The TCE dechlorination rates of PAP-modified NZVIs kept increasing in the third TCE spike (~200 % greater than that of the first spike). PAP-modified NZVIs did not become non-reactive, as occurred for the bare NZVIs, after two cycles of TCE dechlorination. A possible explanation for the increase in the TCE dechlorination rates following the second and third spikes will be discussed in the last section on the desorption of PAP from the surfaces of the NZVIs.
Conceptual model: Donnan potential in the adsorbed PAP layer and NZVI site blocking by PAP trains limit the adverse effect of dissolved solutes in groundwater on TCE dechlorination
The pre-adsorbed PAP layer on the surfaces of NZVIs behaves as a barrier that prevents organic and ionic solutes from effectively blocking and passivating the reactive sites of the NZVIs. The adsorbed PAP layer consists of trains, loops, and tails (Phenrat et al. 2009b). The loops and tails exhibit an extended polymeric layer surrounding the surfaces of the NZVIs, while the trains adsorb directly onto the surfaces of the NZVIs, i.e., they block the NZVI reactive sites. Because of volume exclusion and osmotic pressure effects, the extended PAP layer on the NZVIs decreases the subsequent adsorption of charged macromolecules, such as NOM, onto the surfaces of the NZVIs and, thus, decreases the reactive site blocking by NOM. Similarly, the charged PAP layer exhibits a negative Donnan potential (Phenrat et al. 2008) (Fig. 4), which can decrease the concentration of cationic solutes at the surfaces of NZVIs and possibly reduce their blocking effect compared with that for bare NZVIs. This hypothesis can be theoretically supported by considering the distribution of ionic species with and without the presence of the adsorbed PAP layer (Fig. 5) as discussed below.

Schematic illustrating the ion distribution surrounding a bare and b PAP-modified NZVIs focusing on the effect of the adsorbed PAP layer on ion distribution. Schematic illustrating the electrical potentials surrounding c bare and d PAP-modified NZVIs focusing on the presence of a Donnan potential in the adsorbed PAP layer

Representative distributions of anionic and cation species from the surfaces of a bare and b PAP-modified NZVIs in MI groundwater using the Boltzmann distribution
The adverse effect of ionic species on dechlorination using bare NZVIs is an interfacial phenomenon (Liu et al. 2007) that should be substantially affected by the interfacial concentrations, rather than the bulk concentrations, of ionic species at the surfaces of NZVIs. The interfacial concentration of an ionic species is a function of its valence, its bulk concentration, and the electrical potential in the electrostatic double layer of bare NZVIs (Israelachvili 1992). The concentration of the ionic species (C i *) as a function of distance (x) from the surfaces of the NZVIs can be calculated from the Boltzmann distribution (Israelachvili 1992) (Eq. 3).
where C i0* is the bulk concentration of ionic species i and z i is its valence. For a solution chemistry of interest, the electrical potential (Φ(x)) as a function of the distance (x) from the bare NZVI surface can be approximated using Eq. 4 if the zeta potential of the NZVIs (ζ) and the solution chemistry (which controls the Debye-Hückel parameter (κ)) (Eq. 2) are known.
As shown in Figs. 4a, c and 5a, following the Boltzmann distribution (Eq. 3), cationic species accumulated near the surface of negatively charged NZVIs and their bulk concentration gradually decreases as the distance increases due to the exponential decrease in the electrical potential (Φ(x)). In the Stern layer very close to the surface, an excess of cations neutralizes the surface charge of NZVIs, which might block reactive sites. The degree of cationic accumulation increases with their bulk concentration and valence (i.e., a divalent cation accumulates at the surface to a greater degree than a monovalent cation). In contrast, anionic species are depleted close to the surface in comparison to their bulk concentration (Fig. 5a).
Similar to the case of the bare NZVIs mentioned previously, the adverse effect of ionic species on dechlorination using PAP-modified NZVIs should be governed by the interfacial concentration of ionic species, which is a function of the valence of the ionic species of interest, their bulk concentrations, and the electrical potential in the adsorbed PAP layer on the NZVIs according to the Boltzmann distribution (Eq. 3) (Israelachvili 1992). However, unlike the bare NZVIs, the electrical potential in the adsorbed PAP layer on NZVIs (ψ(x)) is substantially affected by the Donnan potential (ψ DON), which is mostly controlled by the adsorbed layer thickness (d) and charge density in the layer (N) (Phenrat et al. 2008) as shown in Eq. 5 (Fig. 4b, d). The zeta potential of bare NZVIs (ζ) plays only a minor role in the case of polyelectrolyte-modified NZVIs (Eq. 5) in comparison to bare NZVIs (Eq. 4).
The effective Debye-Hückel parameter (κ m ) is expressed in Eq. 6. The corresponding ψ DON and the surface potential (ψ 0) at the boundary between the adsorbed layer and the surrounding solution (not the same as zeta potential) (Fig. 4b, d) are as shown in Eqs. 7 and 8, respectively.
where Z is the valence of the ionized groups on the polyelectrolyte, which is −1 for PAP.
Because of the weak polyelectrolyte nature of PAP (i.e., the relatively low charge density (N) as determined by Ohshima’s soft particle analysis) and its relatively extended layer thickness (d), the electrical potential in the adsorbed PAP layer on the NZVIs (ψ(x)) is low in magnitude but extended (i.e., it has a longer range than the electrical potential of bare NZVIs). As shown in Fig. 5b, according to the distribution (Eq. 3), the ion distribution in the adsorbed PAP layer was relatively unaltered compared with the bulk concentration. Cationic species did not accumulate near the surface of PAP-modified NZVIs as in the case of bare NZVIs. Thus, the site blocking of PAP-modified RNIPs due to cationic species should be less than that of bare NZVIs.
While the loops and tails in the extended PAP layer reduce the adverse effects of groundwater solutes on TCE dechlorination by decreasing the availability of NOM and cationic species at the surfaces of the NZVIs as discussed above, the PAP trains also decrease the blocking effect of NOM or cationic species that reach the surfaces of the NZVIs. Sorption of organic molecules to iron oxide surfaces is typically driven by specific interactions between iron oxide surfaces and carboxylic ligands (Edwards et al. 1996). Since both PAP and NOM have carboxylic groups, the sorption of both macromolecules to iron oxide surfaces of NZVIs is promoted by such oxide surface-ligand complexation. Consequently, the pre-sorption of PAP to NZVIs limits further reactive site blocking of NZVIs by further sorption of NOM and ionic species. For these reasons, NOM and ionic species that reach the reactive sites of bare NZVIs will have a greater impact in blocking the pristine (bare) NZVI reactive sites in comparison to the case of PAP-modified NZVIs, of which substantial amount of sites are already pre-blocked by adsorbed PAP trains.
Desorption of PAP from PAP-modified NZVIs increases the TCE dechlorination rate over time
While the decreases in the adverse effects of groundwater solutes on TCE dechlorination using PAP-modified NZVIs are attributed to the extended, absorbed PAP layers and the adsorbed PAP trains, they cannot explain the observation that the TCE dechlorination rates using PAP-modified NZVIs increased over time from the first to the third spike (Fig. 3b). A possible explanation is that, over time, during the TCE dechlorination in the presence of groundwater solutes, PAP gradually desorbed from the surfaces of the NZVIs, and the PAP-modified NZVIs behaved more similarly to the bare NZVIs in terms of their reactions with TCE. This desorption hypothesis is supported by the EPM of PAP-modified NZVIs recovered from the groundwater samples after the three TCE dechlorination cycles. The EPM of polymer-modified particles as a function of the NaCl concentration provides information on the characteristics of the adsorbed polymer layer (Phenrat et al. 2008). The greater negative charge on the PAP-modified NZVIs, in comparison to bare NZVIs, comes from the charged layers of PAP on the RNIP surface. Figure 1 suggests that the desorption of PAP from the surface of PAP-modified NZVIs after three cycles of TCE dechlorination occurs because the response of the EPM as a function of the NaCl concentration for the PAP-modified NZVIs recovered from MI groundwater is similar to the EPM of bare RNIPs (no polymer on its surface) recovered from MI groundwater, although it is very different from the EPM of fresh PAP-modified NZVIs (fully covered with PAP). The EPM of fresh PAP-modified NZVIs is greater than that of PAP-modified NZVIs or the bare NZVIs recovered from MI groundwater at all NaCl concentrations.
The evidence suggests that PAP desorption is only observed when PAP-modified NZVIs were aged in the presence of groundwater solutes. As shown in Fig. 1, the EPM as a function of the NaCl concentration for PAP-modified NZVIs aged in DI water for the same period of time is similar to that of fresh PAP-modified NZVIs, suggesting that PAP is not desorbed from the NZVI surface in the presence of DI water alone. The relatively insignificant desorption of PAP from NZVIs in DI water is in agreement with the results of a recent study (Kim et al. 2009). In natural groundwater, the desorption of biomacromolecules, such as PAP, can be driven by biotic or abiotic reactions due to microorganisms and abiotic solutes, respectively. Microorganisms might use macromolecules as their carbon source (He et al. 2010; Kirschling et al. 2010, 2011; Xiu et al. 2010), resulting in the removal of macromolecules from the surfaces of NZVIs (Kirschling et al. 2011). However, according to acridine orange counting of bacteria, we observed a low population of bacteria (~104 cells/mL) in all groundwater samples, both for bare and PAP-modified NZVIs. This suggests that there is no biostimulation due to the presence of PAP-modified NZVIs in groundwater, which is in good agreement with the fact that we did not observe any chlorinated by-products, as indicators of biotic dechlorination, during TCE degradation in this study (Xiu et al. 2010). Thus, biodegradation of PAP by microorganisms was unlikely. Consequently, the desorption of PAP was likely driven by either ionic or organic solutes. Normally, under a particular solution chemistry, the adsorption of macromolecules on a substrate is considered to be irreversible due to their multiple-segment attachment nature (Holmberg et al. 2003; Kim et al. 2009). For a macromolecule to desorb from a surface, all polymer segments attached to the surface must be detached at approximately the same time. If only a few segments are detached, there is a high probability that other available segments will occupy the available adsorption sites before the whole polymer desorbs (Holmberg et al. 2003). However, when the solution chemistry is changed due to an increase in ionic concentrations, polyelectrolyte desorption is theoretically possible due to the change in polyelectrolyte confirmation that is induced by the ionic species (de Carvalho 2010; Man et al. 2008). The salt-induced desorption transition of charged macromolecules is an entropically driven phenomenon (de Carvalho 2010). High ionic concentrations cause an electrostatic screening of charged groups in polyelectrolytes, which subsequently decrease the electrostatic volume exclusion of the polyelectrolytes. This leads to an entropic penalty for adsorbed macromolecules, i.e., polyelectrolytes in a desorbed configuration have a lower conformational entropy than polyelectrolytes in an adsorbed configuration. As a result, when a few segments of polyelectrolytes are detached, because of the entropic penalty, other segments might not reattach at the available sites, gradually leading to desorption of the entire polymer. This is hypothesized to occur when mixing polymer-modified NZVIs prepared in a low ionic strength solution (1 mM NaHCO3) with natural groundwater samples (which have a high ionic concentration, as shown in Table 1). This is coupled with the fact that the PAP used in this study is a small macromolecule (MW of 25 kg/mol and 16 monomers per chain), making desorption easier. For this reason, over time, an increasing number of segments of PAP became detached from the NZVI surface and resulted in gradual desorption, as suggested by the increase in the TCE dechlorination rates over time (Fig. 3b). It should be noted that the increase in the TCE dechlorination rates over time was not observed for NZVIs encapsulated in an alginate biopolymer (Bezbaruah et al. 2011). Instead, a gradual decrease of TCE dechlorination rates was evident over time, i.e., a 5–7 % decrease over 5–6 months. Presumably, a relatively thick skin (0.2736 ± 0.0036 mm) of cross-linked Ca-alginate gel, compared with the adsorbed PAP layer in this study (d = 40 nm), makes alginate desorption from NZVIs unlikely.
Implications for applications of NZVIs
The physisorption of polyelectrolytes on NZVI is necessary for effective in situ remediation. However, polymeric surface modification comes with two drawbacks. Firstly, it decreases NZVI reactivity with chlorinated organics such as TCE; adsorbed PAP on the surfaces of NZVIs is reported to decrease the TCE dechlorination rate by 24-fold compared with that of bare NZVIs in DI water (Phenrat et al. 2009b). Secondly, it raises a concern regarding the risk of NZVIs leaching from a treatment zone in the subsurface, which might have unintended ecological effects (Karn et al. 2009; Phenrat et al. 2009c). This study found that although surface modification with PAP decreases NZVI reactivity due to reactive site blocking and decreases the aqueous TCE concentration at the NZVI surface because of the partitioning of TCE to the adsorbed polyelectrolytes (Phenrat et al. 2009a), it subsequently reduces the interaction of NZVIs with non-target groundwater solutes (organic and ionic species), which has been shown to substantially decrease the reactivity of bare NZVIs (Liu et al. 2007). In addition, over an intermediate period of time (30 days), in the presence of groundwater solutes, PAP desorbed, thus restoring the reactivity of NZVIs with TCE. In addition, the desorption of PAP from NZVIs also decreases the chance that NZVIs can leach from the treatment zones to other ecological sites. This suggests that the modification of the NZVI surface with small charged macromolecules, such as PAP, helps to deliver NZVIs to the subsurface, restores NZVI reactivity over time due to a gradual PAP desorption in groundwater, and should not cause a significant, unacceptable risk due to uncontrollable particle migration (Phenrat et al. 2009c).
References
Agrawal A, Ferguson WJ, Gardner BO, Christ JA, Bandstra JZ, Tratnyek PG (2002) Effects of carbonate species on kinetics of dechlorination of 1,1,1-trichloroethane by zero-valent iron. Environ Sci Technol 36:4326–4333
Bennett P, He F, Zhao D, Aiken B, Feldman L (2010) In situ testing of metallic iron nanoparticle mobility and reactivity in a shallow granular aquifer. J Contam Hydrol 116:35–46
Bezbaruah AN, Shanbhogue SS, Simsek S, Khan E (2011) Encapsulation of iron nanoparticles in alginate biopolymer for trichloroethylene remediation. J Nanopart Res 13:6673–6681
Bishop EJ, Fowler DE, Skluzacek JM, Seibel E, Mallouk TE (2010) Anionic homopolymers efficiently target zerovalent iron particles to hydrophobic contaminants in sand columns. Environ Sci Technol 44:9069–9074
Burris DR, Delcomyn CA, Smith MH, Roberts AL (1996) Reductive dechlorination of tetrachloroethylene and trichloroethylene catalyzed by vitamin B12 in homogeneous and heterogeneous systems. Environ Sci Technol 30:3047–3052
de Carvalho SJ (2010) First-order–like transition in salt-induced macroion-polyelectrolyte desorption. EPL 92:18001
Devlin JF, Allin KO (2005) Major anion effects on the kinetics and reactivity of granular iron in glass-encased magnet batch reactor experiments. Environ Sci Technol 39:1868–1874
Edwards M, Benjamin MM, Ryan JN (1996) Role of organic acidity in sorption of natural organic matter (NOM) to oxide surfaces. Colloid Surface A 107:297–307
El-Naggar MM (2006) Effects of Cl−, NO3 −, and SO4 2− anions on the anodic behavior of carbon steel in deaerated 0.50 M NaHCO3 solutions. Appl Surf Sci 252:6179–6194
Evans DF, Wennerstrom H (1999) The colloidal domain; where physics, chemistry, biology, and technology meet. Wiley-VCH, New York
Golas PL, Louie S, Lowry GV, Matyjaszewski K, Tilton RD (2010) Comparative study of polymeric stabilizers for magnetite nanoparticles using ATRP. Langmuir 26:16890–16900
He F, Zhao D (2008) Hydrodechlorination of trichloroethene using stabilized Fe-Pd nanoparticles: reaction mechanism and effects of stabilizers, catalysts, and reaction conditions. Appl Catal B Environ 84:533–540
He F, Zhao D, Paul C (2010) Field assessment of carboxymethyl cellulose stabilized iron nanoparticles for in situ destruction of chlorinated solvents in source zones. Water Res 44:2360–2370
Henn KW, Waddill DW (2006) Utilization of nanoscale zero-valent iron for source remediation—a case study. Remediation J 16:57–77
Holmberg K, Jonsson B, Kronberg B, Lindman B (2003) Surfactants and polymers in aqueous solution, 2nd edn. John Wiley & Sons, Ltd., Chichester, West Sussex, England
Israelachvili JN (1992) Intermolecular and surface forces: with applications to colloidal and biological systems, 2nd edn. Academic Press, New York
Johnson TL, Fish W, Gorby YA, Tratnyek PG (1998) Degradation of carbon tetrachloride by iron metal: complexation effects on the oxide surface. J Contam Hydrol 29:379–398
Karn B, Kuiken T, Otto M (2009) Nanotechnology and in situ remediation: a review of the benefits and potential risks. Environ Health Perspect 117:1823–1831
Kepner RL Jr, Pratt JR (1994) Use of fluorochromes for direct enumeration of total bacteria in environmental samples: past and present. Microbiol Mol Biol R 58:603–615
Kim H-J, Phenrat T, Tilton RD, Lowry GV (2009) Fe0 nanoparticles remain mobile in porous media after aging due to slow desorption of polymeric surface modifiers. Environ Sci Technol 43:3824–3830
Kirschling TL, Gregory KB, Minkley EGJ, Lowry GV, Tilton RD (2010) Impact of nanoscale zero valent iron on geochemistry and microbial populations in trichloroethylene contaminated aquifer materials. Environ Sci Technol 44:3474–3480
Kirschling TL, Golas PL, Unrine JM, Matyjaszewski K, Gregory KB, Lowry GV, Tilton RD (2011) Microbial bioavailability of covalently bound polymer coatings on model engineered nanomaterials. Environ Sci Technol 45:5253–5259
Klausen J, Vikesland PJ, Kohn T, Burris DR, Ball WP, Roberts AL (2003) Longevity of granular iron in groundwater treatment processes: solution composition effects on reduction of organohalides and nitroaromatic compounds. Environ Sci Technol 37:1208–1218
Kocur CM et al (2014) Characterization of nZVI mobility in a field scale test. Environ Sci Technol 48:2862–2869
Kohn T, Livi KJT, Roberts AL, Vikesland PJ (2005) Longevity of granular iron in groundwater treatment processes: corrosion product development. Environ Sci Technol 39:2867–2879
Lim T-T, Zhu B-W (2008) Effects of anions on the kinetics and reactivity of nanoscale Pd/Fe in trichlorobenzene dechlorination. Chemosphere 73:1471–1477
Liu Y, Lowry GV (2006) Effect of particle age (Fe0 content) and solution pH on NZVI reactivity: H2 evolution and TCE dechlorination. Environ Sci Technol 40:6085–6090
Liu Y, Choi H, Dionysiou D, Lowry GV (2005a) Trichloroethene hydrodechlorination in water by highly disordered monometallic nanoiron. Chem Mater 17:5315–5322
Liu Y, Majetich SA, Tilton RD, Sholl DS, Lowry GV (2005b) TCE dechlorination rates, pathways, and efficiency of nanoscale iron particles with different properties. Environ Sci Technol 39:1338–1345
Liu Y, Phenrat T, Lowry GV (2007) Effect of TCE concentration and dissolved groundwater solutes on NZVI-promoted TCE dechlorination and H2 evolution. Environ Sci Technol 41:7881–7887
Lowry GV (2007) Nanomaterials for groundwater remediation. In: Wiesner MR, Bottero J-Y (eds) Environmental nanotechnology: applications and impacts of nanomaterials. McGraw-Hill, New York
Man X, Yang S, Yan D, Shi A-C (2008) Adsorption and depletion of polyelectrolytes in charged cylindrical system within self-consistent field theory. Macromolecules 41:5451–5456
Nurmi JT et al (2005) Characterization and properties of metallic iron nanoparticles: spectroscopy, electrochemistry, and kinetics. Environ Sci Technol 39:1221–1230
Ohshima H (1995) Electrophoresis of soft particles. Adv Colloid Interface Sci 62:189–235
Phenrat T, Saleh N, Sirk K, Tilton R, Lowry GV (2007) Aggregation and sedimentation of aqueous nanoscale zerovalent iron dispersions. Environ Sci Technol 41:284–290
Phenrat T, Saleh N, Sirk K, Kim H-J, Tilton RD, Lowry GV (2008) Stabilization of aqueous nanoscale zerovalent iron dispersions by anionic polyelectrolytes: adsorbed anionic polyelectrolyte layer properties and their effect on aggregation and sedimentation. J Nanopart Res 10:795–814
Phenrat T, Kim H-J, Fagerlund F, Illangasekare T, Tilton RD, Lowry GV (2009a) Particle size distribution, concentration, and magnetic attraction affect transport of polymer-modified Fe0 nanoparticles in sand columns. Environ Sci Technol 43:5079–5085
Phenrat T, Liu Y, Tilton R, Lowry GV (2009b) Adsorbed polyelectrolyte coatings decrease Fe0 nanoparticle reactivity with TCE in water: conceptual model and mechanisms. Environ Sci Technol 43:1507–1514
Phenrat T, Long TC, Lowry GV, Veronesi B (2009c) Partial oxidation (“aging”) and surface modification decrease the toxicity of nanosized zerovalent iron. Environ Sci Technol 43:195–200
Phenrat T, Cihan A, Kim H-J, Mital M, Illangasekare T, Lowry GV (2010) Transport and deposition of polymer-modified Fe0 nanoparticles in 2-D heterogeneous porous media: effects of particle concentration, Fe0 content, and coatings. Environ Sci Technol 44:9086–9093
Phenrat T, Fagerlund F, Illanagasekare T, Lowry GV, Tilton RD (2011) Polymer-modified Fe0 nanoparticles target entrapped NAPL in two dimensional porous media: effect of particle concentration, NAPL saturation, and injection strategy. Environ Sci Technol 45:6102–6109
Sakulchaicharoen N, O’Carroll DM, Herrera JE (2010) Enhanced stability and dechlorination activity of pre-synthesis stabilized nanoscale FePd particles. J Contam Hydrol 118:117–127
Saleh N, Phenrat T, Sirk K, Dufour B, Matyjaszewski K, Tilton RD, Lowry GV (2005) Adsorbed triblock copolymers deliver reactive iron nanoparticles to the oil/water interface. Nano Lett 12:2489–2494
Saleh N et al (2007) Surface modifications enhance nanoiron transport and NAPL targeting in saturated porous media. Environ Eng Sci 24:45–57
Saleh N, Kim H-J, Phenrat T, Matyjaszewski K, Tilton RD, Lowry GV (2008) Ionic strength and composition affect the mobility of surface-modified Fe0 nanoparticles in water-saturated sand columns. Environ Sci Technol 42:3349–3355
Sarathy V et al (2008) Aging of iron nanoparticles in aqueous solution: effects on structure and reactivity. J Phys Chem C 112:2286–2293
Scherer MM, Richter S, Valentine RL, Alvarez PJJ (2000) Chemistry and microbiology of permeable reactive barriers for in situ groundwater clean up. Crit Rev Microbiol 26:221–264
Song H, Carraway ER (2008) Catalytic hydrodechlorination of chlorinated ethenes by nanoscale zero-valent iron. Appl Catal B Environ 78:53–60
Su CM, Puls RW (2004) Nitrate reduction by zerovalent iron: effects of formate, oxalate, citrate, chloride, sulfate, borate, and phosphate. Environ Sci Technol 38:2715–2720
Tratnyek PG, Johnson RL (2006) Nanotechnologies for environmental cleanup. Nano Today 1:44–48
Van Nooten T, Springael D, Bastiaens L (2008) Positive impact of microorganisms on the performance of laboratory-scale permeable reactive iron barriers. Environ Sci Technol 42:1680–1686
Vecchia ED, Luna M, Sethi R (2009) Transport in porous media of highly concentrated iron micro- and nanoparticles in the presence of xanthan gum. Environ Sci Technol 43:8942–8947
Wang W, Zhou M (2010) Degradation of trichloroethylene using solvent-responsive polymer coated Fe nanoparticles. Colloid Surface A 369:232–239
Wang W, Zhou M, Jin Z, Li T (2010) Reactivity characteristics of poly(methyl methacrylate) coated nanoscale iron particles for trichloroethylene remediation. J Hazard Mater 173:724–730
Xiu ZM, Gregory KB, Lowry GV, Alvarez PJ (2010) Effect of bare and coated nanoscale zerovalent iron on tceA and vcrA gene expression in Dehalococcoides spp. Environ Sci Technol 44:7647–7651
Zhang W (2003) Nanoscale iron particles for environmental remediation: an overview. J Nanopart Res 5:323–332
Zhang W-X, Wang C-B, Lien H-L (1998) Treatment of chlorinated organic contaminants with nanoscale bimetallic particles. Catal Today 40:387–395
Acknowledgments
The authors are thankful for financial support from (1) the Thailand Research Fund (TRF) (MRG5680129), (2) the National Nanotechnology Centre (Thailand), a member of the National Science and Technology Development Agency, through grant number P-11-00989, (3) the National Research Council of Thailand (grant no. R2556B070 and R2555C010), and (4) the U.S. EPA (R830898 and R833326), NSF (BES-068646 and EF-0830093), and Department of Defense through the Strategic Environmental Research and Development Program (W912HQ-06-C-0038).
Author information
Authors and Affiliations
Corresponding authors
Additional information
Responsible editor: Philippe Garrigues
Additional file
Below is the link to the electronic supplementary material.
Additional file 1
Simplified TCE reduction pathways, Fe0 content determination, Ohshima’s soft particle analysis and mass balance of the dechlorination studies. This material is available free of charge via the Internet. (DOC 317 kb)
Rights and permissions
About this article

Cite this article
Phenrat, T., Schoenfelder, D., Kirschling, T.L. et al. Adsorbed poly(aspartate) coating limits the adverse effects of dissolved groundwater solutes on Fe0 nanoparticle reactivity with trichloroethylene. Environ Sci Pollut Res 25, 7157–7169 (2018). https://doi.org/10.1007/s11356-015-5092-4
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-015-5092-4