
Abstract
The pathology of cardiovascular disease is multi-faceted, with links to many modifiable and non-modifiable risk factors. Epidemiological evidence now implicates exposure to persistent organic pollutants, such as polychlorinated biphenyls (PCBs), with an increased risk of developing diabetes, hypertension, and obesity; all of which are clinically relevant to the onset and progression of cardiovascular disease. PCBs exert their cardiovascular toxicity either directly or indirectly via multiple mechanisms, which are highly dependent on the type and concentration of PCBs present. However, many PCBs may modulate cellular signaling pathways leading to common detrimental outcomes including induction of chronic oxidative stress, inflammation, and endocrine disruption. With the abundance of potential toxic pollutants increasing globally, it is critical to identify sensible means of decreasing associated disease risks. Emerging evidence now implicates a protective role of lifestyle modifications such as increased exercise and/or nutritional modulation via anti-inflammatory foods, which may help to decrease the vascular toxicity of PCBs. This review will outline the current state of knowledge linking coplanar and non-coplanar PCBs to cardiovascular disease and describe the possible molecular mechanism of this association.
Similar content being viewed by others
Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Polychlorinated biphenyls (PCBs) are a group of odorless and generally colorless synthetic chemicals with no known natural source and are composed of a biphenyl molecule with 2–10 chlorine atoms attached (with a total of 209 possible congeners of the molecule). PCBs were originally manufactured as dielectric and heat transfer fluids, coolants, lubricants, flame-retardants, and plasticizers due to their high thermal conductivity and chemical inertness. These compounds were produced by the Monsanto Corporation, USA, as congener mixtures named Aroclors beginning in 1930 until their production was banned in the USA in 1979 by the United States Congress and later under the Stockholm Convention on Persistent Organic Pollutants in 2001, which aimed to eliminate their production internationally (Porta and Zumeta 2002). The Stockholm Convention included PCBs among the original list of banned persistent organic pollutants (POPs) due to the growing early data linking PCBs with endocrine disruption, neurotoxicity, and environmental bioaccumulation (Agency for Toxic Substances and Disease Registry 2000). Since the original ban in 1979, data have also implicated PCBs in heart and liver disease, developmental abnormalities, and certain cancers (Robertson and Hansen 2001).
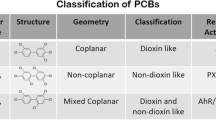
Classification of PCBs is primarily based on their stereochemical differences associated with chlorine binding positions onto the biphenyl molecule, ultimately impacting molecular planarity. Non-ortho or mono-ortho substituted PCBs may assume a planar configuration and are commonly referred to as planar or coplanar congeners, while other congeners are unable to conform to a planar configuration and are referred to as non-planar or non-coplanar (see Fig. 1) (Agency for Toxic Substances and Disease Registry 2000). Planarity of PCB congeners also impacts receptor binding, allowing planar molecules to act similarly to dioxins, a highly toxic class of chemicals that exhibit genotoxic and mutagenic properties (Mandal 2005).

a General structure of a polychlorinated biphenyl with relevant nomenclature highlighted. b Structure of 3,3′,4,4′,5-pentachlorobiphenyl (PCB 126), a coplanar, non-ortho substituted PCB, c Structure of 2,2′,4,4′,5,5′-hexachlorobiphenyl (PCB 153), a non-coplanar, di-ortho substituted PCB
Cardiovascular disease (CVD) is a non-communicable disease that encompasses a group of disorders of the heart and blood vessels including coronary heart disease, cerebrovascular disease, peripheral arterial disease, rheumatic heart disease, congenital heart disease, deep vein thrombosis, and pulmonary embolism (Labarthe 2011). The World Health Organization estimates that 17.3 million people died from CVD in 2008 alone, accounting for 30 % of global deaths and serving as the number one cause of death globally (World Health Organization 2011). Several risk factors and associated diseases including type 2 diabetes, hypertension, obesity, sedentary lifestyle, and over nutrition can contribute to the pathology of CVD. Furthermore, environmental pollutants, such as PCBs, can thus contribute to CVD directly or indirectly via promotion of these and other risk factors and associated diseases. Considering the significant burden that CVDs have on global mortality, it is imperative to explore and assess factors that may promote or exacerbate the pathogenesis of these diseases (World Health Organization 2011). The goal of this review, then, is to summarize current epidemiological and molecular biology findings that link environmental exposures to PCBs with the development of CVD.
It is also important to briefly describe how literature searches were conducted and the criteria for study selection (Woodruff and Sutton 2014). Originally, literature searches were restricted to PCBs and their direct correlation to CVD, but after reviewing the available literature it became important to also examine other disease risk factors affected by PCB exposure, which can indirectly modulate or accelerate the pathogenesis of CVD. Each paper fitting this criteria was individually examined for its’ relevance to the review.
Epidemiological studies linking PCBs and cardiovascular disease risk factors
Hypertension
Of the 17.3 million deaths attributed to CVD in 2008, complications associated with hypertension, or high blood pressure, accounted for roughly half (9.4 million) of those deaths. Even more significant, these 2008 data indicated that 40 % of all adults worldwide (aged 25 and above) had been diagnosed with hypertension, an increase from 600 million reported cases in 1980 to 1 billion cases in 2008. This drastic rise in rates of hypertension has been attributed to a number of factors including poor nutrition, lack of physical activity, and exposure to persistent chemical stressors (World Health Organization 2013).
Growing epidemiological evidence is substantiating a link between exposure to PCBs and increased risk of hypertension. The National Health and Nutrition Examination Survey (NHANES) data set has been a significant source of information on the association between hypertension and PCB exposure. Two recent studies showed that serum PCB levels were significantly associated with hypertension (Peters et al. 2014; Yorita Christensen and White 2011) and that serum PCB levels were, on average, higher among individuals with hypertension (Yorita Christensen and White 2011). Additional studies of NHANES data have indicated that dioxin-like PCB put a person at a higher risk for hypertension than non-dioxin-like PCBs, but that the arrangement of chlorines may also be an important factor (Everett et al. 2007; Ha et al. 2009).
While the NHANES data set has provided a wealth of information, a variety of epidemiological studies involving different cohorts have also found similar associations between PCB exposure and an increased risk of hypertension. Two studies of the Anniston, Alabama cohort, a group of 758 participants residing near the original Monsanto Corporation PCB manufacturing site, showed a correlation between rates of hypertension and serum PCB concentration (Goncharov et al. 2010), and that other than age, total serum PCBs were the strongest determinant of blood pressure level in 394 participants (Goncharov et al. 2011). Additionally, a study of 1,374 Japanese residents is consistent with these findings with their own data indicating that serum levels of dioxin-like PCBs are directly correlated with high blood pressure (Uemura et al. 2009). Finally, hospital discharge rates for hypertension for residents living in upstate New York in zip codes with POP-contaminated sites were increased by 19.2 %, although it is worth noting that these POP sites contained PCBs in addition to other pollutants (Huang et al. 2006).
Studies analyzing the correlation between PCB exposure and rates of hypertension have been performed on a wide array of sample sizes (Akagi and Okumura 1985; Goncharov et al. 2011; Peters et al. 2014; Stehr-Green et al. 1986). Of all the epidemiological studies examined, all but one of the studies found a positive relationship. An important observation is that the studies that found a positive relationship had higher sample sizes, ranging from 394 to 12,200 participants, than the study in disagreement, which had a sample size of 59 participants (Akagi and Okumura 1985), suggesting sample size may have influenced these findings.
Type 2 diabetes
In 2000, an estimated 171 million people were diagnosed with diabetes; this estimate is expected to increase to 366 million by the year 2030 (Wild et al. 2004). Diabetes mellitus is characterized by poorly regulated high blood glucose levels and is primarily categorized into two groups, types 1 and 2. Although most epidemiological studies do not distinguish between types 1 and 2 diabetes, it is estimated that type 2 diabetes accounts for 90–95 % of all diabetes cases (Tang et al. 2014). Among patients with type 2 diabetes, CVD is the major cause of death, with more than 60 % of patient deaths associated with myocardial infarction and stroke (Fox et al. 2007). In addition, adults with diabetes have a 2- to 4-fold increased risk of CVD-related events compared to those without diabetes (Fox et al. 2004) and are at a substantially higher risk for developing CVD (European Association for Cardiovascular et al. 2011).
Increasingly, data and an expanding body of literature are associating PCB exposure with heightened risk and incidence of type 2 diabetes development (Airaksinen et al. 2011; Codru et al. 2007; Everett et al. 2007; Gasull et al. 2012; Grandjean et al. 2011; Hofe et al. 2014; Lee et al. 2006; Persky et al. 2012; Rylander et al. 2005; Silverstone et al. 2012; Tang et al. 2014; Vasiliu et al. 2006). These studies involve a variety of cohorts and subject ages, suggesting that PCB exposure increases the risk of type 2 diabetes development regardless of age or cohort. In addition to type 2 diabetes, PCBs also have been implicated in the development of gestational diabetes. Serum concentrations of PCB 138 and PCB 180 were associated with increased 2-h glucose levels and PCB 180 with increased immunoreactive insulin levels, suggesting that PCBs can contribute to insulin resistance in expecting mothers (Arrebola et al. 2015). Gestational diabetes, characterized by the development of any degree of glucose intolerance in mothers during pregnancy, occurs during approximately 7 % of pregnancies, or 200,000 cases annually. Women who develop gestational diabetes are at an increased risk for the development of type 2 diabetes after pregnancy (American Diabetes 2004).
Obesity
While atherosclerosis and hypertension are commonly associated with the development of CVD, obesity serves as a primary modulator of these conditions and has been implicated heavily in CVD development (Kenchaiah et al. 2002; Poirier et al. 2006; Van Gaal et al. 2006). A limited amount of literature has focused on PCB exposure and its implications on the etiology of obesity development, with primary correlations drawn from laboratory studies (Arsenescu et al. 2008; Ferrante et al. 2014; Kim et al. 2012; Myre and Imbeault 2014) as well as from multiple epidemiological studies that have shown an association that warrants further examination (Donat-Vargas et al. 2014; Gladen et al. 2000; Lee et al. 2012; Verhulst et al. 2009).
Epidemiological studies into the association of PCB exposure and the development of obesity primarily have relied upon the comparison of serum PCB levels and body mass index (BMI) or birth weight, although studies have yielded mixed results. An interesting prospective study of 12,313 non-obese participants in the Seguimiento Universidad de Navarra (SUN) cohort showed that after a median 8.1 years, there were 621 new incidences of obesity among these participants, with a direct correlation seen between increased dietary PCB intake (estimated from an earlier study of dioxin-like PCB concentrations in food) and incidences of obesity (Donat-Vargas et al. 2014). Additional studies of adolescent and adult patients have reported total PCB (Gladen et al. 2000; Lee et al. 2011; Lignell et al. 2013; Valvi et al. 2012; Verhulst et al. 2009) and congener-specific (Dhooge et al. 2010; Glynn et al. 2003; Lee et al. 2012) obesity modulation, while others have found no correlation (Ben Hassine et al. 2014; Karmaus et al. 2009) or even an inverse association (Blanck et al. 2002; Dirinck et al. 2011, 2014; Wu et al. 2011), indicating the complexity of this relationship.
These discrepancies in findings may be related to congener-specific effects of individual PBCs. Studies that did find congener-specific effects (Dhooge et al. 2010; Glynn et al. 2003; Lee et al. 2012) demonstrated that the degree of chlorination of each PCB determined it’s affect on obesity risk. Less chlorinated PCBs were found to be associated with an increased risk of obesity, while more chlorinated PCBs demonstrated a decreased risk. These findings suggest that chlorination, in addition to concentration and planarity, may have a significant impact on the obesity-inducing effects of individual PCBs.
Another explanation for the inconsistencies may also be caused by the apparent protective effects that increased adiposity may have in certain situations, a concept known as the obesity paradox (Lavie et al. 2015). While obesity remains a thoroughly researched and proven risk factor for many chronic conditions, some studies have shown that patients who are obese or overweight have a better or similar prognosis than those with a traditionally healthy weight (Hong et al. 2012; Janssen and Mark 2007; Lavie et al. 2009; Oreopoulos et al. 2009; Romero-Corral et al. 2006). These findings may suggest a protective role of increased adipose deposition in certain situations, which may explain the confusing relationship between PCBs and obesity in the development of CVD.
The protective role of increased adiposity can likely be attributed to the highly lipophilic characteristic of PCBs, which can result in the bioaccumulation of these chemicals in both human and animal fat tissue (Imbeault et al. 2001; Mullerova and Kopecky 2007; Yu et al. 2011). The storage of POPs, such as PCBs, in adipose tissues has been shown to increase the half-life of these chemicals (Longnecker 2006). Although the POPs may be present for a longer period of time, the sequestering of these chemicals into adipose tissues could be less harmful to the individual when comparing the effects of potential exposure to more sensitive target tissues (Hong et al. 2012; La Merrill et al. 2013). Obese and overweight patients have a larger reservoir for POP, and therefore PCB, sequestration, than patients with more ideal body weights, which could help negate some of the harmful effects of PCB exposure. This concept is supported by studies showing that an increase in serum POP levels, which include PCBs, had adverse health effects (Imbeault et al. 2002a, b; Pelletier et al. 2002). Weight loss has also been shown to increase the concentration of PCBs in plasma (Chevrier et al. 2000; Imbeault et al. 2002a). This further illustrates the complexity of obesity and PCB exposure, as weight loss causes a release of toxicants and a temporary increase in plasma toxicant concentration.
The bioaccumulation of PCBs into adipose tissues and differences in recent weight loss may also leave low and/or inconsistent levels of PCB remaining in serum samples for analysis. This suggests that serum PCB concentration measurement may not provide an accurate estimate of a subject’s PCB exposure or body burden. Biomarkers that mirror PCB exposure levels are currently lacking. In a laboratory setting it is routine to compare induction of certain enzymes to PCB exposures, but epidemiological studies do not typically record this information. Ideal analysis of PCB concentrations in human populations would include the measurement of all PCB congeners, in both serum and adipose tissue, while utilizing a common method of normalization between studies. While there are still many questions about the role of PCBs in modulating obesity risk factors, growing epidemiological evidence along with in vitro and in vivo findings have implicated PCBs as a part of a much larger group of environmental pollutants that act as obesogens, or chemicals capable of inappropriately activating molecular pathways that may lead to a predisposition to obesity through dysfunctional weight-control (Baillie-Hamilton 2002; Grun and Blumberg 2006).
Dyslipidemia
Dyslipidemia refers to a wide array of lipid abnormalities that can be causative in the development of CVD. Efforts to prevent CVD have focused significant effort on addressing dyslipidemia because it is readily modifiable by lifestyle changes and drug therapies, most notably statins (Huffman et al. 2013; Martin et al. 2014; Riche and McClendon 2007), which work to prevent cholesterol synthesis by altering the active site of the HMG-CoA reductase enzyme that converts HMG-CoA to mevalonic acid, a cholesterol precursor (Rodriguez-Yanez et al. 2008; Stancu and Sima 2001). There is compelling evidence that decreasing total cholesterol (TC), triglycerides (TG), and low-density lipoproteins (LDL) can help markedly reduce a patient’s risk of CVD (European Association for Cardiovascular et al. 2011; Tenenbaum et al. 2014). Similar to findings on the association between obesity and PCB exposure, the most compelling and complete evidence comes from laboratory studies (Bell et al. 1994; Hitomi and Yoshida 1989; Nagaoka et al. 1986, 1990; Oda et al. 1990). For instance, ApoE (−/−) mice injected with PCB 77 exhibited increased serum cholesterol and atherosclerosis (Arsenescu et al. 2008).
There is a small volume of epidemiological literature directly examining a potential causal relationship between dyslipidemia and PCB exposure is certainly a limiting factor. The difficulty of finding literature examining this is further compounded by the fact that serum lipid measurements are often published within much larger analyses and are often not a point of emphasis, meaning that searching for appropriate PCB literature is complicated. Despite these limitations, though, there is epidemiological evidence suggesting PCB exposure may contribute to dyslipidemia (Goncharov et al. 2008; Lee et al. 2011; Uemura et al. 2009). Among the contributors to dyslipidemia, elevated TG levels has been most consistently associated with PCB exposure (Baker et al. 1980; Chase et al. 1982; Lee et al. 2007; Smith et al. 1982; Stehrgreen et al. 1986; Tokunaga and Kataoka 2003; Uemura et al. 2009). Increases in both LDL (Aminov et al. 2013; Penell et al. 2014) and total cholesterol (Aminov et al. 2013; Stehr-Green et al. 1986; Tokunaga and Kataoka 2003) have also been reported, although there are studies that have shown no correlation (Chase et al. 1982). An inverse relationship between PCB exposure and high-density lipoproteins (HDL) has also been shown (Penell et al. 2014; Smith et al. 1982), which is significant considering that increasing levels of HDL are associated with a decrease in CVD risk (Linsel-Nitschke and Tall 2005).
Limitations of epidemiological study analysis
It should be noted that while epidemiological findings are very useful for determining associations between risk factors, such as PCB exposure, and outcomes, such as hypertension, it is not possible to derive direct causality. The majority of studies presented here is cross-sectional, and simply provide a snapshot of a population and may not adequately describe the complete nature of the association (Levin 2006). The large number of PCB congeners and the variety of molecular mechanism through which PCBs elicit toxicological effects adds additional complexity to these analyses and inhibits direct correlation of a specific risk factor with a more complex outcome. Statistical analysis of PCB concentrations also varied between studies, which is to be expected, but is still a potential source of observed divergence between studies. While most studies used serum levels of lipids to normalize PCB concentrations, the specific method of normalization using serum lipids varied between those studies that did normalize their data.
Potential mechanisms of PCB-induced cardiovascular disease
Coplanar PCBs
The toxicity of coplanar PCBs, such as PCBs 77 and 126, is primarily induced through constant basal activation of the aryl hydrocarbon receptor (AhR), a transcription factor involved in xenobiotic metabolism. As a so-called orphan receptor, AhR has no associated high-affinity endogenous ligand, but rather binds to a wide array of ligands that includes PCBs (Beischlag et al. 2008). Without a ligand present, AhR exists in the cytoplasm of cells as an inactive complex with a heat-shock protein (Hsp90) as well as other co-chaperone proteins such as the p23 protein (Cox and Miller 2004). Once bound to a ligand, AhR translocates into the nucleus, dissociates from both Hsp90s and its co-chaperone proteins, and forms a heterodimer with the aryl hydrocarbon receptor nuclear translocator (ARNT). The AhR-ARNT complex then binds to a xenobiotic responsive element (XRE) in the promoter region of the target genes (Korashy and El-Kadi 2006).
A large number of drug-metabolizing enzymes are induced as a result of AhR activation, including the phase I (oxidation), phase II (conjugation), and transporters of the phase III (excretion) metabolic pathways. The activation of these enzymes results in AhR ligands inducing their own metabolism and clearance from the body (Beischlag et al. 2008). This, combined with the fact that AhR is a relatively ubiquitous protein that is expressed in most bodily tissues, has resulted in AhR being recognized as the body’s primary molecular defense when presented with an environmental toxin (Xiao et al. 2014).
Previous work has demonstrated that coplanar PCBs are capable of acting as AhR agonists (Han et al. 2012; Hennig et al. 2002; Lim et al. 2008; Xiao et al. 2014). The toxicity that is then caused by these coplanar PCBs while stimulating AhR is often attributed to the release of reactive oxygen species (ROS) and the subsequent increase in oxidative stress (Schlezinger et al. 2006). It has been suggested that ROS-induced oxidative stress in conjunction with coplanar PCB exposure is the result of increased expression of, and eventual uncoupling of cytochrome P450 1A subfamily (CYP1A1) (Hennig et al. 2002; Schlezinger et al. 1999, 2006). The metabolism of PCBs by cytochrome P450 enzymes involves a catalytic cycle of reactions that produce metabolites of the parent compound (Guengerich 2008). The metabolism of PCBs depends on the number and position of the chlorines, with the metabolism having an inverse relationship with the number of chlorines on the molecule (Grimm et al. 2015). Although previous literature describes PCB metabolites as being relatively harmless (Schlezinger et al. 2006), emerging evidence suggests that these metabolites may exhibit their own toxicity (Grimm et al. 2015). The relatively slow rate of oxidation of certain PCBs causes the uncoupling of the CYP1A1 catalytic cycle and allows ROS to leak out of the active site, causing oxidative stress. Increased amounts of oxidative stress and inflammation have been shown to be heavily involved in many of the risk factors for CVD (Ceriello and Motz 2004; Marseglia et al. 2014; Ward and Croft 2006), including the development of atherosclerosis (Dhalla et al. 2000). In addition to increasing oxidative stress, ROS also accelerate the degradation of nitric oxide, which impairs vasorelaxation by the endothelium and is often referred to as endothelial dysfunction (Cai and Harrison 2000). Patients with coronary heart disease and increased endothelial dysfunction were shown to have an increased risk of cardiovascular events, demonstrating the importance of oxidative stress in CVD (Heitzer et al. 2001).
In addition to increasing oxidative stress, coplanar PCBs acting as AhR agonists also induce cellular inflammation that is largely mediated by nuclear factor κB (NF-κB) (Hennig et al. 2002; Wu et al. 2014). Previous research has shown that activation of NF-κB drives expression of target genes that activate immune response mechanisms, ultimately resulting in the release of proinflammatory cytokines and adhesion molecules that attract immune cells to modulate pollutant-induced toxicity (Baker et al. 2011). These effects, combined with oxidative stress associated with ROS, have been shown to be atherogenic (Collins and Cybulsky 2001; Mangge et al. 2014). The compensatory mechanism intended to help combat these effects, known as the nuclear factor (erythroid-derived 2)-like 2 (Nrf2) antioxidant response pathway, regulates the response to oxidative stress and, once activated by ROS, Nrf2 protein binds to antioxidant response elements on target genes, leading to their upregulated expression. These target genes include xenobiotic metabolizing enzymes, such as glutathione S-transferases (GSTs), and many cytochrome P450s (CYPs), as well as antioxidant enzymes, NAD(P)H: quinine oxidoreductase-1 (NQO1) (Baird and Dinkova-Kostova 2011; Kansanen et al. 2012; Wakabayashi et al. 2010).
In summary, coplanar PCBs exert their primary cardiovascular toxicity through AhR mediated events. A subsequent increase in cellular ROS results in the upregulation of endogenous defenses such as the Nrf2 antioxidant system, but chronic activation of AhR may overwhelm the body’s natural means of protection leading to a state of chronic inflammation and disease. However, Nrf2 can be activated by bioactive nutrient compounds and certain drugs leading to an increased availability of antioxidant-related enzymes and mediators. Therefore, it is hypothesized Nrf2 activators, such as polyphenols, may prime a physiological system to better defend against the toxicity of PCBs and related environmental pollutants (Newsome et al. 2014).
Non-coplanar PCBs
While a large majority of risk assessment and research into PCBs focuses on coplanar PCBs, non-planar PCBs, such PCB 138, 153, and 180, still predominate in both environmental and biological samples at significant concentrations (Safe 1994). Interestingly, recent studies have shown that PCB 153 is one of the largest contributors for total PCB body burden in humans (Agudo et al. 2009; Axelrad et al. 2009; Moon et al. 2009). Unlike coplanar PCBs, non-coplanar PCBs are not ligands for AhR, but may act as ligands for other nuclear receptors in the body (Al-Salman and Plant 2012; Jacobs et al. 2005). There is some evidence that toxicity caused by non-coplanar PCBs may also be the result of inflammation, mediated by NF-κB (Kwon et al. 2002), but the mechanism of activation remains unresolved.
There has also been significant evidence suggesting that non-coplanar PCBs, or their metabolites, are capable of acting as endocrine disrupting chemicals, which are compounds that are able to mimic, antagonize, alter, or modify normal hormonal activity (Bonefeld-Jorgensen et al. 2001; Connor et al. 1997; De Coster and van Larebeke 2012; Kester et al. 2000; Kretschmer and Baldwin 2005). For instance, PCB 138, 153, and 180 were shown to compete for binding of both the estrogen receptor and androgen receptor (Bonefeld-Jorgensen et al. 2001; Korach et al. 1988). More specifically, lower chlorinated non-coplanar PCBs were shown to be weak estrogen agonists, while higher chlorinated congeners acted as weak estrogen receptor antagonists. In addition to their effects on estrogen receptors, the endocrine disruption of PCBs and their metabolites may also be a result of the inhibition of estrogen sulfotransferase, which then elevates the amount of available estrogen (Diamanti-Kandarakis et al. 2009) and estradiol (Kester et al. 2000). Although non-coplanar PCBs are often associated with being estrogenically active, it is difficult to make a general observation about the structural requirements for estrogenic activity (Arulmozhiraja et al. 2005). The endocrine disruption that non-coplanar PCBs exhibit may have obesity and diabetic-related consequences, both of which increase susceptibility for CVD, although further research is required to determine the exact mechanism of action (Diamanti-Kandarakis et al. 2009). The specific mechanism of endocrine disruption by PCBs and their metabolites are out of the scope of this paper, but are reviewed elsewhere (Kester et al. 2000; Singleton and Khan 2003; Tabb and Blumberg 2006).
Modulation of endocrine levels has been shown to have effects on the cardiovascular system. Increased levels of estrogen can reduce the risk for cardiovascular-related diseases, such as atherosclerosis, improve vascular function, and have an overall protective role over the cardiovascular system in patients with healthy vascular endothelium (Murphy 2011). In patients with a damaged vascular endothelium though, the same estrogen effects that protect healthy vascular endothelium may become harmful (Gouva and Tsatsoulis 2004). Androgens, which include testosterone, have shown similarly divisive results, demonstrating both protective and harmful effects of the hormone (Liu et al. 2003; McGrath et al. 2008).
There is a limited body of literature examining the effect of endocrine disruption by PCBs as it relates to CVD. As demonstrated above, the effect of the endocrine receptors and their associated ligands on cardiovascular disease still remains elusive. Although non-coplanar PCBs have been implicated as endocrine disrupting chemicals, current research does not support a firm conclusion on what the ultimate effect of this disruption is. The ability of some non-coplanar PCBs to act as agonists, while others act as antagonists, further complicates any interpretation. It is important that research studies continue to examine this potential relationship. Understanding any association between PCBs and the endocrine system is imperative considering the volume of literature displaying a link between hormones and their respective receptors, and the development of CVD (Deroo and Korach 2006; Liu et al. 2003; Shearman et al. 2003; Wu and von Eckardstein 2003).
Potential mechanisms of PCB-induced cardiovascular disease risk factors
Hypertension
Many factors have been implicated in the pathogenesis of hypertension, but the overall cause is not currently known, with only 5–10 % of cases having an identifiable cause (Oparil et al. 2003; Touyz 2012). Hypertension can be influenced by a number of environmental, genetic, and organ-specific effects, but renal mechanics likely play a primary role in hypertension development. The renal mechanisms, specifically the renin–angiotensin system, may be modulated by PCBs, providing a link between PCB exposure and hypertension.
The renin–angiotensin system is the result of a series of enzymatic reactions that begins with angiotensinogen, which is converted to angiotensin I by the renin enzyme (also known as angiotensinogenase). The angiotensin-converting enzyme (ACE) then hydrolyzes angiotensin I, resulting in the synthesis of angiotensin II (Oparil et al. 2003). The modulating effects of angiotensin II are mediated by one of two receptors, appropriately named angiotensin type 1 receptor and angiotensin type 2 receptor. In addition to knowing substantially more about the signaling pathway, nearly all of regulatory actions of angiotensin II related to hypertension are attributed to the type 1 receptor (Unger 2002). Effects of angiotensin type 1 receptor activation include, among many others, vasoconstriction as well as aldosterone synthesis and secretion (Unger 2002). This increase in aldosterone synthesis has also been shown to increase blood pressure (Calhoun 2006; Freel and Connell 2004). It has therefore been concluded that angiotensin II, the subsequent activation of the angiotensin type 1 receptor, and the increased production of aldosterone are associated with hypertension (Crowley et al. 2006; Muthalif et al. 2000; Unger 2002), a point further emphasized by the use of ACE inhibitors to treat hypertension (Jones and Hall 2004). Despite the limited body of evidence showing a direct correlation between PCB exposure and the renin–angiotensin system, it is likely that PCB are able to modulate at least a portion of this pathway. Literature has demonstrated that exposure to PCBs increases aldosterone production (Kraugerud et al. 2010; Li and Lin 2007; Li and Wang 2005; Li et al. 2004), which was attributed to increases in CYP11B2 (aldosterone synthase) expression that was independent of AhR activation (Lin et al. 2006). Evidence examining the effects of PCBs on other parts of the renin–angiotensin system is currently lacking.
Another potential mechanism of hypertension development as a result of PCB exposure may be mediated through the AhR. The AhR has been shown to have an important role in both the development and function of the cardiovascular system (Zhang 2011) as well as increased expression in patients with cardiovascular conditions (Huang et al. 2015). Other dioxin chemicals, such as 2,3,7,8-tetrachlorodibenzo-p-dioxin (TCDD) have been shown to increase blood pressure through an AhR mediated pathway (Kopf et al. 2008, 2010), suggesting that coplanar PCBs may be capable of inducing hypertension in a similar manner. Studies demonstrating a clear mechanistic link between PCB exposure and hypertension development are currently lacking. Oxidative stress and inflammation, which have been previously implicated in the development of hypertension (Harrison and Gongora 2009; Savoia and Schiffrin 2006), are increased as a result of PCB exposure, a process that is at least partially mediated by AhR activation (Arzuaga et al. 2007; Han et al. 2012; Petriello et al. 2014a).
It is not surprising that the body of literature demonstrating clear relationships between PCBs and their mechanism of hypertension development is lacking considering the uncertainty surrounding the pathogenesis of the disease in general. The epidemiological link between PCB exposure and hypertension is fairly clear though. This difference in both literature volume and conclusive strength suggests that additional research examining the molecular interactions that mediate the process between PCB exposure and hypertension is required.
Type 2 diabetes
The pathogenesis of type 2 diabetes is the result of a either a decrease in insulin sensitivity or the impairment of the insulin producing β-cells in the pancreas. This would suggest that any chemical associated with this disease acts through these two mechanisms. Exposure to PCBs has been associated with both of these conditions, but the specific mechanism of these PCB-induced augmentations of normal glucose homeostasis remains unclear.
Epidemiological studies have shown an increase in β-cell toxicity, as opposed to a decrease in insulin sensitivity, as a result of PCB exposure (Jensen et al. 2014), as well as no association between exposure and insulin resistance (Jorgensen et al. 2008; Persky et al. 2011, 2012). This may suggest that the observed inverse relationship between PCB exposure and serum insulin (Jensen et al. 2014) is indicative of β-cell toxicity. This impairment of β-cell functionality by PCBs may be related to intracellular levels of Ca2+ (Sargis 2014). Glucose-induced insulin secretion by β-cells is initiated by complex electrochemical interactions between the Ca2+ and K+-channels of the pancreatic β-cells. This information has been previously reviewed elsewhere (Rorsman et al. 2012). Briefly, increased glucose leads to a concentration-dependent decrease in K+ channel activity, which creates an initial action potential firing. This action potential firing activates the voltage-gated Ca2+ channel and subsequent Ca2+ movement into the β-cell, which then stimulates the release of insulin via Ca2+-dependent exocytosis (Rorsman et al. 2012). Earlier studies have shown that PCBs are capable of modulating, and interestingly, increasing intracellular levels of Ca2+ (Fischer et al. 1999), a phenomena observed in studies of other dioxin chemicals (De Tata 2014). The increase in intracellular Ca2+ caused by PCB exposure may cause an initial increase of insulin secretion, but may become cytotoxic and lead to β-cell death (Orrenius et al. 2003). Regardless of the exact mechanism of action, PCBs and similar environmental toxins are capable of augmenting Ca2+ signaling which may lead to a disruption of the insulin secretion pathway of pancreatic β-cells.
Evidence linking PCB exposure to insulin resistance, as with β-cell toxicity, is suggestive at best, but is supported by several of the epidemiological studies previously discussed (Arrebola et al. 2015; Gasull et al. 2012; Lee et al. 2011). There are several theories about how PCBs induce insulin resistance. Modulation of adiponectin levels has been shown to lead to increased rates of type 2 diabetes development (Lim and Jee 2014; Mullerova et al. 2008). Adiponectin is an adipokine produced by adipocytes that is significantly reduced in patients with insulin resistance, and is suggested as a strong predictor of type 2 diabetes (Lim and Jee 2014). Studies have shown a negative correlation between plasma levels of adiponectin and plasma PCB 153 concentrations, which may explain the role of PCB exposure in the development of type 2 diabetes (Lim and Jee 2014; Mullerova et al. 2008). Adiponectin has been shown to decrease basal glucose-levels independent of an increase in insulin levels, suggesting an ability to be a potent insulin enhancer (Berg et al. 2001).
Obesity
The etiology of obesity is not straightforward with genetic, behavioral, environmental, physiological, social, and cultural factors all playing a part in the disease progression. As suggested by the epidemiological evidence, PCBs may act as obesogens and predispose individuals to obesity by modulating existing pathways associated with weight and energy control. Although studies do show possible connections between PCBs and obesogenic effects, the specific molecular pathways of PCB-induced obesity is not clear.
One such connection may be through the regulation proteins secretes by adipocytes, referred to as adipocytokines. These proteins include, leptin, which controls hunger, and adiponectin (Chandran et al. 2003). Decreased levels of leptin or leptin resistance are both possible contributors to obesity development as both of these conditions lead to an inability to signal a reduction in food intake, leading to eventual imbalance in food intake and energy expenditure (Ahima 2008). Exposure to PCBs has been shown to increase leptin levels and gene expression in the longest exposed groups (Ahmed 2013; Ferrante et al. 2014) as well as a reduction in leptin receptor expression (Ferrante et al. 2014). Adiponectin, in addition to type 2 diabetes, has also been implicated in obesity development. The negative association between PCB exposure and adiponectin levels, which has been previously described above, may also contribute to PCB-induced obesity. The effects of PCBs on other biomarkers has been reviewed elsewhere (Ghosh et al. 2014).
Conclusions
Chlorinated organic pollutants, such as PCBs, have been linked to the development or exacerbation of cardiovascular disease. Causally, laboratory and epidemiological studies have shown that PCBs can lead to obesity, diabetes, and lipid abnormalities; all of which are risk factors for developing cardiovascular disease. Populations residing near and around Superfund or related hazardous waste sites, such as the Anniston population in Alabama, allow for some of strongest available correlations to be made between environmental exposures and increased risk for human disease. Further studies of similarly intimately afflicted communities, in the USA and abroad, are necessary to better understand the impacts of one’s environment on non-communicable disease risks.
In addition to examining how PCBs are causing cardiovascular disease, it is also important to identify ways to minimize and hopefully prevent their toxicity. Considering that PCBs are lipid soluble, and readily accumulate in human tissues, ways of decreasing the toxicity of PCBs already in human body is especially important. Recent research has shown that nutritional intervention that promotes a diet rich in antioxidants as well as a healthy lifestyle can help modulate the toxic effects of many of the environmental pollutants present in the human body, including PCBs (Baker et al. 2013; Han et al. 2012; Newsome et al. 2014; Petriello et al. 2014a, b). Also, certain dietary fats may decrease overall body burdens by increasing excretion rates (Jandacek et al. 2010, 2014). Finally, novel evidence now implicates exercise as a mediator of pro-inflammatory pollutants such as PCBs and deserves increased investigation as a modulator of environmental pollutant-induced disease (Murphy et al. 2015). Moving forward, it is imperative that future research continue to critically analyze the toxic effects of PCBs from a molecular to epidemiological level while also promoting positive lifestyle changes that can help combat these effects.
References
Agency for Toxic Substances and Disease Registry (2000): Toxicological profile for polychlorinated biphenyls (PCBs). U.S. Department of Health and Human Services, Public Health Services. http://www.atsdr.cdc.gov/toxprofiles/tp17.pdf
Agudo A, Goni F, Etxeandia A, Vives A, Millan E, Lopez R, Amiano P, Ardanaz E, Barricarte A, Chirlaque MD, Dorronsoro M, Jakszyn P, Larranaga N, Martinez C, Navarro C, Rodriguez L, Sanchez MJ, Tormo MJ, Gonzalez CA (2009) Polychlorinated biphenyls in Spanish adults: determinants of serum concentrations. Environ Res 109:620–628
Ahima RS (2008) Revisiting leptin’s role in obesity and weight loss. J Clin Invest 118:2380–2383
Ahmed RG (2013) Early weaning PCB 95 exposure alters the neonatal endocrine system: thyroid adipokine dysfunction. J Endocrinol 219:205–215
Airaksinen R, Rantakokko P, Eriksson JG, Blomstedt P, Kajantie E, Kiviranta H (2011) Association between type 2 diabetes and exposure to persistent organic pollutants. Diabetes Care 34:1972–1979
Akagi K, Okumura M (1985) Association of blood pressure and PCB level in yusho patients. Environ Health Perspect 59:37–39
Al-Salman F, Plant N (2012) Non-coplanar polychlorinated biphenyls (PCBs) are direct agonists for the human pregnane-X receptor and constitutive androstane receptor, and activate target gene expression in a tissue-specific manner. Toxicol Appl Pharmacol 263:7–13
American Diabetes A (2004) Gestational diabetes mellitus. Diabetes Care 27(Suppl 1):S88–S90
Aminov Z, Haase RF, Pavuk M, Carpenter DO, Anniston Environmental Health Research C (2013) Analysis of the effects of exposure to polychlorinated biphenyls and chlorinated pesticides on serum lipid levels in residents of Anniston, Alabama. Environ Heal : Global Access Sci Source 12:108
Arrebola JP, Gonzalez-Jimenez A, Fornieles-Gonzalez C, Artacho-Cordon F, Olea N, Escobar-Jimenez F, Fernandez-Soto ML (2015) Relationship between serum concentrations of persistent organic pollutants and markers of insulin resistance in a cohort of women with a history of gestational diabetes mellitus. Environ Res 136:435–440
Arsenescu V, Arsenescu RI, King V, Swanson H, Cassis LA (2008) Polychlorinated biphenyl-77 induces adipocyte differentiation and proinflammatory adipokines and promotes obesity and atherosclerosis. Environ Health Perspect 116:761–768
Arulmozhiraja S, Shiraishi F, Okumura T, Iida M, Takigami H, Edmonds JS, Morita M (2005) Structural requirements for the interaction of 91 hydroxylated polychlorinated biphenyls with estrogen and thyroid hormone receptors. Toxicol Sci : Off J Soc Toxicol 84:49–62
Arzuaga X, Reiterer G, Majkova Z, Kilgore MW, Toborek M, Hennig B (2007) PPARalpha ligands reduce PCB-induced endothelial activation: possible interactions in inflammation and atherosclerosis. Cardiovasc Toxicol 7:264–272
Axelrad DA, Goodman S, Woodruff TJ (2009) PCB body burdens in US women of childbearing age 2001–2002: an evaluation of alternate summary metrics of NHANES data. Environ Res 109:368–378
Baillie-Hamilton PF (2002) Chemical toxins: a hypothesis to explain the global obesity epidemic. J Altern Complement Med 8:185–192
Baird L, Dinkova-Kostova AT (2011) The cytoprotective role of the Keap1-Nrf2 pathway. Arch Toxicol 85:241–272
Baker EL Jr, Landrigan PJ, Glueck CJ, Zack MM Jr, Liddle JA, Burse VW, Housworth WJ, Needham LL (1980) Metabolic consequences of exposure to polychlorinated biphenyls (PCB) in sewage sludge. Am J Epidemiol 112:553–563
Baker RG, Hayden MS, Ghosh S (2011) NF-kappaB, inflammation, and metabolic disease. Cell Metab 13:11–22
Baker NA, English V, Sunkara M, Morris AJ, Pearson KJ, Cassis LA (2013) Resveratrol protects against polychlorinated biphenyl-mediated impairment of glucose homeostasis in adipocytes. J Nutr Biochem 24:2168–2174
Beischlag TV, Luis Morales J, Hollingshead BD, Perdew GH (2008) The aryl hydrocarbon receptor complex and the control of gene expression. Crit Rev Eukaryot Gene Expr 18:207–250
Bell FP, Iverson F, Arnold D, Vidmar TJ (1994) Long-term effects of Aroclor 1254 (PCBs) on plasma lipid and carnitine concentrations in rhesus monkey. Toxicology 89:139–153
Ben Hassine S, Hammami B, Ben Ameur W, El Megdiche Y, Barhoumi B, El Abidi R, Driss MR (2014) Concentrations of organochlorine pesticides and polychlorinated biphenyls in human serum and their relation with age, gender, and BMI for the general population of Bizerte, Tunisia. Environ Sci Pollut Res Int 21:6303–6313
Berg AH, Combs TP, Du X, Brownlee M, Scherer PE (2001) The adipocyte-secreted protein Acrp30 enhances hepatic insulin action. Nat Med 7:947–953
Blanck HM, Marcus M, Rubin C, Tolbert PE, Hertzberg VS, Henderson AK, Zhang RH (2002) Growth in girls exposed in utero and postnatally to polybrominated biphenyls and polychlorinated biphenyls. Epidemiology 13:205–210
Bonefeld-Jorgensen EC, Andersen HR, Rasmussen TH, Vinggaard AM (2001) Effect of highly bioaccumulated polychlorinated biphenyl congeners on estrogen and androgen receptor activity. Toxicology 158:141–153
Cai H, Harrison DG (2000) Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ Res 87:840–844
Calhoun DA (2006) Aldosteronism and hypertension. Clin J Am Soc Nephrol: CJASN 1:1039–1045
Ceriello A, Motz E (2004) Is oxidative stress the pathogenic mechanism underlying insulin resistance, diabetes, and cardiovascular disease? The common soil hypothesis revisited. Arterioscler Thromb Vasc Biol 24:816–823
Chandran M, Phillips SA, Ciaraldi T, Henry RR (2003) Adiponectin: more than just another fat cell hormone? Diabetes Care 26:2442–2450
Chase KH, Wong O, Thomas D, Berney BW, Simon RK (1982) Clinical and metabolic abnormalities associated with occupational exposure to polychlorinated biphenyls (PCBs). J Occup Med: Off Publ Ind Med Assoc 24:109–114
Chevrier J, Dewailly E, Ayotte P, Mauriege P, Despres JP, Tremblay A (2000) Body weight loss increases plasma and adipose tissue concentrations of potentially toxic pollutants in obese individuals. Int J Obes Relat Metab Disord: J Int Assoc Study Obes 24:1272–1278
Codru N, Schymura MJ, Negoita S, Akwesasne Task Force on Environment, Rej R, Carpenter DO (2007) Diabetes in relation to serum levels of polychlorinated biphenyls and chlorinated pesticides in adult Native Americans. Environ Health Perspect 115:1442–1447
Collins T, Cybulsky MI (2001) NF-kappaB: pivotal mediator or innocent bystander in atherogenesis? J Clin Invest 107:255–264
Connor K, Ramamoorthy K, Moore M, Mustain M, Chen I, Safe S, Zacharewski T, Gillesby B, Joyeux A, Balaguer P (1997) Hydroxylated polychlorinated biphenyls (PCBs) as estrogens and antiestrogens: structure-activity relationships. Toxicol Appl Pharmacol 145:111–123
Cox MB, Miller CA 3rd (2004) Cooperation of heat shock protein 90 and p23 in aryl hydrocarbon receptor signaling. Cell Stress Chaperones 9:4–20
Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, Kim HS, Smithies O, Le TH, Coffman TM (2006) Angiotensin II causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci U S A 103:17985–17990
De Coster S, van Larebeke N (2012) Endocrine-disrupting chemicals: associated disorders and mechanisms of action. J Environ Public Health 2012:713696
De Tata V (2014) Association of dioxin and other persistent organic pollutants (POPs) with diabetes: epidemiological evidence and new mechanisms of beta cell dysfunction. Int J Mol Sci 15:7787–7811
Deroo BJ, Korach KS (2006) Estrogen receptors and human disease. J Clin Invest 116:561–570
Dhalla NS, Temsah RM, Netticadan T (2000) Role of oxidative stress in cardiovascular diseases. J Hypertens 18:655–673
Dhooge W, Den Hond E, Koppen G, Bruckers L, Nelen V, Van De Mieroop E, Bilau M, Croes K, Baeyens W, Schoeters G, Van Larebeke N (2010) Internal exposure to pollutants and body size in Flemish adolescents and adults: associations and dose–response relationships. Environ Int 36:330–337
Diamanti-Kandarakis E, Bourguignon JP, Giudice LC, Hauser R, Prins GS, Soto AM, Zoeller RT, Gore AC (2009) Endocrine-disrupting chemicals: an Endocrine Society scientific statement. Endocr Rev 30:293–342
Dirinck E, Jorens PG, Covaci A, Geens T, Roosens L, Neels H, Mertens I, Van Gaal L (2011) Obesity and persistent organic pollutants: possible obesogenic effect of organochlorine pesticides and polychlorinated biphenyls. Obesity 19:709–714
Dirinck EL, Dirtu AC, Govindan M, Covaci A, Van Gaal LF, Jorens PG (2014) Exposure to persistent organic pollutants: relationship with abnormal glucose metabolism and visceral adiposity. Diabetes Care 37:1951–1958
Donat-Vargas C, Gea A, Sayon-Orea C, Carlos S, Martinez-Gonzalez MA, Bes-Rastrollo M (2014) Association between dietary intakes of PCBs and the risk of obesity: the SUN project. J Epidemiol Community Health 68:834–841
European Association for Cardiovascular P et al (2011) ESC/EAS Guidelines for the management of dyslipidaemias: the Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Eur Heart J 32:1769–1818
Everett CJ, Frithsen IL, Diaz VA, Koopman RJ, Simpson WM Jr, Mainous AG 3rd (2007) Association of a polychlorinated dibenzo-p-dioxin, a polychlorinated biphenyl, and DDT with diabetes in the 1999–2002 National Health and Nutrition Examination Survey. Environ Res 103:413–418
Ferrante MC, Amero P, Santoro A, Monnolo A, Simeoli R, Di Guida F, Mattace Raso G, Meli R (2014) Polychlorinated biphenyls (PCB 101, PCB 153 and PCB 180) alter leptin signaling and lipid metabolism in differentiated 3T3-L1 adipocytes. Toxicol Appl Pharmacol 279:401–408
Fischer LJ, Wagner MA, Madhukar BV (1999) Potential involvement of calcium, CaM kinase II, and MAP kinases in PCB-stimulated insulin release from RINm5F cells. Toxicol Appl Pharmacol 159:194–203
Fox CS, Coady S, Sorlie PD, Levy D, Meigs JB, D’Agostino RB Sr, Wilson PW, Savage PJ (2004) Trends in cardiovascular complications of diabetes. JAMA 292:2495–2499
Fox CS, Coady S, Sorlie PD, D’Agostino RB Sr, Pencina MJ, Vasan RS, Meigs JB, Levy D, Savage PJ (2007) Increasing cardiovascular disease burden due to diabetes mellitus: the Framingham Heart Study. Circulation 115:1544–1550
Freel EM, Connell JM (2004) Mechanisms of hypertension: the expanding role of aldosterone. Journal of the American Society of Nephrology: JASN 15:1993–2001
Gasull M, Pumarega J, Tellez-Plaza M, Castell C, Tresserras R, Lee DH, Porta M (2012) Blood concentrations of persistent organic pollutants and prediabetes and diabetes in the general population of Catalonia. Environ Sci Technol 46:7799–7810
Ghosh S, Murinova L, Trnovec T, Loffredo CA, Washington K, Mitra PS, Dutta SK (2014) Biomarkers linking PCB exposure and obesity. Curr Pharm Biotechnol 15:1058–1068
Gladen BC, Ragan NB, Rogan WJ (2000) Pubertal growth and development and prenatal and lactational exposure to polychlorinated biphenyls and dichlorodiphenyl dichloroethene. J Pediatr 136:490–496
Glynn AW, Granath F, Aune M, Atuma S, Darnerud PO, Bjerselius R, Vainio H, Weiderpass E (2003) Organochlorines in Swedish women: determinants of serum concentrations. Environ Health Perspect 111:349–355
Goncharov A, Haase RF, Santiago-Rivera A, Morse G, Akwesasne Task Force on the Environment, McCaffrey RJ, Rej R, Carpenter DO (2008) High serum PCBs are associated with elevation of serum lipids and cardiovascular disease in a Native American population. Environ Res 106:226–239
Goncharov A, Bloom M, Pavuk M, Birman I, Carpenter DO (2010) Blood pressure and hypertension in relation to levels of serum polychlorinated biphenyls in residents of Anniston, Alabama. J Hypertens 28:2053–2060
Goncharov A, Pavuk M, Foushee HR, Carpenter DO, Anniston Environmental Health Reseach C (2011) Blood pressure in relation to concentrations of PCB congeners and chlorinated pesticides. Environ Health Perspect 119:319–325
Gouva L, Tsatsoulis A (2004) The role of estrogens in cardiovascular disease in the aftermath of clinical trials. Hormones 3:171–183
Grandjean P, Henriksen JE, Choi AL, Petersen MS, Dalgard C, Nielsen F, Weihe P (2011) Marine food pollutants as a risk factor for hypoinsulinemia and type 2 diabetes. Epidemiology 22:410–417
Grimm FA, Hu D, Kania-Korwel I, Lehmler HJ, Ludewig G, Hornbuckle KC, Duffel MW, Bergman A, Robertson LW (2015): Metabolism and metabolites of polychlorinated biphenyls. Crit Rev Toxicol 45(3):245–272
Grun F, Blumberg B (2006) Environmental obesogens: organotins and endocrine disruption via nuclear receptor signaling. Endocrinology 147:S50–S55
Guengerich FP (2008) Cytochrome p450 and chemical toxicology. Chem Res Toxicol 21:70–83
Ha MH, Lee DH, Son HK, Park SK, Jacobs DR Jr (2009) Association between serum concentrations of persistent organic pollutants and prevalence of newly diagnosed hypertension: results from the National Health and Nutrition Examination Survey 1999–2002. J Hum Hypertens 23:274–286
Han SG, Han SS, Toborek M, Hennig B (2012) EGCG protects endothelial cells against PCB 126-induced inflammation through inhibition of AhR and induction of Nrf2-regulated genes. Toxicol Appl Pharmacol 261:181–188
Harrison DG, Gongora MC (2009) Oxidative stress and hypertension. Med Clin North Am 93:621–635
Heitzer T, Schlinzig T, Krohn K, Meinertz T, Munzel T (2001) Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation 104:2673–2678
Hennig B, Meerarani P, Slim R, Toborek M, Daugherty A, Silverstone AE, Robertson LW (2002) Proinflammatory properties of coplanar PCBs: in vitro and in vivo evidence. Toxicol Appl Pharmacol 181:174–183
Hitomi Y, Yoshida A (1989) Effect of methionine and threonine on the hypercholesterolemia induced by polychlorinated biphenyls in rats fed a nonprotein diet. J Nutr Sci Vitaminol 35:589–596
Hofe CR, Feng L, Zephyr D, Stromberg AJ, Hennig B, Gaetke LM (2014) Fruit and vegetable intake, as reflected by serum carotenoid concentrations, predicts reduced probability of polychlorinated biphenyl-associated risk for type 2 diabetes: National Health and Nutrition Examination Survey 2003–2004. Nutr Res 34:285–293
Hong NS, Kim KS, Lee IK, Lind PM, Lind L, Jacobs DR, Lee DH (2012) The association between obesity and mortality in the elderly differs by serum concentrations of persistent organic pollutants: a possible explanation for the obesity paradox. Int J Obes (Lond) 36:1170–1175
Huang X, Lessner L, Carpenter DO (2006) Exposure to persistent organic pollutants and hypertensive disease. Environ Res 102:101–106
Huang S, Shui X, He Y, Xue Y, Li J, Li G, Lei W, Chen C (2015) AhR expression and polymorphisms are associated with risk of coronary arterial disease in Chinese population. Sci Rep 5:8022
Huffman MD, Lloyd-Jones DM, Ning H, Labarthe DR, Guzman Castillo M, O’Flaherty M, Ford ES, Capewell S (2013) Quantifying options for reducing coronary heart disease mortality by 2020. Circulation 127:2477–2484
Imbeault P, Chevrier J, Dewailly E, Ayotte P, Despres JP, Tremblay A, Mauriege P (2001) Increase in plasma pollutant levels in response to weight loss in humans is related to in vitro subcutaneous adipocyte basal lipolysis. Int J Obes Relat Metab Disord: J Int Assoc Study Obes 25:1585–1591
Imbeault P, Chevrier J, Dewailly E, Ayotte P, Despres JP, Mauriege P, Tremblay A (2002a) Increase in plasma pollutant levels in response to weight loss is associated with the reduction of fasting insulin levels in men but not in women. Metab Clin Exp 51:482–486
Imbeault P, Tremblay A, Simoneau JA, Joanisse DR (2002b) Weight loss-induced rise in plasma pollutant is associated with reduced skeletal muscle oxidative capacity. Am J Physiol Endocrinol Metab 282:E574–E579
Jacobs MN, Nolan GT, Hood SR (2005) Lignans, bacteriocides and organochlorine compounds activate the human pregnane X receptor (PXR). Toxicol Appl Pharmacol 209:123–133
Jandacek RJ, Rider T, Keller ER, Tso P (2010) The effect of olestra on the absorption, excretion and storage of 2,2′,5,5′ tetrachlorobiphenyl; 3,3′,4,4′ tetrachlorobiphenyl; and perfluorooctanoic acid. Environ Int 36:880–883
Jandacek RJ, Heubi JE, Buckley DD, Khoury JC, Turner WE, Sjodin A, Olson JR, Shelton C, Helms K, Bailey TD, Carter S, Tso P, Pavuk M (2014) Reduction of the body burden of PCBs and DDE by dietary intervention in a randomized trial. J Nutr Biochem 25:483–488
Janssen I, Mark AE (2007) Elevated body mass index and mortality risk in the elderly. Obes Rev 8:41–59
Jensen TK, Timmermann AG, Rossing LI, Ried-Larsen M, Grontved A, Andersen LB, Dalgaard C, Hansen OH, Scheike T, Nielsen F, Grandjean P (2014) Polychlorinated biphenyl exposure and glucose metabolism in 9-year-old danish children. J Clin Endocrinol Metab 99:E2643–E2651
Jones DW, Hall JE (2004) Seventh report of the Joint National Committee on Prevention, Detection, Evaluation, and Treatment of High Blood Pressure and evidence from new hypertension trials. Hypertension 43:1–3
Jorgensen ME, Borch-Johnsen K, Bjerregaard P (2008) A cross-sectional study of the association between persistent organic pollutants and glucose intolerance among Greenland Inuit. Diabetologia 51:1416–1422
Kansanen E, Jyrkkanen HK, Levonen AL (2012) Activation of stress signaling pathways by electrophilic oxidized and nitrated lipids. Free Radic Biol Med 52:973–982
Karmaus W, Osuch JR, Eneli I, Mudd LM, Zhang J, Mikucki D, Haan P, Davis S (2009) Maternal levels of dichlorodiphenyl-dichloroethylene (DDE) may increase weight and body mass index in adult female offspring. Occup Environ Med 66:143–149
Kenchaiah S, Evans JC, Levy D, Wilson PW, Benjamin EJ, Larson MG, Kannel WB, Vasan RS (2002) Obesity and the risk of heart failure. N Engl J Med 347:305–313
Kester MH, Bulduk S, Tibboel D, Meinl W, Glatt H, Falany CN, Coughtrie MW, Bergman A, Safe SH, Kuiper GG, Schuur AG, Brouwer A, Visser TJ (2000) Potent inhibition of estrogen sulfotransferase by hydroxylated PCB metabolites: a novel pathway explaining the estrogenic activity of PCBs. Endocrinology 141:1897–1900
Kim MJ, Pelloux V, Guyot E, Tordjman J, Bui LC, Chevallier A, Forest C, Benelli C, Clement K, Barouki R (2012) Inflammatory pathway genes belong to major targets of persistent organic pollutants in adipose cells. Environ Health Perspect 120:508–514
Kopf PG, Huwe JK, Walker MK (2008) Hypertension, cardiac hypertrophy, and impaired vascular relaxation induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin are associated with increased superoxide. Cardiovasc Toxicol 8:181–193
Kopf PG, Scott JA, Agbor LN, Boberg JR, Elased KM, Huwe JK, Walker MK (2010) Cytochrome P4501A1 is required for vascular dysfunction and hypertension induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin. Toxicol Sci: Off J Soc Toxicol 117:537–546
Korach KS, Sarver P, Chae K, McLachlan JA, McKinney JD (1988) Estrogen receptor-binding activity of polychlorinated hydroxybiphenyls: conformationally restricted structural probes. Mol Pharmacol 33:120–126
Korashy HM, El-Kadi AO (2006) The role of aryl hydrocarbon receptor in the pathogenesis of cardiovascular diseases. Drug Metab Rev 38:411–450
Kraugerud M, Zimmer KE, Dahl E, Berg V, Olsaker I, Farstad W, Ropstad E, Verhaegen S (2010) Three structurally different polychlorinated biphenyl congeners (Pcb 118, 153, and 126) affect hormone production and gene expression in the human H295R in vitro model. J Toxicol Environ Health A 73:1122–1132
Kretschmer XC, Baldwin WS (2005) CAR and PXR: xenosensors of endocrine disrupters? Chem Biol Interact 155:111–128
Kwon O, Lee E, Moon TC, Jung H, Lin CX, Nam KS, Baek SH, Min HK, Chang HW (2002) Expression of cyclooxygenase-2 and pro-inflammatory cytokines induced by 2,2′,4,4′,5,5′-hexachlorobiphenyl (PCB 153) in human mast cells requires NF-kappa B activation. Biol Pharm Bull 25:1165–1168
La Merrill M, Emond C, Kim MJ, Antignac JP, Le Bizec B, Clement K, Birnbaum LS, Barouki R (2013) Toxicological function of adipose tissue: focus on persistent organic pollutants. Environ Health Perspect 121:162–169
Labarthe D (2011) Epidemiology and prevention of cardiovascular diseases: a global challenge. Jones and Bartlett Publishers, Sudbury, xvii, 709 p. pp
Lavie CJ, Milani RV, Artham SM, Patel DA, Ventura HO (2009) The obesity paradox, weight loss, and coronary disease. Am J Med 122:1106–1114
Lavie CJ, De Schutter A, Milani RV (2015) Healthy obese versus unhealthy lean: the obesity paradox. Nat Rev Endocrinol 11:55–62
Lee DH, Lee IK, Song K, Steffes M, Toscano W, Baker BA, Jacobs DR Jr (2006) A strong dose–response relation between serum concentrations of persistent organic pollutants and diabetes: results from the National Health and Examination Survey 1999–2002. Diabetes Care 29:1638–1644
Lee DH, Lee IK, Porta M, Steffes M, Jacobs DR Jr (2007) Relationship between serum concentrations of persistent organic pollutants and the prevalence of metabolic syndrome among non-diabetic adults: results from the National Health and Nutrition Examination Survey 1999–2002. Diabetologia 50:1841–1851
Lee DH, Steffes MW, Sjodin A, Jones RS, Needham LL, Jacobs DR Jr (2011) Low dose organochlorine pesticides and polychlorinated biphenyls predict obesity, dyslipidemia, and insulin resistance among people free of diabetes. PLoS One 6:e15977
Lee DH, Lind L, Jacobs DR Jr, Salihovic S, van Bavel B, Lind PM (2012) Associations of persistent organic pollutants with abdominal obesity in the elderly: The Prospective Investigation of the Vasculature in Uppsala Seniors (PIVUS) study. Environ Int 40:170–178
Levin KA (2006) Study design III: cross-sectional studies. Evid Based Dent 7:24–25
Li LA, Lin TC (2007) Interacting influence of potassium and polychlorinated biphenyl on cortisol and aldosterone biosynthesis. Toxicol Appl Pharmacol 220:252–261
Li LA, Wang PW (2005) PCB126 induces differential changes in androgen, cortisol, and aldosterone biosynthesis in human adrenocortical H295R cells. Toxicol Sci : Off J Soc Toxicol 85:530–540
Li LA, Wang PW, Chang LW (2004) Polychlorinated biphenyl 126 stimulates basal and inducible aldosterone biosynthesis of human adrenocortical H295R cells. Toxicol Appl Pharmacol 195:92–102
Lignell S, Aune M, Darnerud PO, Hanberg A, Larsson SC, Glynn A (2013) Prenatal exposure to polychlorinated biphenyls (PCBs) and polybrominated diphenyl ethers (PBDEs) may influence birth weight among infants in a Swedish cohort with background exposure: a cross-sectional study. Environ Health 12:44
Lim JE, Jee SH (2014) Association between serum levels of adiponectin and polychlorinated biphenyls in Korean men and women. Endocrine 48(1):211–217
Lim EJ, Majkova Z, Xu S, Bachas L, Arzuaga X, Smart E, Tseng MT, Toborek M, Hennig B (2008) Coplanar polychlorinated biphenyl-induced CYP1A1 is regulated through caveolae signaling in vascular endothelial cells. Chem Biol Interact 176:71–78
Lin TC, Chien SC, Hsu PC, Li LA (2006) Mechanistic study of polychlorinated biphenyl 126-induced CYP11B1 and CYP11B2 up-regulation. Endocrinology 147:1536–1544
Linsel-Nitschke P, Tall AR (2005) HDL as a target in the treatment of atherosclerotic cardiovascular disease. Nat Rev Drug Discov 4:193–205
Liu PY, Death AK, Handelsman DJ (2003) Androgens and cardiovascular disease. Endocr Rev 24:313–340
Longnecker MP (2006) Pharmacokinetic variability and the miracle of modern analytical chemistry. Epidemiology 17:350–351
Mandal PK (2005) Dioxin: a review of its environmental effects and its aryl hydrocarbon receptor biology. J Comp Physiol B Biochem Syst Environ Physiol 175:221–230
Mangge H, Becker K, Fuchs D, Gostner JM (2014) Antioxidants, inflammation and cardiovascular disease. World J Cardiol 6:462–477
Marseglia L, Manti S, D’Angelo G, Nicotera A, Parisi E, Di Rosa G, Gitto E, Arrigo T (2014) Oxidative stress in obesity: a critical component in human diseases. Int J Mol Sci 16:378–400
Martin SS, Blaha MJ, Blankstein R, Agatston A, Rivera JJ, Virani SS, Ouyang P, Jones SR, Blumenthal RS, Budoff MJ, Nasir K (2014) Dyslipidemia, coronary artery calcium, and incident atherosclerotic cardiovascular disease: implications for statin therapy from the multi-ethnic study of atherosclerosis. Circulation 129:77–86
McGrath KC, McRobb LS, Heather AK (2008) Androgen therapy and atherosclerotic cardiovascular disease. Vasc Health Risk Manag 4:11–21
Moon HB, Kim HS, Choi M, Yu J, Choi HG (2009) Human health risk of polychlorinated biphenyls and organochlorine pesticides resulting from seafood consumption in South Korea, 2005–2007. Food Chem Toxicol : Int J Publ Br Ind Biol Res Assoc 47:1819–1825
Mullerova D, Kopecky J (2007) White adipose tissue: storage and effector site for environmental pollutants. Physiol Res / Academia Scientiarum Bohemoslovaca 56:375–381
Mullerova D, Kopecky J, Matejkova D, Muller L, Rosmus J, Racek J, Sefrna F, Opatrna S, Kuda O, Matejovic M (2008) Negative association between plasma levels of adiponectin and polychlorinated biphenyl 153 in obese women under non-energy-restrictive regime. Int J Obes (Lond) 32:1875–1878
Murphy E (2011) Estrogen signaling and cardiovascular disease. Circ Res 109:687–696
Murphy MO, Petriello MC, Han SG, Sunkara M, Morris AJ, Esser K, Hennig B (2015): Exercise protects against PCB-induced inflammation and associated cardiovascular risk factors. Environ Sci Pollut Res. doi:10.1007/s11356-014-4062-6
Muthalif MM, Karzoun NA, Gaber L, Khandekar Z, Benter IF, Saeed AE, Parmentier JH, Estes A, Malik KU (2000) Angiotensin II-induced hypertension: contribution of Ras GTPase/Mitogen-activated protein kinase and cytochrome P450 metabolites. Hypertension 36:604–609
Myre M, Imbeault P (2014) Persistent organic pollutants meet adipose tissue hypoxia: does cross-talk contribute to inflammation during obesity? Obes Rev: Off J Int Assoc Study Obes 15:19–28
Nagaoka S, Kato M, Aoyama Y, Yoshida A (1986) Comparative studies on the hypercholesterolaemia induced by excess dietary tyrosine or polychlorinated biphenyls in rats. Br J Nutr 56:509–517
Nagaoka S, Miyazaki H, Aoyama Y, Yoshida A (1990) Effects of dietary polychlorinated biphenyls on cholesterol catabolism in rats. Br J Nutr 64:161–169
Newsome BJ, Petriello MC, Han SG, Murphy MO, Eske KE, Sunkara M, Morris AJ, Hennig B (2014) Green tea diet decreases PCB 126-induced oxidative stress in mice by up-regulating antioxidant enzymes. J Nutr Biochem 25:126–135
Oda H, Matsushita N, Hirabayashi A, Yoshida A (1990) Hyperlipoproteinemia in rats fed polychlorinated biphenyls. J Nutr Sci Vitaminol 36:117–122
Oparil S, Zaman MA, Calhoun DA (2003) Pathogenesis of hypertension. Ann Intern Med 139:761–776
Oreopoulos A, Kalantar-Zadeh K, Sharma AM, Fonarow GC (2009) The obesity paradox in the elderly: potential mechanisms and clinical implications. Clin Geriatr Med 25:643–659, viii
Orrenius S, Zhivotovsky B, Nicotera P (2003) Regulation of cell death: the calcium-apoptosis link. Nat Rev Mol Cell Biol 4:552–565
Pelletier C, Doucet E, Imbeault P, Tremblay A (2002) Associations between weight loss-induced changes in plasma organochlorine concentrations, serum T(3) concentration, and resting metabolic rate. Toxicol Sci : Off J Soc Toxicol 67:46–51
Penell J, Lind L, Salihovic S, van Bavel B, Lind PM (2014) Persistent organic pollutants are related to the change in circulating lipid levels during a 5 year follow-up. Environ Res 134:190–197
Persky V, Piorkowski J, Turyk M, Freels S, Chatterton R Jr, Dimos J, Bradlow HL, Chary LK, Burse V, Unterman T, Sepkovic D, McCann K (2011) Associations of polychlorinated biphenyl exposure and endogenous hormones with diabetes in post-menopausal women previously employed at a capacitor manufacturing plant. Environ Res 111:817–824
Persky V, Piorkowski J, Turyk M, Freels S, Chatterton R Jr, Dimos J, Bradlow HL, Chary LK, Burse V, Unterman T, Sepkovic DW, McCann K (2012) Polychlorinated biphenyl exposure, diabetes and endogenous hormones: a cross-sectional study in men previously employed at a capacitor manufacturing plant. Environ Heal : Global Access Sci Source 11:57
Peters JL, Fabian MP, Levy JI (2014) Combined impact of lead, cadmium, polychlorinated biphenyls and non-chemical risk factors on blood pressure in NHANES. Environ Res 132:93–99
Petriello MC, Newsome B, Hennig B (2014a) Influence of nutrition in PCB-induced vascular inflammation. Environ Sci Pollut Res Int 21:6410–6418
Petriello MC, Newsome BJ, Dziubla TD, Hilt JZ, Bhattacharyya D, Hennig B (2014b) Modulation of persistent organic pollutant toxicity through nutritional intervention: emerging opportunities in biomedicine and environmental remediation. Sci Total Environ 491–492:11–16
Poirier P, Giles TD, Bray GA, Hong Y, Stern JS, Pi-Sunyer FX, Eckel RH, American Heart A, Obesity Committee of the Council on Nutrition PA, Metabolism (2006) Obesity and cardiovascular disease: pathophysiology, evaluation, and effect of weight loss: an update of the 1997 American Heart Association Scientific Statement on Obesity and Heart Disease from the Obesity Committee of the Council on Nutrition, Physical Activity, and Metabolism. Circulation 113:898–918
Porta M, Zumeta E (2002) Implementing the Stockholm Treaty on Persistent Organic Pollutants. Occup Environ Med 59:651–652
Riche DM, McClendon KS (2007) Role of statins for the primary prevention of cardiovascular disease in patients with type 2 diabetes mellitus. Am J Health Syst Pharm : AJHP : Off J Am Soc Health Syst Pharm 64:1603–1610
Robertson LW, Hansen LG (2001) PCBs : recent advances in environmental toxicology and health effects. University Press of Kentucky, Lexington, xxx, 461 p. pp
Rodriguez-Yanez M, Agulla J, Rodriguez-Gonzalez R, Sobrino T, Castillo J (2008) Statins and stroke. Ther Adv Cardiovasc Dis 2:157–166
Romero-Corral A, Montori VM, Somers VK, Korinek J, Thomas RJ, Allison TG, Mookadam F, Lopez-Jimenez F (2006) Association of bodyweight with total mortality and with cardiovascular events in coronary artery disease: a systematic review of cohort studies. Lancet 368:666–678
Rorsman P, Braun M, Zhang Q (2012) Regulation of calcium in pancreatic alpha- and beta-cells in health and disease. Cell Calcium 51:300–308
Rylander L, Rignell-Hydbom A, Hagmar L (2005) A cross-sectional study of the association between persistent organochlorine pollutants and diabetes. Environ Heal : Global Access Sci Source 4:28
Safe SH (1994) Polychlorinated biphenyls (PCBs): environmental impact, biochemical and toxic responses, and implications for risk assessment. Crit Rev Toxicol 24:87–149
Sargis RM (2014) The hijacking of cellular signaling and the diabetes epidemic: mechanisms of environmental disruption of insulin action and glucose homeostasis. Diabetes Metab J 38:13–24
Savoia C, Schiffrin EL (2006) Inflammation in hypertension. Curr Opin Nephrol Hypertens 15:152–158
Schlezinger JJ, White RD, Stegeman JJ (1999) Oxidative inactivation of cytochrome P-450 1A (CYP1A) stimulated by 3,3′,4,4′-tetrachlorobiphenyl: production of reactive oxygen by vertebrate CYP1As. Mol Pharmacol 56:588–597
Schlezinger JJ, Struntz WD, Goldstone JV, Stegeman JJ (2006) Uncoupling of cytochrome P450 1A and stimulation of reactive oxygen species production by co-planar polychlorinated biphenyl congeners. Aquat Toxicol 77:422–432
Shearman AM, Cupples LA, Demissie S, Peter I, Schmid CH, Karas RH, Mendelsohn ME, Housman DE, Levy D (2003) Association between estrogen receptor alpha gene variation and cardiovascular disease. JAMA 290:2263–2270
Silverstone AE, Rosenbaum PF, Weinstock RS, Bartell SM, Foushee HR, Shelton C, Pavuk M (2012) Polychlorinated biphenyl (PCB) exposure and diabetes: results from the Anniston Community Health Survey. Environ Health Perspect 120:727–732
Singleton DW, Khan SA (2003) Xenoestrogen exposure and mechanisms of endocrine disruption. Front Biosci: J Virtual Libr 8:s110–s118
Smith AB, Schloemer J, Lowry LK, Smallwood AW, Ligo RN, Tanaka S, Stringer W, Jones M, Hervin R, Glueck CJ (1982) Metabolic and health consequences of occupational exposure to polychlorinated biphenyls. Br J Ind Med 39:361–369
Stancu C, Sima A (2001) Statins: mechanism of action and effects. J Cell Mol Med 5:378–387
Stehrgreen PA, Welty E, Steele G, Steinberg K (1986) Evaluation of potential health-effects associated with serum polychlorinated biphenyl levels. Environ Health Perspect 70:255–259
Stehr-Green PA, Ross D, Liddle J, Welty E, Steele G (1986) A pilot study of serum polychlorinated biphenyl levels in persons at high risk of exposure in residential and occupational environments. Arch Environ Health 41:240–244
Tabb MM, Blumberg B (2006) New modes of action for endocrine-disrupting chemicals. Mol Endocrinol 20:475–482
Tang M, Chen K, Yang F, Liu W (2014) Exposure to organochlorine pollutants and type 2 diabetes: a systematic review and meta-analysis. PLoS One 9:e85556
Tenenbaum A, Klempfner R, Fisman EZ (2014) Hypertriglyceridemia: a too long unfairly neglected major cardiovascular risk factor. Cardiovasc Diabetol 13:159
Tokunaga S, Kataoka K (2003) A longitudinal analysis on the association of serum lipids and lipoproteins concentrations with blood polychlorinated biphenyls level in chronic “Yusho” patients. Fukuoka igaku zasshi = Hukuoka acta medica 94:110–117
Touyz RM (2012) New insights into mechanisms of hypertension. Curr Opin Nephrol Hypertens 21:119–121
Uemura H, Arisawa K, Hiyoshi M, Kitayama A, Takami H, Sawachika F, Dakeshita S, Nii K, Satoh H, Sumiyoshi Y, Morinaga K, Kodama K, Suzuki T, Nagai M, Suzuki T (2009) Prevalence of metabolic syndrome associated with body burden levels of dioxin and related compounds among Japan’s general population. Environ Health Perspect 117:568–573
Unger T (2002) The role of the renin-angiotensin system in the development of cardiovascular disease. Am J Cardiol 89:3A–9A, discussion 10A
Valvi D, Mendez MA, Martinez D, Grimalt JO, Torrent M, Sunyer J, Vrijheid M (2012) Prenatal concentrations of polychlorinated biphenyls, DDE, and DDT and overweight in children: a prospective birth cohort study. Environ Health Perspect 120:451–457
Van Gaal LF, Mertens IL, De Block CE (2006) Mechanisms linking obesity with cardiovascular disease. Nature 444:875–880
Vasiliu O, Cameron L, Gardiner J, Deguire P, Karmaus W (2006) Polybrominated biphenyls, polychlorinated biphenyls, body weight, and incidence of adult-onset diabetes mellitus. Epidemiology 17:352–359
Verhulst SL, Nelen V, Hond ED, Koppen G, Beunckens C, Vael C, Schoeters G, Desager K (2009) Intrauterine exposure to environmental pollutants and body mass index during the first 3 years of life. Environ Health Perspect 117:122–126
Wakabayashi N, Slocum SL, Skoko JJ, Shin S, Kensler TW (2010) When NRF2 talks, who’s listening? Antioxid Redox Signal 13:1649–1663
Ward NC, Croft KD (2006) Hypertension and oxidative stress. Clin Exp Pharmacol Physiol 33:872–876
Wild S, Roglic G, Green A, Sicree R, King H (2004) Global prevalence of diabetes: estimates for the year 2000 and projections for 2030. Diabetes Care 27:1047–1053
Woodruff TJ, Sutton P (2014) The navigation guide systematic review methodology: a rigorous and transparent method for translating environmental health science into better health outcomes. Environ Health Perspect 122:1007–1014
World Health Organization (2011): Global status report on noncommunicable diseases 2010. http://whqlibdoc.who.int/publications/2011/9789240686458_eng.pdf?ua=1
World Health Organization (2013): A global brief on hypertension: silent killer, global public health crisis. http://apps.who.int/iris/bitstream/10665/79059/1/WHO_DCO_WHD_2013.2_eng.pdf
Wu FC, von Eckardstein A (2003) Androgens and coronary artery disease. Endocr Rev 24:183–217
Wu KS, Xu XJ, Liu JX, Guo YY, Huo X (2011) In utero exposure to polychlorinated biphenyls and reduced neonatal physiological development from Guiyu, China. Ecotoxicol Environ Saf 74:2141–2147
Wu F, Zheng Y, Gao J, Chen S, Wang Z (2014) Induction of oxidative stress and the transcription of genes related to apoptosis in rare minnow (Gobiocypris rarus) larvae with Aroclor 1254 exposure. Ecotoxicol Environ Saf 110:254–260
Xiao L, Zhang Z, Luo X (2014) Roles of xenobiotic receptors in vascular pathophysiology. Circ J : Off J Jpn Circ Soc 78:1520–1530
Yorita Christensen KL, White P (2011) A methodological approach to assessing the health impact of environmental chemical mixtures: PCBs and hypertension in the National Health and Nutrition Examination Survey. Int J Environ Res Public Health 8:4220–4237
Yu GW, Laseter J, Mylander C (2011) Persistent organic pollutants in serum and several different fat compartments in humans. J Environ Public Health 2011:417980
Zhang N (2011) The role of endogenous aryl hydrocarbon receptor signaling in cardiovascular physiology. J Cardiovasc Dis Res 2:91–95
Acknowledgments
Supported by grant number P42ES007380 from the National Institute of Environmental Health Sciences. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Environmental Health Sciences or the National Institute of Health.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Philippe Garrigues
Rights and permissions
About this article

Cite this article
Perkins, J.T., Petriello, M.C., Newsome, B.J. et al. Polychlorinated biphenyls and links to cardiovascular disease. Environ Sci Pollut Res 23, 2160–2172 (2016). https://doi.org/10.1007/s11356-015-4479-6
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-015-4479-6