
Abstract
The main aim of this study was to determine the sorption and biodegradation parameters of trichloroethene (TCE) and tetrachloroethene (PCE) as input data required for their fate and transport modelling in a Quaternary sandy aquifer. Sorption was determined based on batch and column experiments, while biodegradation was investigated using the compound-specific isotope analysis (CSIA). The aquifer materials medium (soil 1) to fine (soil 2) sands and groundwater samples came from the representative profile of the contaminated site (south-east Poland). The sorption isotherms were approximately linear (TCE, soil 1, K d = 0.0016; PCE, soil 1, K d = 0.0051; PCE, soil 2, K d = 0.0069) except for one case in which the best fitting was for the Langmuir isotherm (TCE, soil 2, K f = 0.6493 and S max = 0.0145). The results indicate low retardation coefficients (R) of TCE and PCE; however, somewhat lower values were obtained in batch compared to column experiments. In the column experiments with the presence of both contaminants, TCE influenced sorption of PCE, so that the R values for both compounds were almost two times higher. Non-significant differences in isotope compositions of TCE and PCE measured in the observation points (δ13C values within the range of −23.6 ÷ −24.3 ‰ and −26.3 ÷−27.7 ‰, respectively) indicate that biodegradation apparently is not an important process contributing to the natural attenuation of these contaminants in the studied sandy aquifer.
Similar content being viewed by others


Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Dissolved chlorinated solvents, such as trichloroethene (TCE) and tetrachloroethene (PCE), are among the most frequently detected organic contaminants in groundwater worldwide and thus pose a significant threat to drinking water supply systems (Stroo and Ward 2010; Wiedemeier et al. 1999). After the Water Framework Directive (2000/60/EC) had been implemented into the Polish law, consequently, new drinking water standards were applied; hence, a number of sites in Poland groundwater contamination of TCE and PCE were detected (Kret et al. 2013). Consequently, there is an increasing need to investigate these sites with a special emphasis on description and prediction of the contaminants’ transport and fate in groundwater systems from the source to the defined receptor(s). This can be facilitated by using numerical modelling, representing the physical phenomena occurring in groundwater systems (Bear and Cheng 2010; Van der Heijde et al. 1980). Numerical modelling is a valuable tool for simulating contaminants’ migration along the transport pathway, risk assessment, as well as planning and designing effective groundwater remediation systems (Kitanidis and McCarty 2012). However, a successful model development relies largely on appropriate estimates for the aquifer parameters (Rifai et al. 2010). Such parameters can be obtained with a series of experimental methods, estimated using prediction methods or found in the literature (Dello Site 2001). Parameters’ estimation is particularly important and may be difficult in the case of TCE and PCE, as they belong to DNAPLs (dense non-aqueous phase liquids) and may sink at the bottom of an aquifer as a separate phase and then spread out in the direction of groundwater flow along the preferential pathways (Kueper et al. 2003).
The aim of this paper is to estimate sorption (using batch and column tests) and biodegradation (by means of the compound-specific isotope analysis—CSIA) as the input parameters for a numerical fate and transport model of TCE and PCE in the sandy aquifer in south-east Poland, where their concentrations in groundwater exceed drinking water standards more than 1000 times (Kret et al. 2011). Such model is currently being developed and applied to assess the risk associated with TCE and PCE contamination, as well as to select and design the effective groundwater remediation strategy.
Transport parameters
To fully represent TCE and PCE migration in a numerical model, it is required to know physicochemical properties of the contaminants, as well as hydrogeological and geochemical conditions of the site, such as hydraulic conductivity (k), hydraulic gradient (H), dry bulk density (ρ s), porosity/effective porosity (θ t/θ), longitudinal and transverse dispersivity (α L, α v), distribution coefficient (K), biodegradation rate (usually incorporated in the model as a first order decay), initial concentrations (C s) and source size (Strickland and Korleski 2007). Along the transport pathway, a variety of physical, chemical and/or biological processes may influence contaminants’ migration (Dridi et al. 2009; Wiedemeier et al. 1999), from which the most important are dispersion, dilution, volatilization, sorption and biodegradation. The last two mostly influence the TCE and PCE migration velocity in an aquifer (Dridi et al. 2009). All of these processes are incorporated to the model by single or a set of governing equations. Three dimensional transports of contaminants in groundwater, with constant porosity, can be described by the partial differential equation (Zheng and Wang 1999):
where C—the concentration of contaminant dissolved in groundwater (ML−3); t—time (t); x i —the distance along the respective Cartesian coordinate axis (L); D ij —the hydrodynamic dispersion coefficient tensor (L2T−1); v i —the seepage or linear pore water velocity (LT−1); q s —the volumetric flow rate per unit volume of aquifer representing fluid sources (positive) and sinks (negative) (T−1); C s —the concentration of the source or sink flux for contaminant (ML—3); θ—porosity of the porous medium (−); R n —the chemical reaction term, in this example representing sorption (retardation factor R) and first-order rate reaction (λ) (ML−3 T−1)
Sorption
The term sorption refers to physical and/or chemical attachment of a compound to a solid surface, i.e. porous medium solid (Abulaban et al. 1998). Sorption parameters may be estimated based on (Fig. 1) the literature, laboratory experiments (including batch and column tests) or field investigations. In the case of organic contaminants, the most common sorption isotherms are (Table 1) Henry’s (linear), Freundlich’s (logarithmic) and Langmuir’s (including the maximum sorption capacity) (Limousin et al. 2007). To characterize TCE and PCE sorption for higher concentrations, the linear model can be used, while for lower concentrations, the Freundlich model usually fits better (Akyol et al. 2011). An appropriate sorption model may be chosen by fitting a theoretical curve to experimental data, while the accuracy of fitting may be evaluated by a determination coefficient (r 2) (Karickhoff et al. 1979). Sorption models, however, do not include the effects of speciation, pH and redox potential, thus the results of batch experiments should be regarded as approximates. Column experiments are more widely used to estimate sorption parameters by fitting breakthrough curve (BTC) (Tang et al. 2009). Flow through techniques have the advantage of approximating real conditions more closely, i.e. by allowing the solids to be rested relative to the mobile solutes and by maintaining appropriate solid/solution ratios (Benker et al. 1998). Sorption of TCE and PCE depends mainly on the organic carbon content and the aquifer material’s grain-size distribution (the content of clay and silt fractions). It is also controlled by the geochemical conditions (Cwiertny and Scherer 2010; Kueper et al. 2003).

Methods of determining the sorption parameters (Dowgiałło 2002, modified)
Sorption parameters have already been tested in several studies using different methods and diverse soil types. The list of results presented in the literature with references is shown in Table 2. Authors usually postulate that for sandy materials, the retardation factor ranges from 1.04 to 2.95 (TCE) and from 1.2 to 3.6 (PCE) depending on TOC content, soil type, and the type of experiment.
Biodegradation
Biodegradation is a process that leads to reduction of TCE and PCE loads in groundwater. It involves the breakdown of organic compounds, either through biotransformation into less complex metabolites, or through mineralization into inorganic minerals (Singh and Ward 2004; Wiedemeier et al. 1998). TCE and PCE may be biodegraded firstly by reductive dechlorination in anaerobic conditions or secondly by co-metabolism in aerobic conditions. Whether the biodegradation occurs at the specific site depends on numerous environmental conditions (pH, temperature, Eh, etc.). In the literature (e.g. Van Breukelen et al. 2005; Suarez and Rifai 1999), the wide range of TCE and PCE biodegradation rates is presented. For instance, the first-order rate constant amounts to 1.1 ÷ 3.7 and 0.9 ÷ 1.5 1/year for PCE and TCE, respectively. It is recommended to use multiple methods providing more than one line of evidence for the biodegradation assessment (Nijenhuis et al. 2007).
To assess the rates of TCE and PCE biodegradation, several methods can be used, e.g. (Bombach et al. 2010) geochemical analyses, microbial and molecular methods, tracer tests, metabolite analysis, compound-specific isotope analysis (CSIA) and in situ microcosms. Recently, stable isotope fractionation approaches have been developed and implemented at many sites (e.g. Imfeld et al. 2008; Nijenhuis et al. 2007, 2013). The CSIA takes advantage that during biodegradation, molecules with lighter isotopes (e.g. C12) react preferentially over the heavier isotope (C13) fractions, which results in an enrichment of the heavier stable isotope in the residuum (Cichocka et al. 2007). The isotope composition and residual concentration are analysed, and enrichment in the heavy isotope indicates biodegradation (Imfeld et al. 2008). The carbon isotope composition is reported in δ notation (‰) relative to the Vienna Pee Dee Belemnite standard (V-PDB, IAEA-Vienna) (Hunkeler et al. 2008):
Based on the change in isotope ratio, the biodegradation rate constant may be calculated, what is the input parameter in contaminants’ transport modelling. The isotope fractionation may be incorporated into reactive transport models and used for verification or calibration (Van Breukelen et al. 2005; Hunkeler and Aravena 2010).
Materials and methods
Materials
Two types of the aquifer materials for the sorption parameters estimation were collected from a representative profile at the studied site, namely medium (soil 1) and fine (soil 2) sands. Physicochemical properties of the aquifer materials were analysed and the results are listed in Table 3.
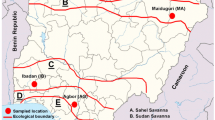
For the biodegradation study, seven sampling points (piezometers or wells) were chosen, located at the site along the contamination plume pathway, starting from the potential source of TCE and PCE (the former metalwork). The location of the wells is presented in Fig. 2. Groundwater samples were taken using a peristaltic pump (Ejkelkamp). To ensure representative sampling, field parameters (pH, redox potential, conductivity, temperature and dissolved oxygen content) were monitored in situ. The samples were taken after obtaining stabilization of the parameters mentioned above. Two replicate samples were collected in autoclaved 250-mL glass vials containing ca. 90 g of solid NaCl (to inhibit further microbial activity) with ca. 50-mL headspace. The samples were sealed with Teflon-coated septa.

Location of monitoring wells
Batch tests
The experiment was conducted for both aquifer materials and proceeded to two stages. During the first stage, the time needed to reach equilibrium for both contaminants between sorbed and dissolved phases was estimated. The aim of the second stage was to estimate partition coefficients (K) based on identified sorption isotherms and to calculate retardation factors (R) using an appropriate formula (see Table 1).
Samples of 5.0 g (±0.1 g) of dry, uncontaminated aquifer material were placed into 50-mL borosilicate glass vials. Then, the model solutions of distilled water spiked with TCE and/or PCE were added to obtain the contaminated water (L) to solid phase (S) ratio of L/S = 5:1 (v/v). To exclude possible biodegradation (biotransformation) of TCE and PCE, 0.15 g/L of sodium azide (NaN3) was added. Bottles were closed immediately after filling, so that no headspace was left. Vials were shaken for 72 h with 450 rph and placed afterwards in a dark place to reach the equilibrium. The experiment was conducted at room temperature (ca. 22 °C).
In the first stage, TCE and PCE concentrations (Table 4) were selected based on their maximum values observed in groundwater at the contaminated site in 2010. TCE and PCE concentrations (depending on the sample) were measured at specified time intervals to determine time of reaching the equilibrium. The equilibrium was assumed to be reached when a difference between two successive TCE and PCE concentration measurements in the solutions does not exceed ±15 % (a measurement’s error for the detection method used, i.e. gas chromatography).
In the second stage, sorption isotherms and partition coefficients were estimated and, consequently, retardation factors R were calculated. Five solutions with different concentrations of TCE and PCE were prepared for both soils (Table 4), analogously as at the first stage. Samples were measured for TCE and PCE concentrations in the solutions after reaching the equilibrium (as determined in the first stage). TCE and PCE concentrations were measured using gas chromatography method (GC) at WESSLING Polska sp. z o.o. licenced laboratory in Krakow, Poland, according to PN-EN ISO 10301:202 and EN 1484 standards.
The sorbed mass of TCE/PCE related to the mass of a sorbent (aquifer material) was calculated from the equation based on mass balance:
where S—the concentration of a contaminant sorbed by an aquifer material (mg/kg), C 0—the initial concentration of a contaminant in solution (mg/L), Ceq—the concentration of a contaminant in solution after reaching the equilibrium (mg/L), V—volume of solution used in the experiment (L), m—mass of sorbent (aquifer material) (g).
From the estimated partition coefficients (K) retardation factors (R) were calculated using the equations from Table 1 selected based on determination coefficients (r 2) of the fitted sorption models.
Column experiment
A soil sample was placed in a special cylinder made from galvanized steel (length of 10.8 cm and 6.35 cm in diameter) and carried out under saturated conditions. The columns were equipped with a vent and filled in with the wet aquifer material progressively from the bottom of the column to avoid trapping air bubbles. Above and below the material a paper filter and steel wool were placed to avoid clogging of the inlet and outlet ports.
Water/solution was provided to the column by a peristaltic pump with a velocity of 0.102–0.114 cm/min, similar to the actual groundwater flow rate at the site. The conservative/reactive tracers were introduced to the system by short, 5 min, injections. The system was equipped with a valve allowing for fast changes between tracer and water injections. The installation was set up to minimize migration pathways between its parts. The experiment was conducted at room temperature (ca. 22 °C). The experimental setup is shown in Fig. 3.

The experimental setup: a stage 1, b stage 2
Groundwater from an uncontaminated well at the studied area was used with 15 g/L of sodium azide (NaN3) added to avoid TCE/PCE biodegradation (biotransformation). Chloride was used as a conservative tracer, while TCE and PCE served as reactive tracers. The chloride input concentration was of 500 mg/L, while TCE or/and PCE concentrations were selected based on the actual values measured in groundwater at the site (about 4 and 3 mg/L, respectively).
The experiment was conducted for both materials (soil 1 and 2) and proceeded in two stages. Throughout the first stage, the conservative tracer was injected. Before that, an equilibrium was reached in the column between the solution and sorption complex through stabilization of conductivity. Subsequently, the conservative tracer was injected and again, the valve was changed. Concentrations of the conservative tracer were measured in the outlet solution at specific times. Moreover, for the duration of the first stage an online conductivity measurement was carried out. The time of total water exchange in the column was also determined as approximately 1 h. The second stage was conducted analogously to the first one, except that the reactive tracers (TCE and PCE) were injected. The outlet solution was collected every 8 min (minimum time to reach the volume of 20 mL required for laboratory analyses). The second stage ran for 2 h, i.e. two times longer than the first stage.
The interpretation was based on breakthrough curves (BTC) showing changes of contaminant (reactive tracer) concentrations in the output solution in the function of time. Based on observed differences in conservative/reactive tracer concentrations in output solutions, it was possible to estimate the R coefficient. The CXTFIT/Excel was used to determine BTCs using equilibrium convection-dispersion equation (CDE) (Tang et al. 2010). In column experiment, a size of the column can influence on sorption parameters’ uncertainty, but it has no significant influence on estimation of R coefficient (Marciniak et al. 2006). Despite the small column diameter used in the experiment, estimation of R factor based on fitted BTC was done with sufficient accuracy.
TCE and PCE concentrations were measured using gas chromatography method (GC) as described in “Batch tests”. Concentration of chlorides were measured by the titration method.
Biodegradation studies
To assess TCE and PCE biodegradation rates, CSIA was applied. Concentrations of TCE and PCE in groundwater samples were analysed with gas chromatography with flame ionization detection (GC-FID) (Varian Chromack CP-3800 equipped with a 30 m × 0.53 mm GS-Q column, J&W Scientific). The carrier gas was helium; the FID was operated at 250 °C. The injection was automated using an HP 7694 headspace auto sampler (Hewlett Packard) adding 0.5-mL headspace to 10-mL vials flushed with helium, closed with a Teflon-coated butyl rubber septum and crimped. Gas chromatography combustion isotope ratio mass spectrometer (GC-C-IRMS) was applied in order to determine the stable carbon isotope composition of the chlorinated ethenes as described previously (e.g. Imfeld et al. 2008; Kästner et al. 2010). The GC-C-IRMS system consisted of a gas chromatograph (model 6890, Agilent Technology) coupled via Conflow III interface (ThermoFinnigan) to a MAT 252 mass spectrometer (ThermoFinnigan). Samples were measured in at least three replicates. The carbon isotope ratio measurements were conducted via headspace analysis. Aliquots (500 to 1000 μL) of headspace samples were injected into a gas chromatograph in splitless or split mode (split 1:1) using split/splitless injector at 250 °C. Column used for the separation was PoraBOND (50 m × 0.32 mm × 0.5 μm, Agilent Technologies). The temperature programme was as follows: 40 °C (5 min), 20 °C∙min−1 to 250 °C, 250 °C (6 min).
For further analysis of data, it was assumed that the total analytical uncertainty is approximately ±0.5 ‰, so the observed fractionation must be at the minimum ±2 ‰ to confirm biodegradation (to ensure reliable interpretation, as presented in, e.g. Hunkeler et al. 2008). The isotope fractionation during volatilization, dissolution, diffusion and sorption is typically small and does not influence significantly the isotopic composition of the contaminants (Hunkeler et al. 2008). Hence, it was assumed that the isotope fractionation is caused solely by biodegradation.
Results and discussion
Batch tests
The time to reach equilibrium for soil 1 was of 30 and 40 days for TCE and PCE, respectively, i.e. 10 days shorter than for soil 2. The concentrations of TCE and PCE in solutions were fitted to three sorption models: Henry’s, Freundlich’s and Langmuir’s (Fig. 4). The determination coefficients (r 2) for considered sorption models are listed in Table 5. In the case of TCE, a sufficient fitting was obtained for the linear and Langmuir’s models: r 2 = 0.88 (soil 1) and for the Langmuir model: r 2 = 0.74 (soil 2). In the case of PCE, the best correlations were for the linear model: r 2 = 0.96 (soil 1) and r 2 = 0.98 (soil 2). Based on the fitted sorption models, retardation factors were calculated for both contaminants and aquifer materials (soils 1 and 2) (Table 6).

Sorption isotherms of TCE and PCE for investigated soils 1—Henry’s, 2—Langmuir’s, 3—Freunlich’s, 4—observed (measured) values
Column experiments
In the column experiment, 11 BTCs were registered (Fig. 5). Based on them, retardation coefficients (R) for both contaminants and aquifer materials were calculated (see Table 6). Since the samples were taken every ca. 8 min, the R values were calculated for the average time of sampling. The calculated R values for soil 2 are higher than for soil 1. They are also higher comparing to the batch test results. Furthermore, when both contaminants were present in the column, TCE influenced on PCE, so that the R values of both contaminants are significantly higher (Table 6). Column experiments are more accurate, thus the results represent better sorption processes in the aquifer.

Observation (circles) and nonlinear least squares fits (lines) for soil 1 and 2
Batch and column studies indicate that both materials (medium and fine sand) are characterized by higher retardation of PCE than of TCE in the studied sandy aquifer. Moreover, higher R values can be observed for fine sand (soil 2), which is probably due to higher content of grains with small diameter in comparison to medium sand (soil 1). Thus, sorption of TCE and PCE on both aquifer materials can be classified as ‘small sorption capacity’ according to the classification presented by Witczak et al. (2013) (Tables 7). Only when both contaminants are present together, the sorption capacity is between ‘small’ and ‘medium’. The results obtained are within the range of those reported in literature (see Table 2).
Biodegradation studies
The maximum concentrations in analysed samples reached 807 and 387 [μg/L] for TCE and PCE, respectively. In two monitoring points (H-1, S-9) no TCE and PCE and in two others (S4c, S6b) no PCE were detected; therefore, no isotope analysis was performed in these points. For the remaining points. the measured average δ13C values were for TCE within the range of 23.6 ÷ −24.3 ‰ and for PCE of −26.3 ÷ 27.7 ‰ (Table 8, Fig. 6). In the case of TCE, the difference between the maximum and minimum δ13C may not be considered as significant, because it is less than ±1 ‰. For PCE, the difference is about ±1.4 ‰, still too low to clearly indicate biodegradation to occur. Nevertheless, PCE concentrations were mostly relatively low; therefore, the mean δ13C was calculated with higher standard deviation, so the reliability of δ13C is not sufficient enough (allowed standard deviation is ≤0,5 ‰). Neither the correlation of δ13C and the concentration of chlorinated solvents nor the distance from source was observed. Therefore, it was concluded that there is inadequate evidence for biodegradation of TCE and PCE via reductive dehalogenation to occur at the studied site. Hence, the main processes leading to the observed decrease of TCE and PCE concentrations in groundwater are dilution and sorption.

Box plots of δ13C
Conclusions
Two experiments (batch and column) were used to estimate sorption of TCE and PCE in the sandy aquifer. Results obtained indicate that sorption (and consequently retardation) of both contaminants are low. Batch tests resulted in lower K d and R values compared to the column experiment. Generally, TCE is characterized by lower retardation than PCE in the studied aquifer. Furthermore, in presence of both contaminants simultaneously in the column TCE influenced sorption of PCE, resulting in almost two times higher R values for both contaminants.
Estimated sorption parameters reflect the results described in literature for the similar types of experiments and soils. They indicate that transport of TCE and PCE in groundwater is controlled by properties of the aquifer materials, such as particle size and TOC.
There is an inadequate evidence for biodegradation of TCE and PCE via reductive dehalogenation to occur in the studied aquifer; thus, this parameter may be neglected in the modelling studies.
The results of the study allow for better understanding of TCE and PCE migration in the studied aquifer. They provide input parameters for numerical contaminant transport modelling to estimate risk for the receptors (municipal waterworks) and support the decision-making process for selecting an effective remediation strategy.
Results obtained in the presented study are used in fate and transport model that is recently being built using Visual MODFLOW software. MTD3MS code was selected in our study to model transport processes of chlorinated solvents in groundwater. For this code, sorption and reaction parameters are assigned on a cell-by-cell basis. As input parameters to characterize TCE and PCE transport in the aquifer, the following parameters were specified:
-
(i)
The sorption isotherm type—Henry’s
-
(ii)
the partition coefficient (K d) (calculated from R factor based on an isotherm type and corresponding equation), varying from 3.7°10−11 to 8.5°10−11 L/μg and from 1.21°10−11 to 2.10°10−11 L/μg for TCE and PCE, respectively (depending on the layer)
-
(iii)
the first-order reaction (biodegradation) rate for the dissolved (or mobile) phase—omitted from the fate and transport model
References
Abulaban A, Nieberb JL, Misra D (1998) Modeling plume behavior for nonlinearly sorbing solutes in saturated homogeneous porous media. Adv Water Res 21:487–498
Akyol NH, Yolcubal I, Yüksel DI (2011) Sorption and transport of trichloroethylene in caliche soil. Chemosphere 82:809–16
Bear J, Cheng A (2010) Modeling groundwater flow and contaminant transport. Springer Netherlands, Dordrecht
Benker E, Davis GB, Barry DA (1998) Estimating the retardation coefficient of trichloroethene for a sand aquifer low in sediment organic carbon—a comparison of methods. J Contam Hydrol 30:157–178
Bombach P, Richnow HH, Kästner M, Fischer A (2010) Current approaches for the assessment of in situ biodegradation. Appl Microbiol Biotechnol 86:839–52
Bourg ACM, Philippe D, Mouvet C, Sauty JP (1993) Migration of chlorinated hydrocarbon solvents through Coventry sandstone rock columns. J Hydrol 149:183–207
Brusseau ML (1992) Non equilibrium transport of organic chemicals: the impact of porewater velocity. J Contam Hydrol 9:353–368
Brusseau ML, Larsen T, Christensen TH (1991) Rate-limited sorption and nonequilibrium transport of organic chemicals in low organic carbon aquifer materials. Water Resour Res 27:1137–1145
Brusseau ML, Schnaar G, Johnson GR, Russo AE (2012) Nonideal transport of contaminants in heterogeneous porous media: 10. Impact of co-solutes on sorption by porous media with low organic-carbon contents. Chemosphere 89:1302–6
Cichocka D, Siegert M, Imfeld G, Andert J, Beck K, Diekert G, Richnow H-H, Nijenhuis I (2007) Factors controlling the carbon isotope fractionation of tetra- and trichloroethene during reductive dechlorination by Sulfurospirillum ssp. and Desulfitobacterium sp. strain PCE-S. FEMS Microbiol. Ecol 62:98–107
Curtis GP, Roberts PV, Reinhard M (1986) A natural gradient experiment on solute transport in a sand aquifer. 4. Sorption of organic solutes and its influence on mobility. Water Resources Research 22:2059–67
Cwiertny DM, Scherer MM (2010) Abiotic Processes affecting the remediation of chlorinated solvents. In: Stroo HF, Ward CH (eds) In situ remediation of chlorinated solvent plumes. Springer, New York, pp 69–108
Delle Site A (2001) Factors affecting sorption of organic compounds in natural sorbent/water systems and sorption coefficients for selected pollutants. A review. J Phys Chem Ref Data 30(1):187–253
Dowgiałło J (ed) (2002) Hydrogeological Lexicon. Polish Geological Survey, Warszawa (in Polish)
Dridi L, Poller I, Razakarisoa O, Schäfer G (2009) Characterization of a DNAPL source zone in a porous aquifer using the partitioning interwell tracer test and an inverse modeling approach. J Contam Hydrol 107:22–44
European Union Directive. Directive 2000/60/EC of the European parliament and the Council of 23 October 2000 establishing a framework for Community action in the field of water policy. Official journal L 327, 22.12.2000
Hellerich LA, Nikolaidis NP (2005) Sorption studies of mixed chromium and chlorinated ethenes at the field and laboratory scales. J Environ Manag 75:77–88
Hinz C (2001) Description of sorption data with isotherm equations. Geoderma 99:225–243
Hunkeler A, Aravena R (2010) Investigating the origin and fate of organic contaminants in groundwater using stable isotope analysis. In: Aeolion CM, Höhener P, Hunkeler D, Aravena R (eds) Environmental isotopes in biodegradation and bioremediation. CRC Press, Boca Raton, pp 249–291
Hunkeler D, Meckenstock RU, Sherwood Lollar B, Schmidt TC, Wilson JT (2008) A Guide for Assessing Biodegradation and Source Identification of Organic Ground Water Contaminants using Compound Specific Isotope Analysis (CSIA). US EPA, Ada
Imfeld G, Nijenhuis I, Nikolausz M, Zeiger S, Paschke H, Drangmeister J, Grossmann J, Richnow HH, Weber S (2008) Assessment of in situ degradation of chlorinated ethenes and bacterial community structure in a complex contaminated groundwater system. Water Res 42(4–5):871–82
Jo Y-J, Lee J-Y, Yi M-J, Kim H-S, Lee K-K (2010) Soil contamination with TCE in an industrial complex: contamination levels and implication for groundwater contamination. Geosci J 14:313–320
Karickhoff SW, Brown DS, Scott TA (1979) Sorption of hydrophobic pollutants on natural sediments. Water Res 13(3):241–248
Kästner M, Fischer A, Nijenhuis I, Geyer R, Stelzer N, Bombach P, Tebbe CC, Richnow HH (2010) Assessment of microbial in situ activity in contaminated aquifers. Eng Life Sci 6(3):234–251
Kitanidis PK, McCarty PL (eds) (2012) Delivery and mixing in the subsurface: processes and design principles for in situ remediation, SERDP/ESTCP environmental remediation technology. Springer, New York
Kret E, Kiecak A, Malina G, Szklarczyk T (2011) Evaluation of Quaternary groundwater chemical status exploited by waterworks in Nowa Dęba. Bull Polish Geological Inst 445:329–336 (in Polish)
Kret E, Grajales Mesa SJ, Kiecak A, Malina G (2013) Tri- and tetrachloroethylene in groundwater exploited by a municipal waterworks: numerical modelling and mitigative measures. In: Borchers U, Gray J, Thompson KC (eds) Water contamination emergencies: managing the threats. RSC Publishing, Cambridge, pp 71–85
Kueper BH, Wealthall GP, Smith JWN, Leharne SA, Lerner DN (2003) An illustrated handbook of DNAPL transport and fate in the subsurface. Environmental Agency, Bristol
Larsen T, Kjeldsen P, Christensen TH, Skov B, Refstrup M (1989) Sorption of specific organics in low concentrations on aquifer materials of low organic carbon content: laboratory experiments. In: Kobus HE, Kinzelbach W (eds) Contaminant Transport in Groundwater. Balkema, Rotterdam, pp 133–140
Limousin G, Gaudet J-P, Charlet L, Szenknect S, Barthès V, Krimissa M (2007) Sorption isotherms: a review on physical bases, modeling and measurement. Appl Geochem 22:249–275
Ma C, Wu Y, Sun C, Lee L (2007) Adsorption characteristics of perchloroethylene in natural sandy materials with low organic carbon content. Environ Geol 52:1511–1519
Marciniak M, Małoszewski P, Okońska M (2006) The influence of column experiment scale effect on the tracer migration parameter identification by the methods of analytical solutions and numerical modelling. Geologos 10:168–187 (in Polish)
Nijenhuis I, Nikolausz M, Köth A, Felföldi T, Weiss H, Drangmeister J, Grossmann J, Kästner M, Richnow HH (2007) Assessment of the natural attenuation of chlorinated ethenes in an anaerobic contaminated aquifer in the Bitterfeld/Wolfen area using stable isotope techniques, microcosm studies and molecular biomarkers. Chemosphere 67(2):300–11
Nijenhuis I, Schmidt M, Pellegatti E, Paramatti E, Richnow HH, Gargini A (2013) A stable isotope approach for source apportionment of chlorinated ethene plumes at a complex multi-contamination events urban site. J Contam Hydrol 153:92–105
Rifai HS, Borden RC, Newell CJ, Bedient PB (2010) Modeling remediation of chlorinated solvent plumes. In: Stroo HF, Ward CH (eds) In situ remediation of chlorinated solvent plumes. Springer, New York, pp 145–184
Rivett MO, Allen-King RM (2003) A controlled field experiment on groundwater contamination by a multicomponent DNAPL: dissolved-plume retardation. J Contam Hydrol 66:117–46
Ruffino B, Zanetti M (2009) Adsorption study of several hydrophobic organic contaminants on an aquifer material. Am J Environ Sci 5:507–515
Rüttinger S, Tobschall HJ, Breiter R, Hirsch K, Avila S, Neeße T, Bayer M (2006) Natural Attenuation-Untersuchungen an einem mit LCKW kontaminierten Altdeponiestandort. Grundwasser 11:184–193
Salaices Avila MA, Breiter R, Neeße Th (2002) Column studies for the analysis of VOC sorption in soils at low concentrations. http://www.altlasten-bayern.de/download/NA2002_TP3_2_Poster_European_Conference.pdf
Schuler G, Eberl J, Martens S (2006) Dimensionierung wasserwirtschaftlicher Vorranggebiete in Kies-Grundwasserleitern unter Berücksichtigung von Schadstofftransport. Grundwasser 11:276–285
Singh A, Ward OP (eds) (2004) Biodegradation and bioremediation. Springer, Berlin
Strickland T, Korleski C (2007) Technical Guideance Manual for Groundwater Investigations,.Chapter 14. Ground Water Flow and Fate and Transport Modeling. State of Ohio Environmental Protection Agency, Division of Drinking and Ground Waters, Ohio, USA
Stroo HF, Ward CH (eds) (2010) In situ remediation of chlorinated solvent plumes. Springer, New York
Suarez MP, Rifai HS (1999) Biodegradation rates for fuel hydrocarbons and chlorinated solvents in groundwater. Bioremediat J 3(4):337–362
Tang G, Mayes MA, Parker JC, Yin XL, Watson DB, Jardine PM (2009) Improving parameter estimation for column experiments by multi-model evaluation and comparison. J Hydrol 376:567–578
Tang G, Mayes MA, Parker JC, Jardine PM (2010) CXTFIT/Exel—a modular adaptable code for parameter estimation, sensitivity analysis and uncertainty analysis for laboratory and field tracer experiments. Comput Geosci 36(9):1200–1209
Van Breukelen BM, Hunkeler D, Volkering F (2005) Quantification of sequential chlorinated ethene degradation by use of a reactive transport model incorporating isotope fractionation. Environ Sci Technol 39(11):4189–97
Van der Heijde P, Bachmat Y, Bredehoeft J, Andrews B, Holtz D, Sebastian S (1980) Groundwater Management: The Use of Numerical Models, Water Resources Monograph, AGU, Washington, DC
Wiedemeier TH, Swanson MA, Moutoux A, Gordon K, Wilson JT, Wilson BH, Kampbell DH, Haas PE, Miller RN, Hansen JE, Chapelle FH (1998) Technical Protocol for Evaluating Natural Attenuation of Chlorinated Solvents in Ground Water. National Risk Management Research Laboratory, Office of Research and Development, U.S. Environmental Protection Agency. EPA/600/R-98/128, Cincinnati
Wiedemeier TH, Rifai HS, Newell CJ, Wilson JT (1999) Natural attenuation of fuels and chlorinated solvents in the subsurface. Wiley, New York
Wilson JT, Enfield CG, Dunlap WJ, Cosby RL, Foster DA, Baskin LB (1981) Transport and fate of selected organic pollutants in a sandy soil. J Environ Qual 10(4):501–506
Witczak S, Kania J, Kmiecik E (2013) Guidebook on selected physical and chemical indicators of groundwater contamination and methods of their determination. Inspectorate for Environmental Protection, Warszawa (in Polish)
Zhao X, Wallace RB, Hyndman DW, Dybas MJ, Voice TC (2005) Heterogeneity of chlorinated hydrocarbon sorption properties in a sandy aquifer. J Contam Hydrol 78:327–42
Zheng C, Wang PP (1999) MT3DMS. A modular three-dimensional multispecies transport model for a simulation of advection, dispersion and chemical reactions of contaminants in groundwater systems. U.S. Army of Corps of engineers, Washington, DC
Acknowledgments
The financial support for this study was provided by the State Committee for Scientific Research of the Ministry of Scientific Research and Information Technology (No. NN525349238) and partially by AGH University of Science and Technology (No. 11.11.140.026) and Deutsche Bundesstiftung Umwelt (DBU Scholarship Exchange Programme with CEE Countries). We wish to thank WESSLING Polska Sp. z o.o., particularly Ms. A. Raj for her help with the experimental design, as well as S.J. Grajales Mesa and A. Kawecka for their assistance with the column experiments.
Author information
Authors and Affiliations
Corresponding author
Additional information
Responsible editor: Philippe Garrigues
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article

Cite this article
Kret, E., Kiecak, A., Malina, G. et al. Identification of TCE and PCE sorption and biodegradation parameters in a sandy aquifer for fate and transport modelling: batch and column studies. Environ Sci Pollut Res 22, 9877–9888 (2015). https://doi.org/10.1007/s11356-015-4156-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11356-015-4156-9